General
All melting points were determined on a Büchi B-540 (Flawil, Switzerland) capillary melting point apparatus and are uncorrected. IR spectra were obtained on a Bruker ALPHA FT-IR spectrometer (Billerica, MA, USA) in KBr pellets of as a film. 1H-NMR, 13C-NMR and 19F-NMR spectra were recorded at 295 K on a Bruker Avance III HD 600 (Billerica, MA, USA) (600, 150 and 564.7 MHz for 1H-, 13C- and 19F-NMR spectra, respectively) or at ambient temperature on a Bruker Avance III 400 (Billerica, MA, USA) (400 and 100 MHz for 1H and 13C-NMR spectra, respectively) spectrometer. CDCl3 or CD3OD was used as the solvent, tetramethylsilane (TMS) for 1H, 13C-NMR or trichlorofluoromethane (CFCl3) for 19F-NMR as the internal standard. Chemical shifts (δ) and coupling constants (J) are given in ppm and in Hz, respectively. Mass spectra were recorded on a Bruker O-TOF MAXIS Impact mass spectrometer (Billerica, MA, USA) coupled with a Dionex Ultimate 3000 RS HPLC (Sunnyvale, CA, USA) system with a diode array detector. The reactions were followed by analytical thin-layer chromatography on silica gel 60 F254 (Darmstadt, Germany) and HPLC-MS on a Shimadzu LC-20 HPLC equipment (Kyoto, Japan). Purifications by flash chromatography were carried out using Merck 107736 silica gel 60 H (Darmstadt, Germany) using a hexane–ethyl acetate or dichloromethane–methanol solvent system. All reagents were purchased from commercial sources.
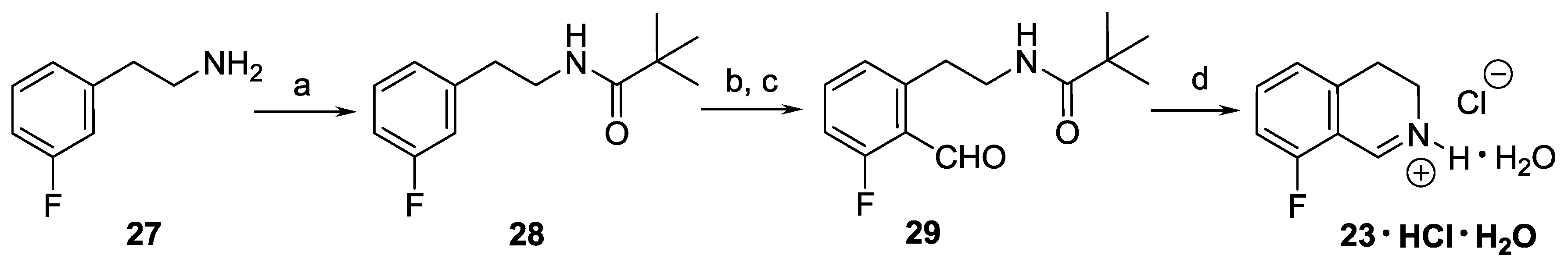
N-[2-(3-Fluorophenyl)ethyl]-2,2-dimethylpropanamide (28). Pivaloyl chloride (24.3 mL, 23.8 g, 198 mmol) in dichloromethane (40 mL) was added to a solution of 27 (23.5 mL, 25.0 g, 180 mmol) and triethylamine (30.0 mL, 21.8 g, 216 mmol) in dichloromethane (160 mL) at 0 °C. After stirring for 1 h at room temperature, the mixture was washed with an aqueous sodium hydrogencarbonate solution (5%, 3 × 70 mL). The organic layer was dried over MgSO4 and evaporated. The solid residue was recrystallized from heptane to afford the title compound (37.2 g, 93%) as a white solid. Mp 69–70 °C (heptane). IR (KBr): ν = 3348, 1633, 1535 cm−1. 1H-NMR (400 MHz, CDCl3): δ = 7.27 (td, JHH = 7.8 Hz, JHF = 6.1 Hz, 1H), 6.99–6.87 (m, 3H), 5.62 (br s, 1H), 3.49 (td, JCH2–CH2 = 6.9 Hz, JCH2–NH = 5.9 Hz, 2H), 2.82 (t, J = 6.9 Hz, 2H), 1.15 (s, 9H). 13C-NMR (150 MHz, CDCl3): δ = 178.4, 162.9 (d, JCF = 246 Hz), 141.6 (d, JCF = 7.1 Hz), 130.0 (d, JCF = 8.3 Hz), 124.5 (d, JCF = 2.7 Hz), 115.6 (d, JCF = 21.0 Hz), 113.4 (d, JCF = 20.9 Hz), 40.4, 38.6, 35.4 (d, JCF = 1.7 Hz), 27.5. 19F-NMR (564.7 MHz, CDCl3): δ = −113.7 (ddd, JFH = 9.8, 8.9, 6.1 Hz). HRMS calcd. for C13H19FNO+ ([M + H]+): 224.1445, found: 224.1446.
