Traceless Solid-Phase Synthesis of [6,7,8 + 5,6,7]-Fused Molecular Frameworks


Abstract
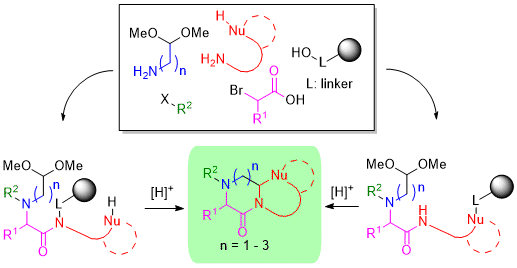
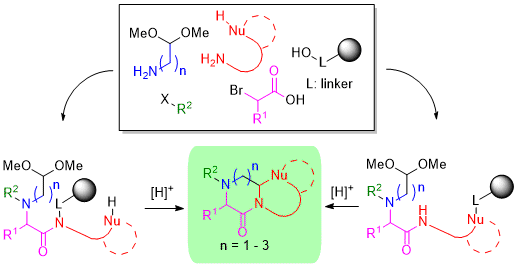
:
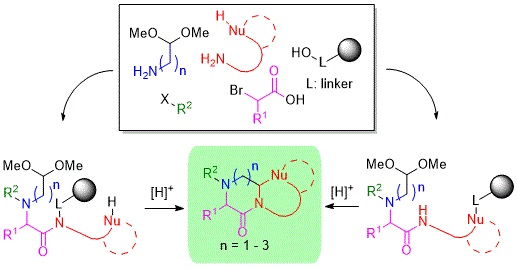
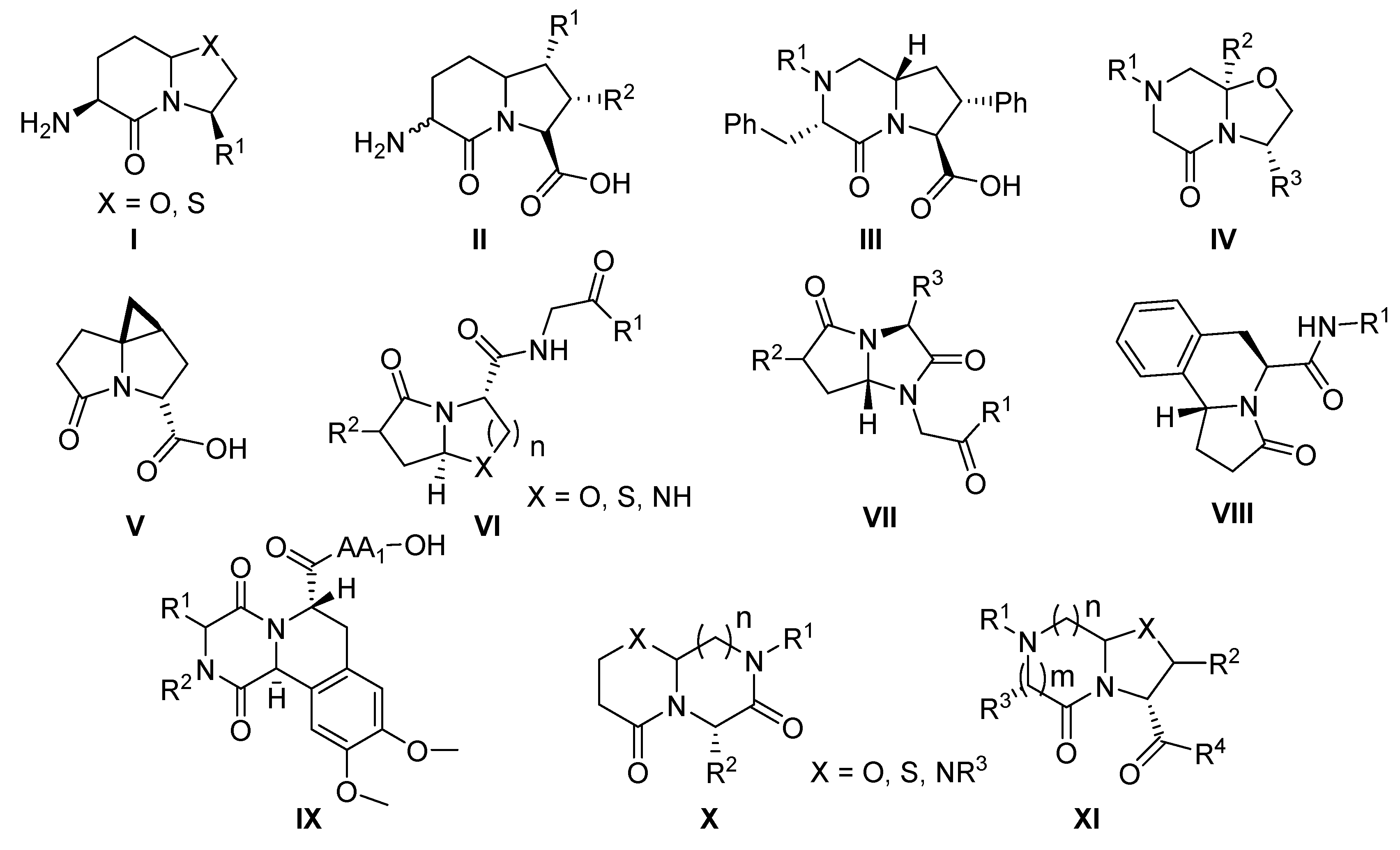
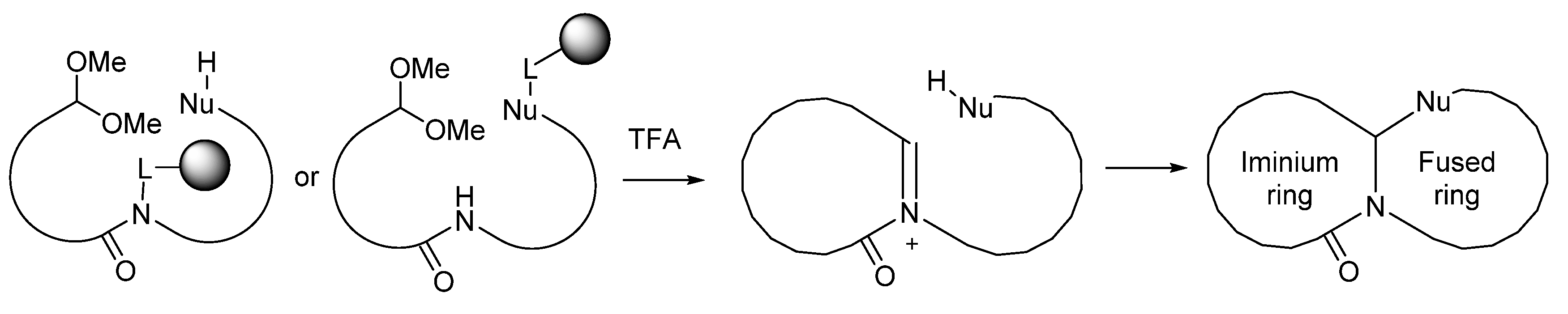
1. Introduction
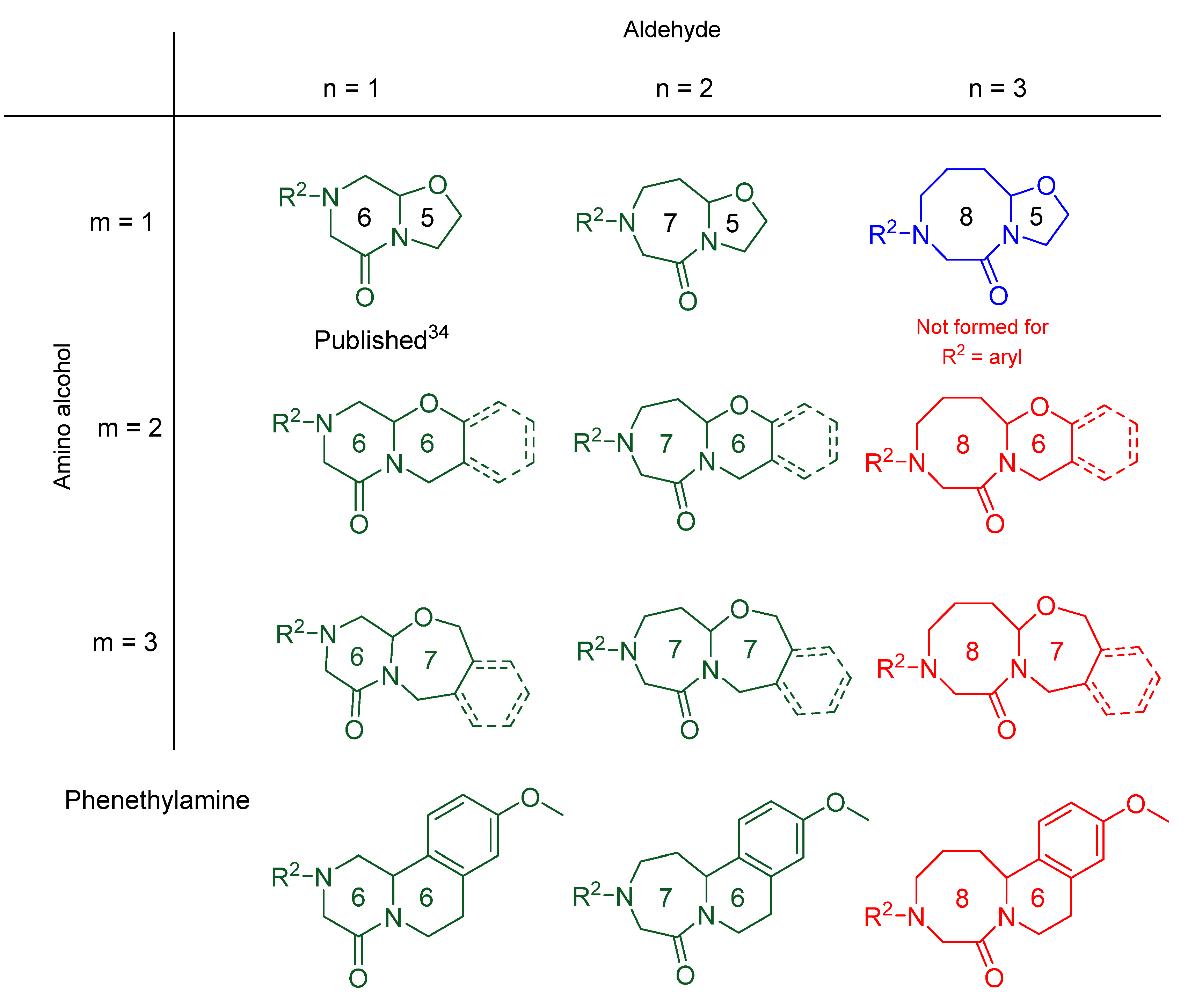
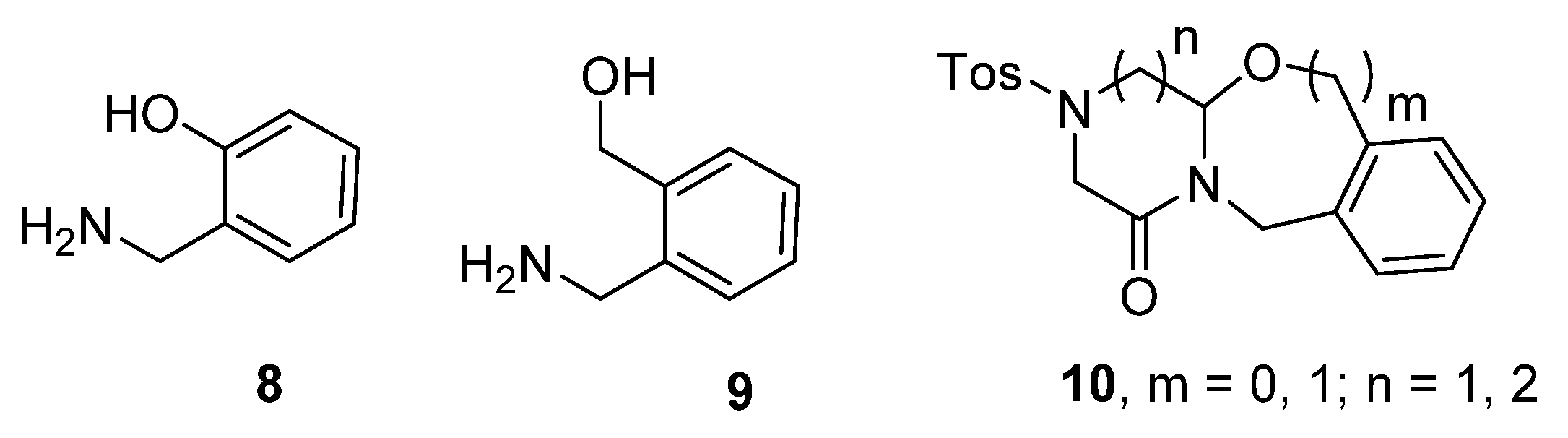
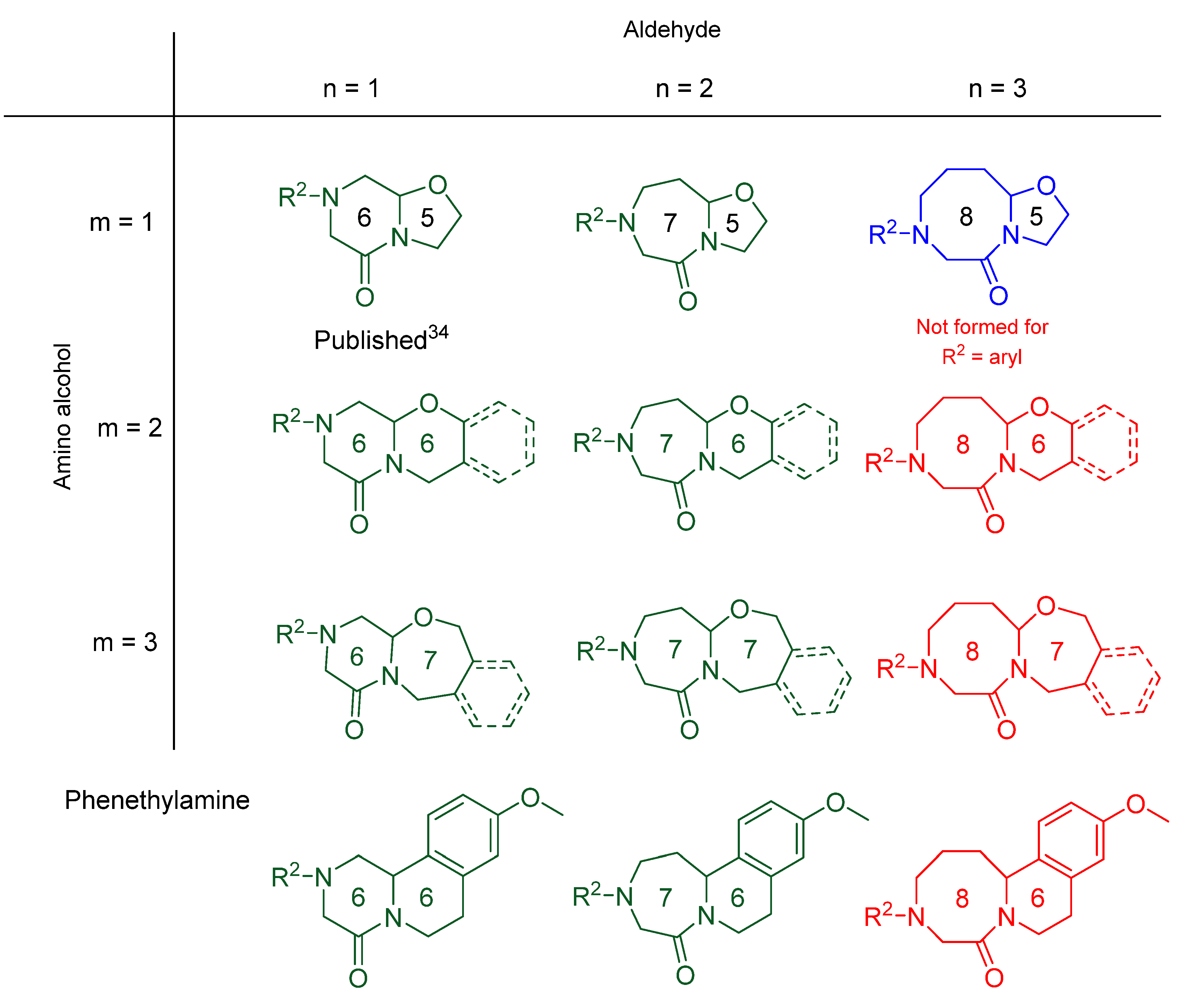
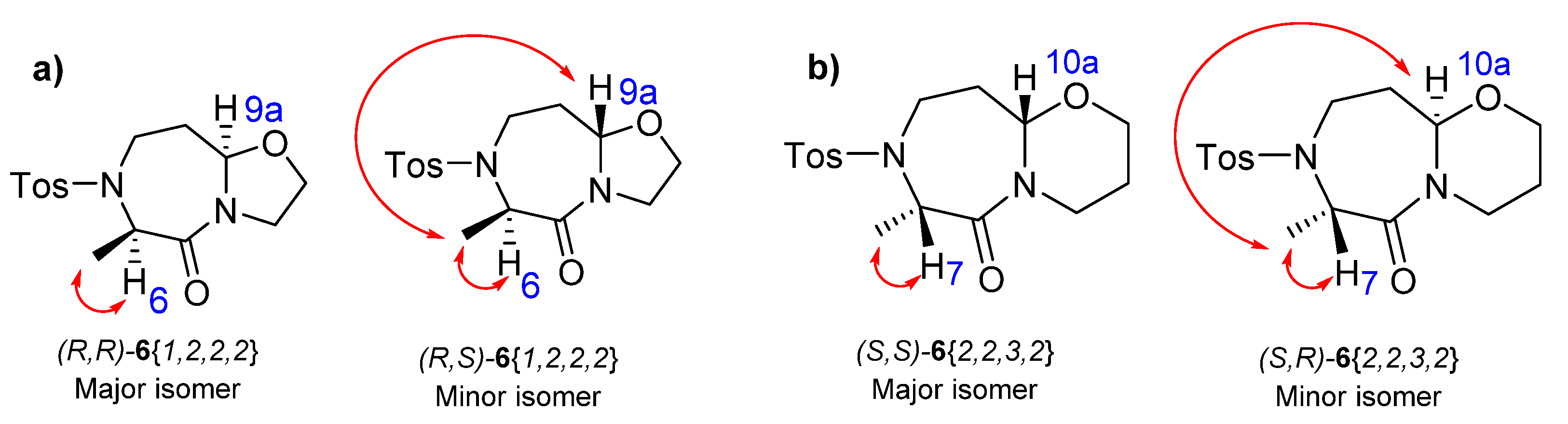
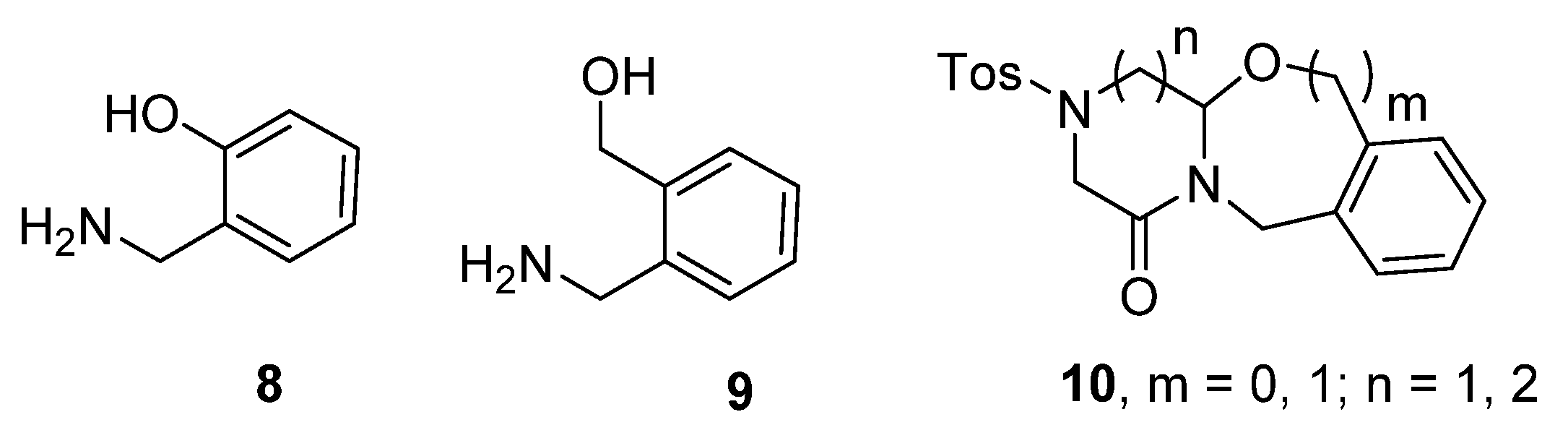
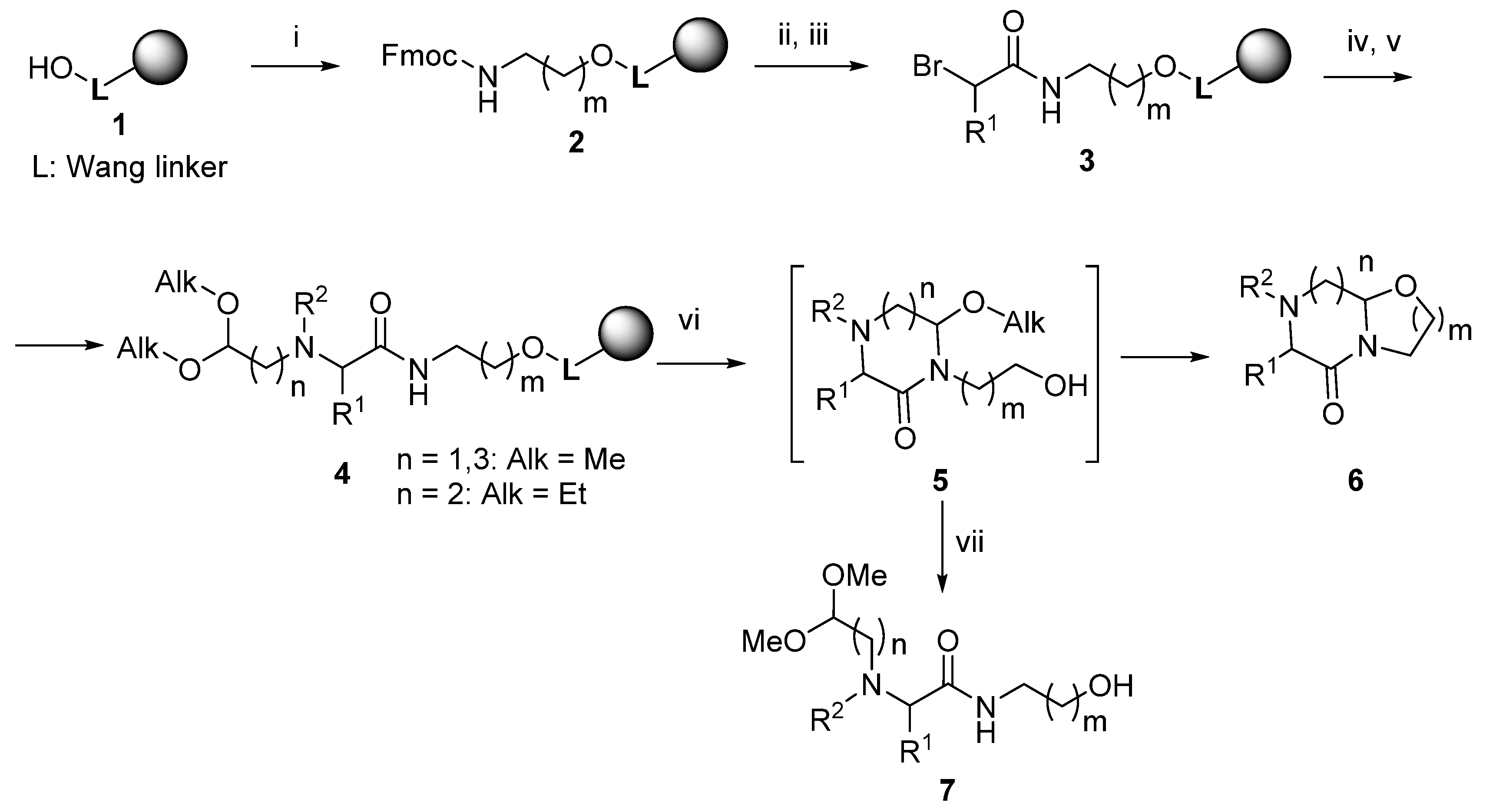
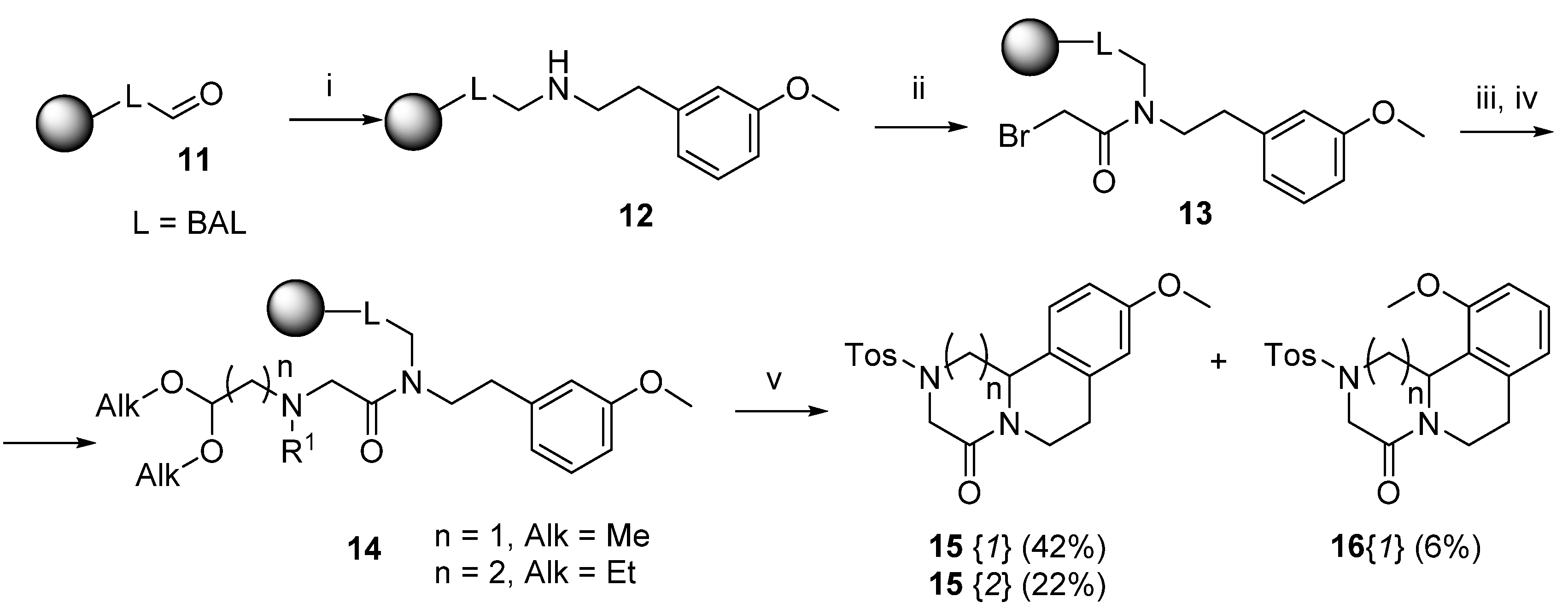
2. Results
3. Materials and Methods
3.1. General
3.2. Reaction of Wang Resin 1 with Trichloroacetonitrile and Fmoc-amino Alcohol (Resin 2)
3.3. Acylation with α-Bromocarboxylic Acids (Resins 3 And 13)
3.4. Reaction with Amino-aldehyde Dialkyl Acetal and N-Derivatization (Resin 4)
3.5. Acid-Mediated Cleavage, Cyclization and Isolation (Compounds 6 and 7)
3.6. Reductive Amination of the BAL Resin (Resin 12)
3.7. Reaction with Amino-aldehyde Dialkyl Acetal and N-Derivatization (Resin 14) Acid-mediated Cleavage, Cyclization and Isolation (Compounds 15 and 16)
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M.; Snader, K.M. The Influence of Natural Products Upon Drug Discovery. Nat. Prod. Rep. 2000, 17, 215–234. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M.; Snader, K.M. Natural Products as Sources of New Drugs Over the Period 1981–2002. J. Nat. Prod. 2003, 66, 1022–1037. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Vallejo, F.; Giulianotti, M.A.; Houghten, R.A.; Medina-Franco, J.L. Expanding the Medicinally Relevant Chemical Space With Compound Libraries. Drug Discov. Today 2012, 17, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Lovering, F.; Bikker, J.; Humblet, C. Escape From Flatland: Increasing Saturation As an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, S.; Bon, R.S.; Kumar, K.; Waldmann, H. Biology-Oriented Synthesis. Angew. Chem. Int. Ed. 2011, 50, 10800–10826. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.; Wetzel, S.; Kumar, K.; Waldmann, H. Biology-Inspired Synthesis of Compound Libraries. Cell. Mol. Life Sci. 2008, 65, 1186–1201. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.J.; Beckmann, H.S.G.; Spring, D.R. Diversity-Oriented Synthesis: Producing Chemical Tools for Dissecting Biology. Chem. Soc. Rev. 2012, 41, 4444–4456. [Google Scholar] [CrossRef] [PubMed]
- La Venia, A.; Ventosa-Andrés, P.; Krchnak, V. Peptidomimetics via Iminium Chemistry on Solid Phase: Single, Fused, and Bridged Heterocycles. Peptidomimetics II Top. Heterocycl. Chem. 2017, 49, 105–126. [Google Scholar]
- Maryanoff, B.E.; Zhang, H.C.; Cohen, J.H.; Turchi, I.J.; Maryanoff, C.A. Cyclizations of N-Acyliminium Ions. Chem. Rev. 2004, 104, 1431–1628. [Google Scholar] [CrossRef] [PubMed]
- Yazici, A.; Pyne, S.G. Intermolecular Addition Reactions of N-Acyliminium Ions (Part I). Synthesis 2009, 3, 339–368. [Google Scholar]
- Yazici, A.; Pyne, S.G. Intermolecular Addition Reactions of N-Acyliminium Ions (Part II). Synthesis 2009, 4, 513–541. [Google Scholar]