N-[2-(3-Fluoro-2-formylphenyl)ethyl]-2,2-dimethylpropanamide (29). A solution of BuLi (1.6 M in hexane, 42.1 mL, 67.4 mmol) was added to a solution of 28 (5.01 g, 22.5 mmol) in THF (70 mL) at −78 °C. After stirring for 2 h at −78 °C, DMF (10.4 mL, 9.84 g, 134.8 mmol) was added. The mixture was stirred for 1 h. After warming to ambient temperature, the reaction mixture was diluted with a saturated aqueous solution of ammonium chloride (40 mL), and extracted with ethyl acetate (30 and 2 × 10 mL). The combined organic layer was washed with brine (40 mL), and dried over MgSO4. The solvents were evaporated, the residue was purified by flash chromatography (5–30% ethyl acetate in hexane) to afford the title compound (3.03 g, 54%) as a pale yellow solid. Mp 86–87 °C (hexane/diethyl ether). IR (KBr): ν = 3336, 1698, 1626, 1538 cm−1. 1H-NMR (600 MHz, CDCl3): δ = 10.53 (s, 1H), 7.50 (ddd, JHH = 8.3, 7.7 Hz, JHF = 5.8 Hz, 1H), 7.10 (br d, J = 7.7 Hz, 1H), 7.07 (ddd, JHF = 10.7 Hz, JHH = 8.3, 0.9 Hz, 1H), 6.08 (br s, 1H), 3.50 (td, JCH2–CH2 = 6.9 Hz, JCH2–NH = 5.6 Hz, 2H), 3.19 (t, J = 6.9 Hz, 2H), 1.13 (s, 9H). 13C-NMR (150 MHz, CDCl3): δ = 189.9 (d, JCF = 11.8 Hz), 178.6, 166.5 (d, JCF = 258 Hz), 143.3, 135.5 (d, JCF = 10.5 Hz), 127.8 (d, JCF = 3.2 Hz), 122.7 (d, JCF = 5.2 Hz), 114.5 (d, JCF = 21.7 Hz), 40.6, 38.6, 32.8, 27.5. 19F-NMR (564.7 MHz, CDCl3): δ = −121.2 (dd, JFH = 10.7, 5.8 Hz). HRMS calcd. for C14H19FNO2+ ([M + H]+): 252.1394, found: 252.1394.
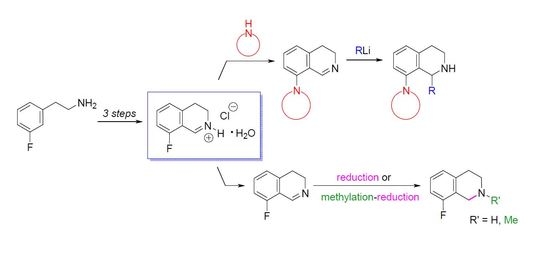
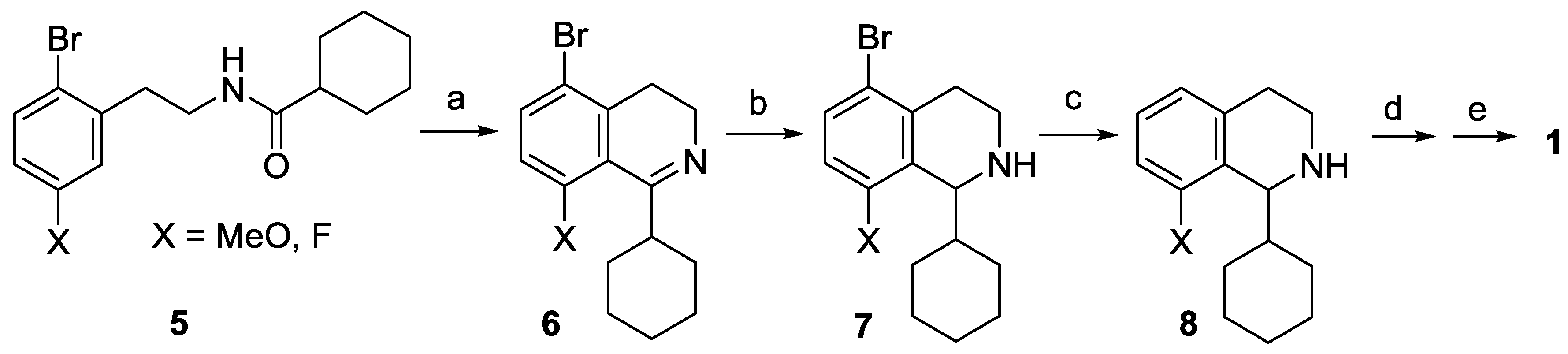
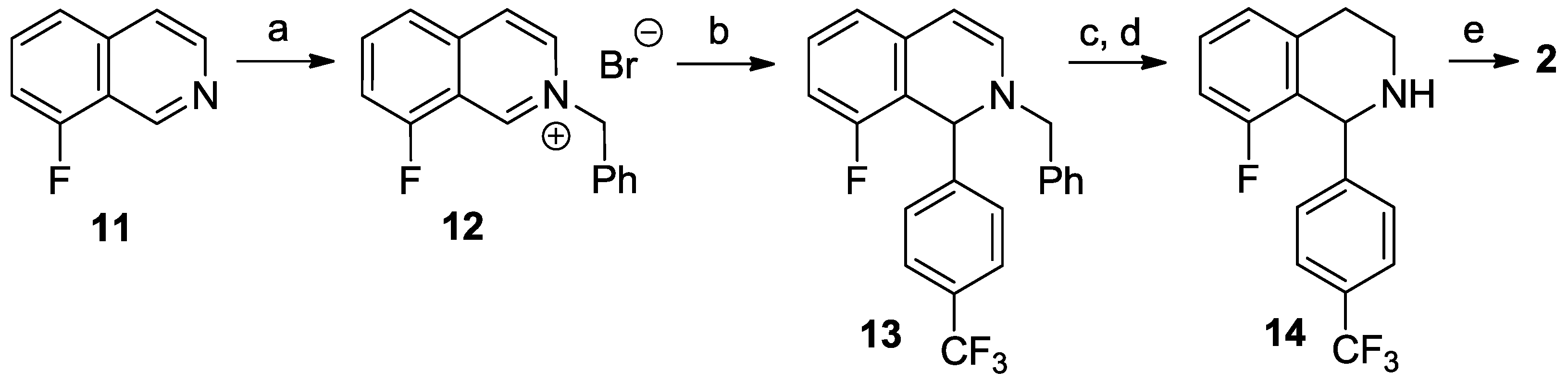
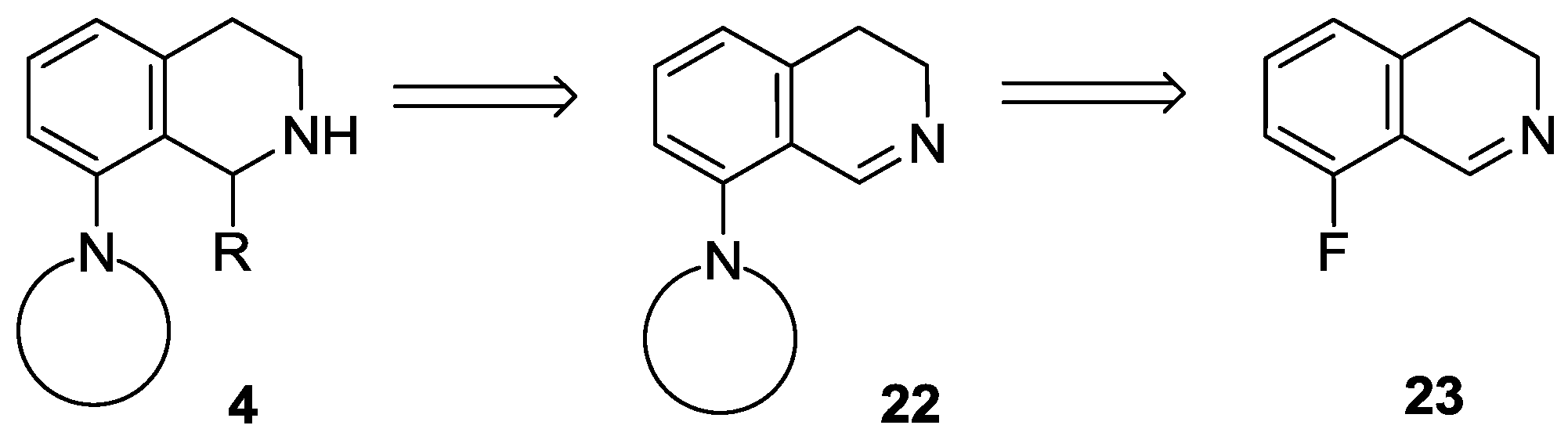
8-Fluoro-3,4-dihydroisoquinoline hydrochloride hydrate (23 ∙ HCl ∙ H2O). A solution of 29 (3.29 g, 13.1 mmol) in dichloromethane (25 mL) and aqueous hydrochloric acid (15%, 60 mL) were vigorously stirred for 24 h at 25 °C. The aqueous layer was extracted with diethyl ether (35 mL), and the