- Le Quement, S.T.; Nielsen, T.E.; Meldal, M. Scaffold Diversity Through Intramolecular Cascade Reactions of Solid-Supported Cyclic N-Acyliminium Intermediates. J. Comb. Chem. 2007, 9, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Ventosa-Andres, P.; La-Venia, A.; Ripoll, C.A.B.; Hradilova, L.; Krchnak, V. Synthesis of Nature-Inspired Medium-Sized Fused Heterocycles From Amino Acids. Chem. Eur. J. 2015, 21, 13112–13119. [Google Scholar] [CrossRef] [PubMed]
- Hanessian, S.; McNaughton-Smith, G.; Lombart, H.G.; Lubell, W.D. Design and Synthesis of Conformationally Constrained Amino Acids As Versatile Scaffolds and Peptide Mimetics. Tetrahedron 1997, 53, 12789–12854. [Google Scholar] [CrossRef]
- Nielsen, T.E.; Le Quement, S.; Meldal, M. Solid-Phase Synthesis of Bicyclic Dipeptide Mimetics by Intramolecular Cyclization of Alcohols, Thiols, Amines, and Amides with N-Acyliminium Intermediates. Org. Lett. 2005, 7, 3601–3604. [Google Scholar] [CrossRef] [PubMed]
- Airiau, E.; Spangenberg, T.; Girard, N.; Schoenfelder, A.; Salvadori, J.; Taddei, M.; Mann, A. A General Approach to Aza-Heterocycles by Means of Domino Sequences Driven by Hydroformylation. Chem. Eur. J. 2008, 14, 10938–10948. [Google Scholar] [CrossRef] [PubMed]
- Bencsik, J.R.; Kercher, T.; O’Sulliva, M.; Josey, J.A. Efficient, Stereoselective Synthesis of Oxazolo[3,2-a]Pyrazin-5-Ones: Novel Bicyclic Lactam Scaffolds From the Bicyclocondensation of 3-Aza-1,5-Ketoacids and Amino Alcohols. Org. Lett. 2003, 5, 2727–2730. [Google Scholar] [CrossRef] [PubMed]
- Ciofi, L.; Morvillo, M.; Sladojevich, F.; Guarna, A.; Trabocchi, A. Skeletal Diversity by Sequential One-Pot and Stepwise Routes Using Morpholine Ester Scaffolds. Tetrahedron Lett. 2010, 51, 6282–6285. [Google Scholar] [CrossRef]
- Estiarte, M.A.; Rubiralta, M.; Diez, A.; Thormann, M.; Giralt, E. Oxazolopiperidin-2-Ones As Type II’ Beta-Turn Mimetics: Synthesis and Conformational Analysis. J. Org. Chem. 2000, 65, 6992–6999. [Google Scholar] [CrossRef] [PubMed]
- Wirt, U.; Frohlich, R.; Wunsch, B. Asymmetric Synthesis of 1-Substituted Tetrahydro-3-Benzoazepines. Tetrahedron: Asymmetry 2005, 16, 2199–2202. [Google Scholar] [CrossRef]
- Vagner, J.; Qu, H.; Hruby, V.J. Peptidomimetics, a Synthetic Tool of Drug Discovery. Curr. Opin. Chem. Biol. 2008, 12, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Lawandi, J.; Toumieux, S.; Seyer, V.; Campbell, P.; Thielges, S.; Juillerat-Jeanneret, L.; Moitessier, N. Constrained Peptidomimetics Reveal Detailed Geometric Requirements of Covalent Prolyl Oligopeptidase Inhibitors. J. Med. Chem. 2009, 52, 6672–6684. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yang, J.; Ying, J.; Xiong, C.; Zhang, J.; Cai, C.; Hruby, V.J. Stereoselective Synthesis of Dipeptide Beta-Turn Mimetics: 7-Benzyl and 8-Phenyl Substituted Azabicyclo[4.3.0]Nonane Amino Acid Esters. J. Org. Chem. 2002, 67, 6353–6360. [Google Scholar] [PubMed]
- Tong, Y.; Fobian, Y.; Wu, M.; Boyd, N.D.; Moeller, K.D. Conformationally Constrained Substance P Analogues: The Total Synthesis of a Constrained Peptidomimetic for the Phe7-Phe8 Region. J. Org. Chem. 2000, 65, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Hanessian, S.; Buckle, R.; Bayrakdarian, M. Design and Synthesis of a Novel Class of Constrained Tricyclic Pyrrolizidinone Carboxylic Acids As Carbapenem Mimics. J. Org. Chem. 2002, 67, 3387–3397. [Google Scholar] [CrossRef] [PubMed]