organic layer was extracted with water (15 mL). The combined aqueous layer was evaporated, and the residue was recrystallized from ethanol/diethyl ether to afford the title compound (2.08 g, 78%) as a pale yellow solid. Mp 103–105 °C (ethanol/diethyl ether). IR (KBr): ν = 3471, 1664 cm−1. 1H-NMR (400 MHz, CDCl3): δ = 16.00 (br s, 1H), 9.12 (br s, 1H), 7.78 (ddd, JHH = 8.4, 7.6 Hz, JHF = 5.7 Hz, 1H), 7.23–7.18 (m, 2H), 4.11 (t, J = 8.1 Hz, 2H), 3.24 (t, J = 8.1 Hz, 2H). 13C-NMR (150 MHz, CDCl3), δ = 163.0 (d, JCF = 266 Hz), 159.2 (d, JCF = 6.4 Hz), 140.3 (d, JCF = 9.9 Hz), 138.3, 124.5 (d, JCF = 3.3 Hz), 115.6 (d, JCF = 19.5 Hz), 112.9 (d, JCF = 11.8 Hz), 41.3, 24.3 (d, JCF = 2.2 Hz). 19F-NMR (564.7 MHz, CDCl3): δ = −113.1 (dd, JFH = 8.8, 5.7 Hz). Anal. calcd. for C9H11ClFNO (203.64): Cl 17.41, N 6.88%. Found: Cl 17.83, N 7.02%.
8-Fluoro-3,4-dihydroisoquinoline (23). To a vigorously stirred mixture of 23 ∙ HCl ∙ H2O (5.89 g, 28.9 mmol) in dichloromethane (100 mL) and water (50 mL) aqueous sodium carbonate (10%, 20 mL) was added. The layers were separated and the aqueous layer was extracted with dichloromethane (3 × 25 mL). The combined organic layer was extracted with water (2 × 50 mL) and brine (50 mL) and dried over MgSO4. The solvent was evaporated to afford the title compound (3.99 g, 93%) as a pale brown oil. IR (film): ν = 1671 cm−1. 1H-NMR (400 MHz, CDCl3): δ = 8.65 (t, J = 2.2 Hz, 1H), 7.32 (ddd, JHH = 8.2, 7.5 Hz, JHF = 5.7 Hz, 1H), 6.97 (dd, JHF = 9.4 Hz, JHH = 8.2 Hz, 1H), 6.94 (d, J = 7.5 Hz, 1H), 3.78 (m, 2H), 2.74 (m, 2H). 13C-NMR (150 MHz, CDCl3), δ = 160.0 (d, JCF = 254 Hz), 153.3 (d, JCF = 5.2 Hz), 138.6 (d, JCF = 2.8 Hz), 132.3 (d, JCF = 8.8 Hz), 122.9 (d, JCF = 3.4 Hz), 116.2 (d, JCF = 12.6 Hz), 114.0 (d, JCF = 20.6 Hz), 46.9, 24.6 (d, JCF = 2.7 Hz). 19F-NMR (564.7 MHz, CDCl3): δ = −123.9 (dd, JFH = 9.4, 5.7 Hz). HRMS calcd. for C9H9FN+ ([M + H]+): 150.0714, found: 150.0723.