- Cox, E.D.; Cook, J.M. The Pictet-Spengler Condensation: A New Direction for an Old Reaction. Chem. Rev. 1995, 95, 1797–1842. [Google Scholar] [CrossRef]
- Nielsen, T.E.; Meldal, M. Solid-Phase Synthesis of Pyrroloisoquinolines Via the Intramolecular N-Acyliminium Pictet-Spengler Reaction. J. Comb. Chem. 2005, 7, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.E.; Meldal, M. Solid-Phase Intramolecular N-Acyliminium Pictet-Spengler Reactions As Crossroads to Scaffold Diversity. J. Org. Chem. 2004, 69, 3765–3773. [Google Scholar] [CrossRef] [PubMed]
- Diness, F.; Beyer, J.; Meldal, M. Solid-Phase Synthesis of Tetrahydro-Beta-Carbolines and Tetrahydroisoquinolines by Stereoselective Intramolecular N-Carbamyliminium Pictet-Spengler Reactions. Chem. Eur. J. 2006, 12, 8056–8066. [Google Scholar] [CrossRef] [PubMed]
- Petersen, R.; LeQuement, S.T.; Nielsen, T.E. Synthesis of a Natural Product-Like Compound Collection Through Oxidative Cleavage and Cyclization of Linear Peptides. Angew. Chem. Int. Ed. 2014, 53, 11778–11782. [Google Scholar] [CrossRef] [PubMed]
- Schutznerova, E.; Oliver, A.G.; Zajicek, J.; Krchnak, V. Polymer-Supported Stereoselective Synthesis of (1S,5S)-6-Oxa-3,8-Diazabicyclo[3.2.1]Octanes. Eur. J. Org. Chem. 2013, 3158–3165. [Google Scholar] [CrossRef]
- La Venia, A.; Lemrova, B.; Krchnak, V. Regioselective Incorporation of Backbone Constraints Compatible With Traditional Solid-Phase Peptide Synthesis. ACS Comb. Sci. 2013, 15, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Ventosa-Andres, P.; Barea Ripoll, C.A.; La-Venia, A.; Krchnak, V. Solid-Phase Synthesis of Fused 1,4-Diazepanone Peptidomimetics Via Tandem N-Iminium Ion Cyclization-Nucleophilic Addition. Tetrahedron Lett. 2015, 56, 5424–5428. [Google Scholar] [CrossRef]