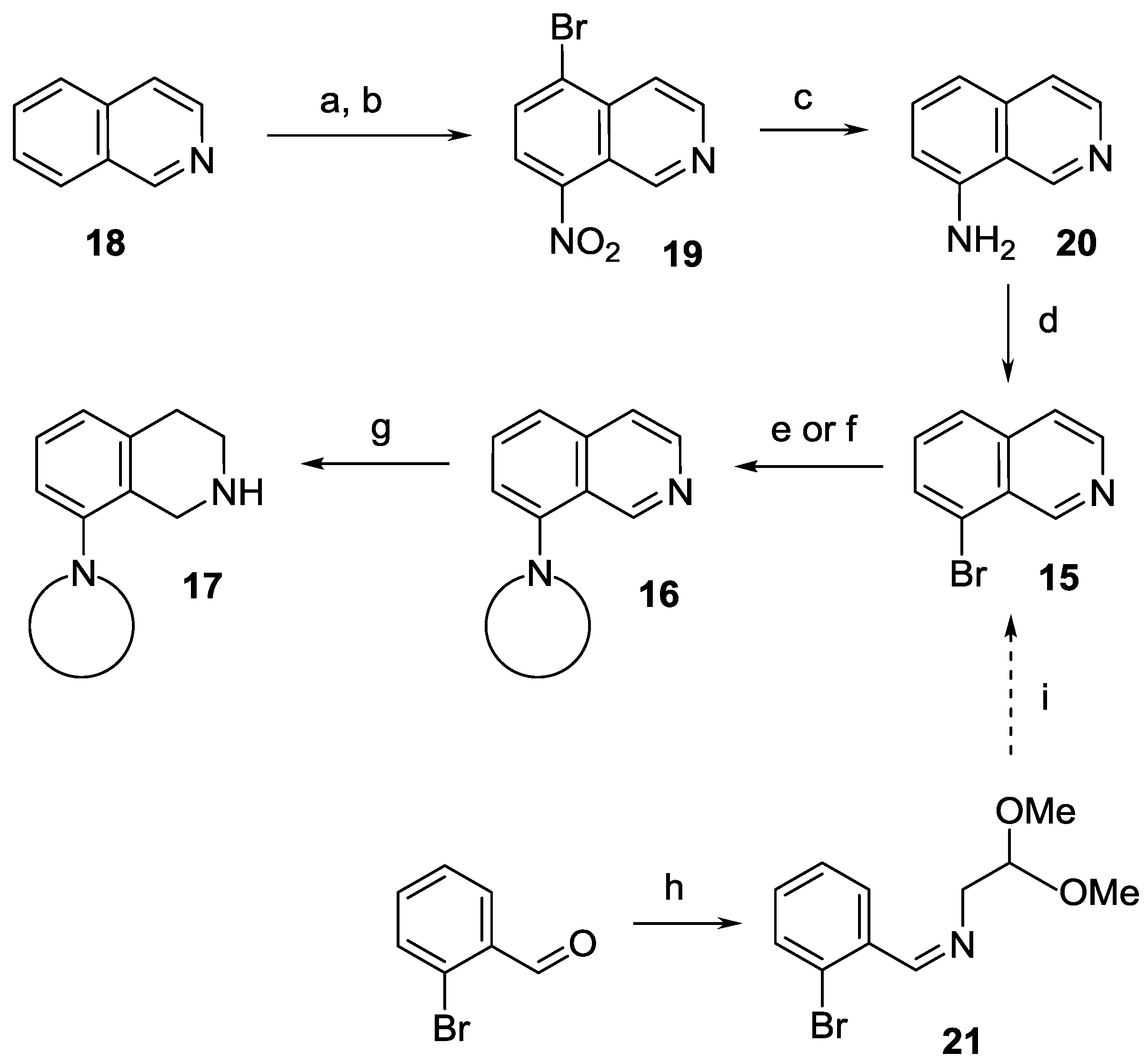
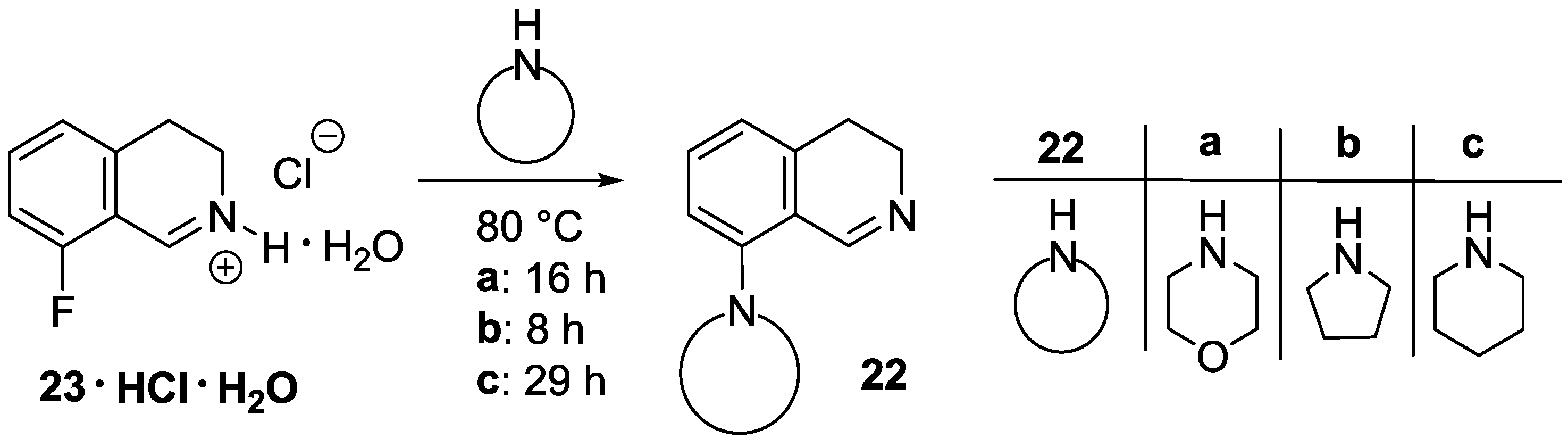
8-(Morpholin-4-yl)-3,4-dihydroisoquinoline (22a). A mixture of 23 ∙ HCl ∙ H2O (3.45 g, 16.9 mmol) and morpholine (4.42 mL, 4.42 g, 50.8 mmol) was stirred for 16 h at 80 °C in a sealed tube. After the reaction mixture was cooled, dichloromethane (60 mL) was added, and the resulting mixture was extracted with water (3 × 20 mL). The combined organic layer was dried over MgSO4, and evaporated. The residue was purified by flash chromatography (0–2% methanol in dichloromethane) to afford the title compound (1.86 g, 51%) as a brown oil. IR (film): ν = 2955, 1619, 1238 cm−1. 1H-NMR (400 MHz, CDCl3): δ = 8.63 (br s, 1H), 7.32 (dd, J = 8.1, 7.4 Hz, 1H), 6.93 (d, J = 8.1 Hz, 1H), 6.87 (d, J = 7.4 Hz, 1H), 3.88 (m, 4H), 3.67 (br t, J = 7.3 Hz, 2H), 3.01 (m, 4H), 2.69 (br t, J = 7.3 Hz, 2H). 13C-NMR (150 MHz, CDCl3): δ = 157.4, 151.0, 139.0, 131.6, 121.7, 121.3, 116.5, 67.0, 53.4, 46.5, 29.6, 25.5. HRMS calcd. for C13H17N2O+ ([M+H]+): 217.1335, found: 217.1341.