- La Venia, A.; Dolensky, B.; Krchnak, V. Polymer-Supported Stereoselective Synthesis of Tetrahydro-2H-Oxazolo[3,2-a]Pyrazin-5(3H)-Ones From N-(2-Oxo-Ethyl)-Derivatized Dipeptides Via Eastbound Iminiums. ACS Comb. Sci. 2013, 15, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Hanessian, S.; Xie, F. Polymer-Bound P-Alkoxybenzyl Trichloracetimidates: Reagents for the Protection of Alcohols as Benzyl Ethers on Solid-Phase. Tetrahedron Lett. 1998, 39, 733–736. [Google Scholar] [CrossRef]
- Cayley, A.N.; Gallagher, K.A.; Menard-Moyon, C.; Schmidt, J.P.; Diorazio, L.J.; Taylor, R.J.K. Preparation of Novel Polycyclic Heterocycles Using a Tin(II) Chloride Dihydrate-Mediated Deacetalisation-Bicyclisation Sequence. Synthesis 2008, 3846–3856. [Google Scholar]
- Reid, M.; Taylor, R.J.K. A Stannous Chloride-Induced Deacetalisation–Cyclisation Process to Prepare the ABC Ring System of ‘Upenamide. Tetrahedron Lett. 2004, 45, 4181–4183. [Google Scholar] [CrossRef]
- Ladziat, V. Recent Applications of Rare-Earth Metal(III) Triflates in Cycloaddition and Cyclization Reactions. Arkivoc 2014, 2014, 307–336. [Google Scholar] [CrossRef]
- Marti-Centelles, V.; Pandey, M.D.; Burguete, M.I.; Luis, S.V. Macrocyclization Reactions: The Importance of Conformational, Configurational, and Template-Induced Preorganization. Chem. Rev. 2015, 115, 8736–8834. [Google Scholar] [CrossRef] [PubMed]
- Vassilikogiannakis, G.; Margaros, I.; Tofi, M. Olefin Metathesis: Remote Substituents Governing the Stereoselectivity of 11-Membered-Ring Formation. Org. Lett. 2004, 6, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.J.; Alsina, J.; Songster, M.F.; Vagner, J.; Albericio, F.; Barany, G. Backbone Amide Linker (BAL) Strategy for Solid-Phase Synthesis of C-Terminal-Modified and Cyclic Peptides 1, 2, 3. J. Am. Chem. Soc. 1998, 120, 5441–5452. [Google Scholar] [CrossRef]
- Krchnak, V.; Padera, V. The Domino Blocks: A Simple Solution for Parallel Solid Phase Organic Synthesis. Bioorg. Med. Chem. Lett. 1998, 22, 3261–3264. [Google Scholar] [CrossRef]
Sample Availability: Not available. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry * | Compound | m | n | R1 | R2 | Rings | Purity [%] a | Yield [%] b |
|---|---|---|---|---|---|---|---|---|
| 1 | 6{1,2,1,1} | 1 | 2 | H | Ns | 7 + 5 | 94 | 70 |
| 2 | 6{1,2,1,2} | 1 | 2 | H | Tos | 7 + 5 | 93 | 70 |
| 3 | 6{1,2,1,3} | 1 | 2 | H | 2-NO2-4-CF3-C6H3 | 7 + 5 | 68 | 64 |
| 4 | 6{1,3,1,1} | 1 | 3 | H | Ns | 8 + 5 | 86 | 66 |
| 5 | 6{1,3,1,2} | 1 | 3 | H | Tos | 8 + 5 | 53 | 21 |
| 6 | 6{2,1,1,3} | 2 | 1 | H | 2-NO2-4-CF3-C6H3 | 6 + 6 | 97 | 62 |
| 7 | 6{2,2,1,1} | 2 | 2 | H | Ns | 7 + 6 | 98 | 83 |
| 8 | 6{2,2,1,2} | 2 | 2 | H | Tos | 7 + 6 | 97 | 90 |
| 9 | 6{2,2,1,3} | 2 | 2 | H | 2-NO2-4-CF3-C6H3 | 7 + 6 | 95 | 80 |
| 10 | 6{3,1,1,3} | 3 | 1 | H | 2-NO2-4-CF3-C6H3 | 6 + 7 | 86 | 84 |
| 11 | 6{3,2,1,2} | 3 | 2 | H | Tos | 7 + 7 | 72 | 54 |
| 12 | 6{3,2,1,3} | 3 | 2 | H | 2-NO2-4-CF3-C6H3 | 7 + 7 | 88 | 44 |