8-(Pyrrolidin-1-yl)-3,4-dihydroisoquinoline (22b). A mixture of 23 ∙ HCl ∙ H2O (3.50 g, 17.2 mmol) and pyrrolidine (4.23 mL, 3.66 g, 51.6 mmol) was stirred for 8 h at 80 °C in a sealed tube. After the reaction mixture was cooled, dichloromethane (60 mL) was added, and the resulting mixture was extracted with water (3 × 20 mL). The combined organic layer was dried over MgSO4, and evaporated. The residue was purified by flash chromatography (0–5% methanol in dichloromethane) to afford the title compound (1.68 g, 49%) as an orange oil. IR (film): ν = 2945, 1608 cm−1. 1H-NMR (400 MHz, CDCl3), δ = 8.63 (br s, 1H), 7.19 (dd, J = 8.3, 7.3 Hz, 1H), 6.69 (d, J = 8.3 Hz, 1H), 6.59 (d, J = 7.3 Hz, 1H), 3.64 (br t, J ≈ 7 Hz, 2H), 3.39 (m, 4H), 2.67 (t, J = 7.2 Hz, 2H), 1.96 (m, 4H). 13C-NMR (150 MHz, CDCl3): δ = 159.2, 148.7, 139.4, 131.1, 116.9, 112.8, 52.7, 46.0, 26.5, 25.6. HRMS calcd. for C13H17N2+ ([M + H]+): 201.1386, found: 201.1393.
8-(Pyrrolidin-1-yl)-3,4-dihydroisoquinoline hydrochloride (22b ∙ HCl). Base 22b (1.68 g, 8.4 mmol) was dissolved in toluene (10 mL) and a solution of hydrochloric acid gas in isopropyl alcohol was added dropwise. The precipitate was filtered off to afford the title compound (1.98 g, 99%) as an orange solid. Mp 218–220 °C (ethanol/diethyl ether). IR (KBr): ν = 2858, 1612 cm−1. 1H-NMR (600 MHz, CDCl3): δ = 13.68 (br s, 1H), 9.00 (d, J = 8.4 Hz, 1H), 7.40 (dd, J = 8.8, 7.1 Hz, 1H), 6.72 (br d, J = 8.8 Hz, 1H), 6.57 (br d, J = 7.1 Hz, 1H), 3.84 (td, JCH2–CH2 = 7.5 Hz, JCH2–NH = 3.0 Hz, 2H), 3.60 (m, 4H), 3.04 (t, J = 7.5 Hz, 2H), 2.06 (m, 4H). 13C-NMR (150 MHz, CDCl3): δ = 161.1, 152.5, 138.3, 137.9, 116.0, 114.6, 109.4, 53.4, 39.7, 27.1, 25.8. Anal. calcd. for C13H17ClN2 (236.74): Cl 14.97, N 11.83%. Found: Cl 14.89, N 11.57%.
8-(Piperidin-1-yl)-3,4-dihydroisoquinoline (22c). A mixture of 23 ∙ HCl ∙ H2O (1.05 g, 5.2 mmol) and piperidine (1.53 mL, 1.31 g, 15.5 mmol) was stirred for 29 h at 80 °C in a sealed tube. After the reaction mixture was cooled, dichloromethane (20 mL) was added, and the resulting mixture was extracted with water (3 × 6 mL). The combined organic layer was dried over MgSO4, and evaporated. The residue was purified by flash chromatography (0–1% methanol in dichloromethane) to afford the title compound (188 mg, 17%) as a brown oil. IR (film): ν = 2935, 1620 cm−1. 1H-NMR (400 MHz, CDCl3): δ = 8.59 (br s, 1H), 7.27 (dd, J = 8.2, 7.4 Hz, 1H), 6.90 (br d, J = 8.2 Hz, 1H), 6.79 (br d, J = 7.4 Hz, 1H), 3.66 (m, 2H), 2.96 (m, 4H), 2.67 (m, 2H), 1.75 (m, 4H), 1.59 (m, 2H). 13C-NMR (150 MHz, CDCl3): δ = 158.0, 152.6, 138.7, 131.4, 120.7, 116.6, 54.7, 46.5, 26.3, 25.7, 24.1. HRMS calcd. for C14H19N2+ ([M + H]+): 215.1543, found: 215.1549.
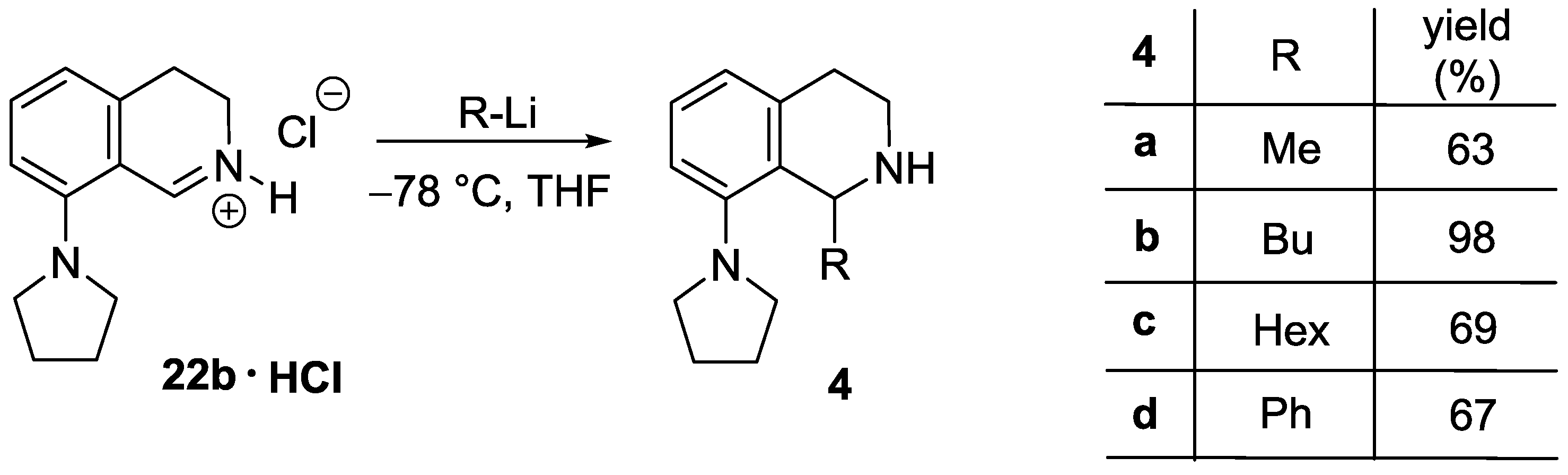
1-Methyl-8-(pyrrolidin-1-yl)-1,2,3,4-tetrahydroisoquinoline (4a). A solution of MeLi (1.6 M in diethyl ether, 2.67 mL, 4.28 mmol) was added to a suspension of 22b ∙ HCl (506 mg, 2.14 mmol) in THF (15 mL) at −78 °C. The mixture was stirred for 30 min at room temperature. The reaction mixture was diluted with a saturated aqueous solution of ammonium chloride (5 mL) and water (5 mL). The layers were separated and the aqueous layer was extracted with diethyl ether (2 × 8 mL). The combined organic layer was dried over MgSO4. The solvents were evaporated, the residue was purified by flash chromatography (3–6% methanol in dichloromethane) to afford the title compound (291 mg, 63%) as a pale brown oil. IR (film): ν = 2963, 1583 cm−1. 1H-NMR (400 MHz, CDCl3): δ = 7.09 (dd, J = 7.9, 7.5 Hz, 1H), 6.92 (br d, J = 7.9 Hz, 1H), 6.74 (br d, J = 7.5 Hz, 1H), 4.47 (q, J = 6.7 Hz, 1H), 3.35–3.23 (m, 3H), 3.09 (dt, Jgem = 12.8 Hz, J = 5.4 Hz, 1H), 2.97–2.78 (m, 4H), 2.03–1.79 (m, 4H), 1.47 (d, J = 7.1 Hz, 3H). 13C-NMR (150 MHz, CDCl3): δ = 148.0, 135.7, 133.4, 126.2, 123.0, 116.0, 51.8, 48.9, 38.8, 30.3, 21.2, 19.9. HRMS calcd. for C14H21N2+ ([M + H]+): 217.1699, found: 217.1705.
1-Butyl-8-(pyrrolidin-1-yl)-1,2,3,4-tetrahydroisoquinoline (4b). A solution of BuLi (1.6 M in hexane, 2.65 mL, 4.25 mmol) was added to a suspension of 22b ∙ HCl (502 mg, 2.12 mmol) in THF (15 mL) at −78 °C. The mixture was stirred for 30 min at room temperature. The reaction mixture was diluted with a saturated aqueous solution of ammonium chloride (5 mL) and water (5 mL). The layers were separated and the aqueous layer was extracted with diethyl ether (2 × 8 mL). The combined organic layer was dried over MgSO4. The solvents were evaporated to afford the title compound (537 mg, 98%) as a pale brown oil. IR (film): ν = 2955, 1583 cm−1. 1H-NMR (400 MHz, CDCl3): δ = 7.07 (dd, J = 8.1, 7.5 Hz, 1H), 6.94 (dd, J = 8.1, 1.4 Hz, 1H), 6.75 (dd, J = 7.5, 1.4 Hz, 1H), 4.21 (dd, J = 9.8, 2.5 Hz, 1H), 3.29 (m, 2H), 3.21 (m, 1H), 3.00 (dt, Jgem = 12.6 Hz, J = 5.2 Hz, 1H), 2.91–2.82 (m, 3H), 2.76 (dt, Jgem = 16.4 Hz, J = 5.2 Hz, 1H), 2.00–1.78 (m, 5H), 2.10 (br s, 1H), 1.60 (m, 1H), 1.49 (m, 1H), 1.43–1.27 (m, 3H), 0.91 (t, J = 7.1 Hz, 3H). 13C-NMR (150 MHz, CDCl3): δ = 148.1, 134.7, 131.1, 126.9, 123.4, 117.1, 53.1, 52.2, 38.2, 32.5, 28.6, 28.4, 25.0, 22.4, 13.8. HRMS calcd. for C17H27N2+ ([M + H]+): 259.2169, found: 259.2171.
1-Hexyl-8-(pyrrolidin-1-yl)-1,2,3,4-tetrahydroisoquinoline (4c). A solution of hexyllithium (2.5 M in hexane, 1.73 mL, 4.31 mmol) was added to a suspension of 22b ∙ HCl (510 mg, 2.16 mmol) in THF (15 mL) at −78 °C. The mixture was stirred for 30 min at room temperature. The reaction mixture was diluted with a saturated aqueous solution of ammonium chloride (5 mL) and water (5 mL). The layers were separated and the aqueous layer was extracted with diethyl ether (2 × 8 mL). The combined organic layer was dried over MgSO4. The solvents were evaporated, the residue was purified by flash chromatography (1–5% methanol in dichloromethane) to afford the title compound (427 mg, 69%) as a pale brown oil. IR (film): ν = 2924, 1583 cm−1. 1H-NMR (600 MHz, CDCl3): δ = 7.06 (dd, J = 8.0, 7.4 Hz, 1H), 6.93 (br d, J = 8.0 Hz, 1H), 6.75 (dd, J = 7.4, 1.1 Hz, 1H), 4.16 (dd, J = 9.9, 2.5 Hz, 1H), 3.28 (m, 2H), 3.17 (m, 1H), 2.96 (m, 1H), 2.96 (ddd, Jgem = 12.7 Hz, J = 5.7, 4.7 Hz, 1H), 2.87–2.80 (m, 3H), 2.72 (dt, Jgem = 16.4 Hz, J = 4.9 Hz, 1H), 1.98–1.91 (m, 2H), 1.89–1.80 (m, 3H), 1.57 (m, 1H), 1.49 (m, 1H), 1.41–1.25 (m, 7H), 0.88 (t, J = 6.9 Hz, 3H). 13C-NMR (150 MHz, CDCl3): δ = 148.0, 136.0, 134.5, 126.1, 123.6, 116.6, 53.1, 52.2, 38.4, 32.9, 31.7, 30.3, 29.2, 26.8, 25.0, 22.6, 14.1. HRMS calcd. for C19H31N2+ ([M + H]+): 287.2482, found: 287.2483.