| Entry | Compound | m | n | R1 | Rings | Purity [%] a | Yield [%] b |
|---|---|---|---|---|---|---|---|
| 1 | (R,R)-6{1,2,2,2} | 1 | 2 | (R)-Me | 7 + 5 | 85 | 49 |
| 2 | (R,S)-6{1,2,2,2} | 1 | 2 | (R)-Me | 7 + 5 | 15 | 6 |
| 3 | (S,S)-6{1,2,3,2} | 1 | 2 | (S)-Me | 7 + 5 | 85 | 44 |
| 4 | (S,R)-6{1,2,3,2} | 1 | 2 | (S)-Me | 7 + 5 | 15 | 8 |
| 5 | (S,S)-6{2,2,3,2} | 2 | 2 | (S)-Me | 7 + 6 | 55 | 32 |
| 6 | (S,R)-6{2,2,3,2} | 2 | 2 | (S)-Me | 7 + 6 | 45 | 17 |
| Entry | Compound | m | n | Purity [%] a | Yield [%] b |
|---|---|---|---|---|---|
| 1 | 10{1,1} | 0 | 1 | 76 | 41 |
| 2 | 10{1,2} | 0 | 2 | 81 | 43 |
| 3 | 10{2,1} | 1 | 1 | 97 | 90 |
| 4 | 10{2,2} | 1 | 2 | 96 | 81 |
| Entry | Compound | n | Purity [%] a | Yield [%] b |
| 1 | 15{1} | 1 | 83 | 42 |
| 2 | 16{1} | 1 | 17 | 6 |
| 3 | 15{2} | 2 | 77 | 22 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giménez-Navarro, V.; Krchňák, V. Traceless Solid-Phase Synthesis of [6,7,8 + 5,6,7]-Fused Molecular Frameworks. Molecules 2018, 23, 1090. https://doi.org/10.3390/molecules23051090
Giménez-Navarro V, Krchňák V. Traceless Solid-Phase Synthesis of [6,7,8 + 5,6,7]-Fused Molecular Frameworks. Molecules. 2018; 23(5):1090. https://doi.org/10.3390/molecules23051090
Chicago/Turabian StyleGiménez-Navarro, Vanesa, and Viktor Krchňák. 2018. "Traceless Solid-Phase Synthesis of [6,7,8 + 5,6,7]-Fused Molecular Frameworks" Molecules 23, no. 5: 1090. https://doi.org/10.3390/molecules23051090
APA StyleGiménez-Navarro, V., & Krchňák, V. (2018). Traceless Solid-Phase Synthesis of [6,7,8 + 5,6,7]-Fused Molecular Frameworks. Molecules, 23(5), 1090. https://doi.org/10.3390/molecules23051090