1-Phenyl-8-(pyrrolidin-1-yl)-1,2,3,4-tetrahydroisoquinoline (4d). A solution of PhLi (1.9 M in dibutyl ether, 2.24 mL, 4.25 mmol) was added to a suspension of 22b ∙ HCl (503 mg, 2.13 mmol) in THF (15 mL) at −78 °C. The mixture was stirred for 30 min at room temperature. The reaction mixture was diluted with a saturated aqueous solution of ammonium chloride (5 mL) and water (5 mL). The layers were separated and the aqueous layer was extracted with diethyl ether (2 × 8 mL). The combined organic layer was dried over MgSO4. The solvents were evaporated, the residue was purified by flash chromatography (1–5% methanol in dichloromethane) to afford the title compound (396 mg, 67%) as a pale brown oil. IR (film): ν = 2959, 1584 cm−1. 1H-NMR (600 MHz, CDCl3): δ = 7.22 (m, 2H), 7.17 (m, 2H), 7.11 (m, 2H), 6.93 (br d, J = 8.0 Hz, 1H), 6.87 (br d, J = 7.5 Hz, 1H), 5.37 (br s, 1H), 3.03 (m, 1H), 2.94–2.87 (m, 4H), 2.79 (m, 1H), 2.56 (m, 2H), 1.93 (br s, 1H), 1.66 (m, 2H), 1.53 (m, 2H). 13C-NMR (150 MHz, CDCl3): δ = 148.5, 144.9, 136.9, 132.8, 127.9, 127.7, 126.9, 126.3, 123.6, 117.5, 57.3, 52.0, 38.7, 29.6, 24.7. HRMS calcd. for C19H23N2+ ([M + H]+): 279.1856, found: 279.1860.
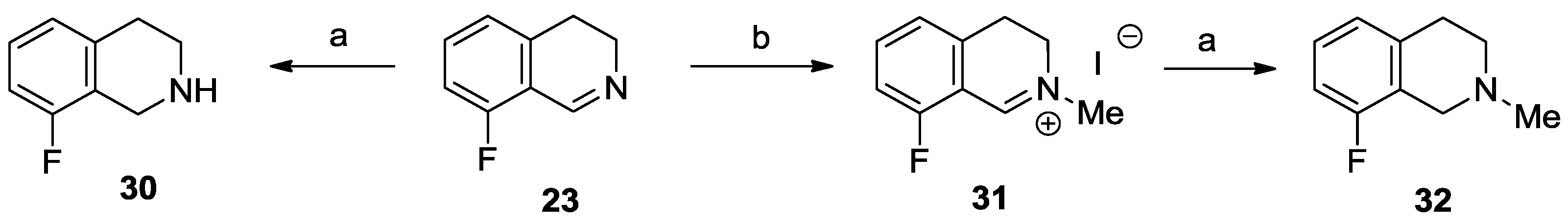
8-Fluoro-1,2,3,4-tetrahydroisoquinoline (30). Sodium borohydride (153 mg, 4.04 mmol) was added to a solution of 23 (502 mg, 3.37 mmol) in methanol (10 mL), and the reaction mixture was cooled with an ice/water bath. After stirring for 1 h at room temperature, water (5 mL) was added, and the resulting mixture was extracted with dichloromethane (3 × 8 mL). The combined organic layer was dried over MgSO4, and evaporated to afford the title compound (478 mg, 94%) as a yellow oil. IR (film): ν = 3299, 1463, 1241 cm−1. 1H-NMR (600 MHz, CDCl3): δ = 7.09 (ddd, JHH = 8.2, 7.6 Hz, JHF = 5.7 Hz, 1H), 6.88 (d, J = 7.6 Hz, 1H), 6.83 (dd, JHF = 9.7 Hz, JHH = 8.2 Hz, 1H), 4.03 (br s, 2H), 3.11 (t, J = 5.9 Hz, 2H), 2.79 (br t, J = 5.9 Hz, 2H), 1.77 (br s, 1H). 13C-NMR (150 MHz, CDCl3): δ = 159.5 (d, JCF = 244 Hz), 137.4 (d, JCF = 5.0 Hz), 126.8 (d, JCF = 8.6 Hz), 124.6 (d, JCF = 3.1 Hz), 123.4 (d, JCF = 17.1 Hz), 112.0 (d, JCF = 21.3 Hz), 43.3, 42.2 (d, JCF = 5.1 Hz), 28.8 (d, JCF = 2.8 Hz). 19F-NMR (564.7 MHz, CDCl3): δ = −121.2 (dd, JFH = 9.7, 5.7 Hz). HRMS calcd. for C9H11FN+ ([M + H]+): 152.0870, found: 152.0874.
8-Fluoro-2-methyl-3,4-dihydroisoquinolin-2-ium iodide (31). Methyl iodide (0.43 mL, 970 mg, 6.88 mmol) was added to a solution of 23 (513 mg, 3.44 mmol) in dichloromethane (10 mL). After stirring for 24 h at room temperature the reaction mixture was filtered to afford the title compound (726 mg, 73%) as a yellow solid. Mp 230–232 °C (ethanol/diethyl ether). IR (KBr): ν = 2991, 1679, 1621 cm−1. 1H-NMR (400 MHz, CD3OD): δ = 9.34 (br s, 1H), 7.85 (ddd, JHH ≈ 8.5, 7.5 Hz, JHF = 5.8 Hz, 1H), 7.35–7.27 (m, 2H), 4.11 (t, J = 8.1 Hz, 2H), 3.85 (s, 3H), 3.34 (t, J = 8.1 Hz, 2H). 13C-NMR (150 MHz, CD3OD): δ = 164.1 (d, JCF = 264 Hz), 162.6 (br s), 141.5 (d, JCF = 9.9 Hz), 139.2, 125.5 (d, JCF = 3.4 Hz), 116.2 (d, JCF = 19.8 Hz), 115.0 (d, JCF = 11.7 Hz), 51.2, 48.7, 25.9 (d, JCF = 2.2 Hz). 19F-NMR (564.7 MHz, CD3OD): δ = −114.7 (dd, JFH = 9.6, 5.8 Hz). HRMS calcd. for C10H11FN+ ([M + H]+): 164.0870, found: 164.0876.
8-Fluoro-2-methyl-1,2,3,4-tetrahydroisoquinoline (32). Sodium borohydride (80 mg, 2.09 mmol) was added to a solution of 31 (508 mg, 1.75 mmol) in methanol (14 mL), and the reaction mixture was cooled with an ice/water bath. After stirring for 1 h at room temperature, water (6 mL) was added, and the resulting mixture was extracted with dichloromethane (3 × 8 mL). The combined organic layer was dried over MgSO4, and evaporated. The residue was triturated in hexane and filtered. The filtrate was evaporated to afford the title compound (251 mg, 87%) as a colorless oil. IR (film): ν = 2924, 1468 cm−1. 1H-NMR (400 MHz, CDCl3): δ = 7.10 (ddd, JHH = 8.2, 7.7 Hz, JHF = 5.8 Hz, 1H), 6.90 (d, J = 7.7 Hz, 1H), 6.83 (dd, JHF = 9.7 Hz, JHH = 8.2 Hz, 1H), 3.59 (s, 2H), 2.93 (t, J = 5.9 Hz, 2H), 2.67 (t, J = 5.9 Hz, 2H), 2.49 (s, 3H). 13C-NMR (150 MHz, CDCl3): δ = 159.4 (d, JCF = 244 Hz), 136.5 (d, JCF = 4.7 Hz), 126.9 (d, JCF = 8.6 Hz), 124.0 (d, JCF = 3.2 Hz), 122.5 (d, JCF = 16.3 Hz), 111.9 (d, JCF = 20.9 Hz), 52.2, 51.6 (d, JCF = 5.5 Hz), 46.1, 29.0 (d, JCF = 2.5 Hz). 19F-NMR (564.7 MHz, CDCl3): δ = −121.3 (dd, JFH = 9.7, 5.8 Hz). HRMS calcd. for C10H13FN+ ([M + H]+): 166.1027, found: 166.1032.




{kind=link}
{kind=link}
{kind=link}
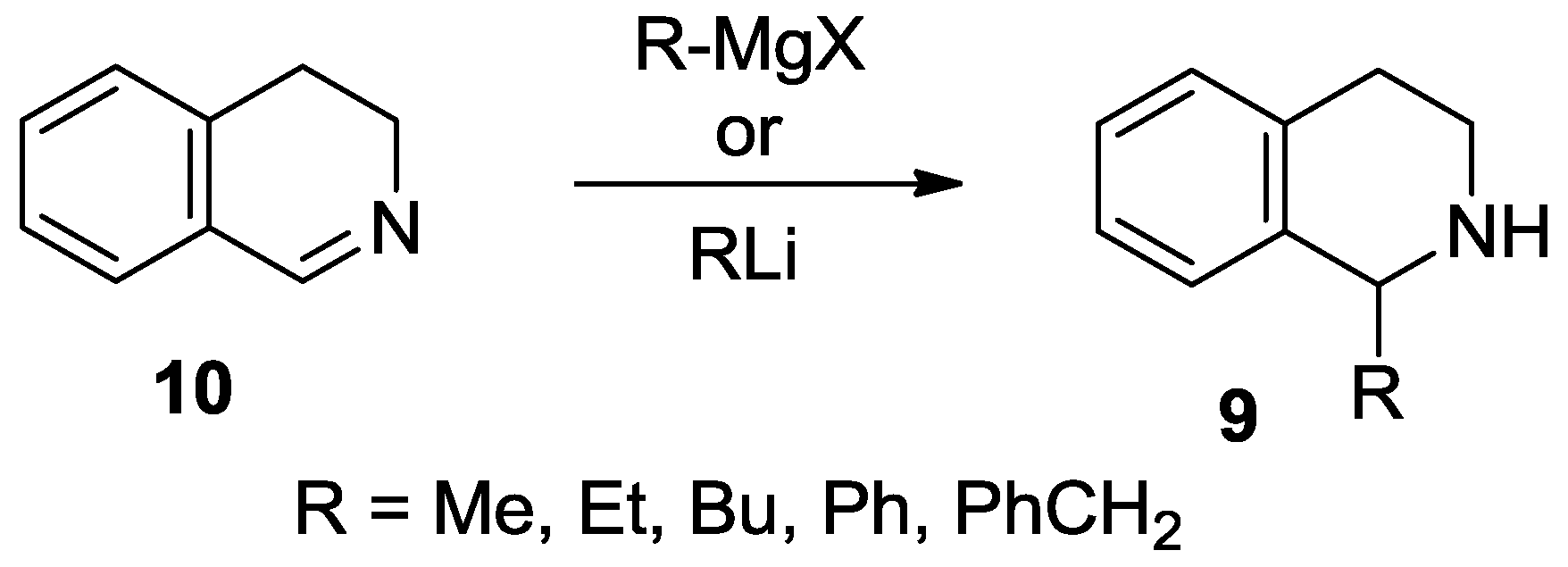{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










