Design, Synthesis, and Evaluation of a New Series of Thiazole-Based Anticancer Agents as Potent Akt Inhibitors





Abstract
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Chemistry
3.2. General Procedure for the Synthesis of the Compounds
4-(4-Cyanophenoxy)benzaldehyde thiosemicarbazone (A)
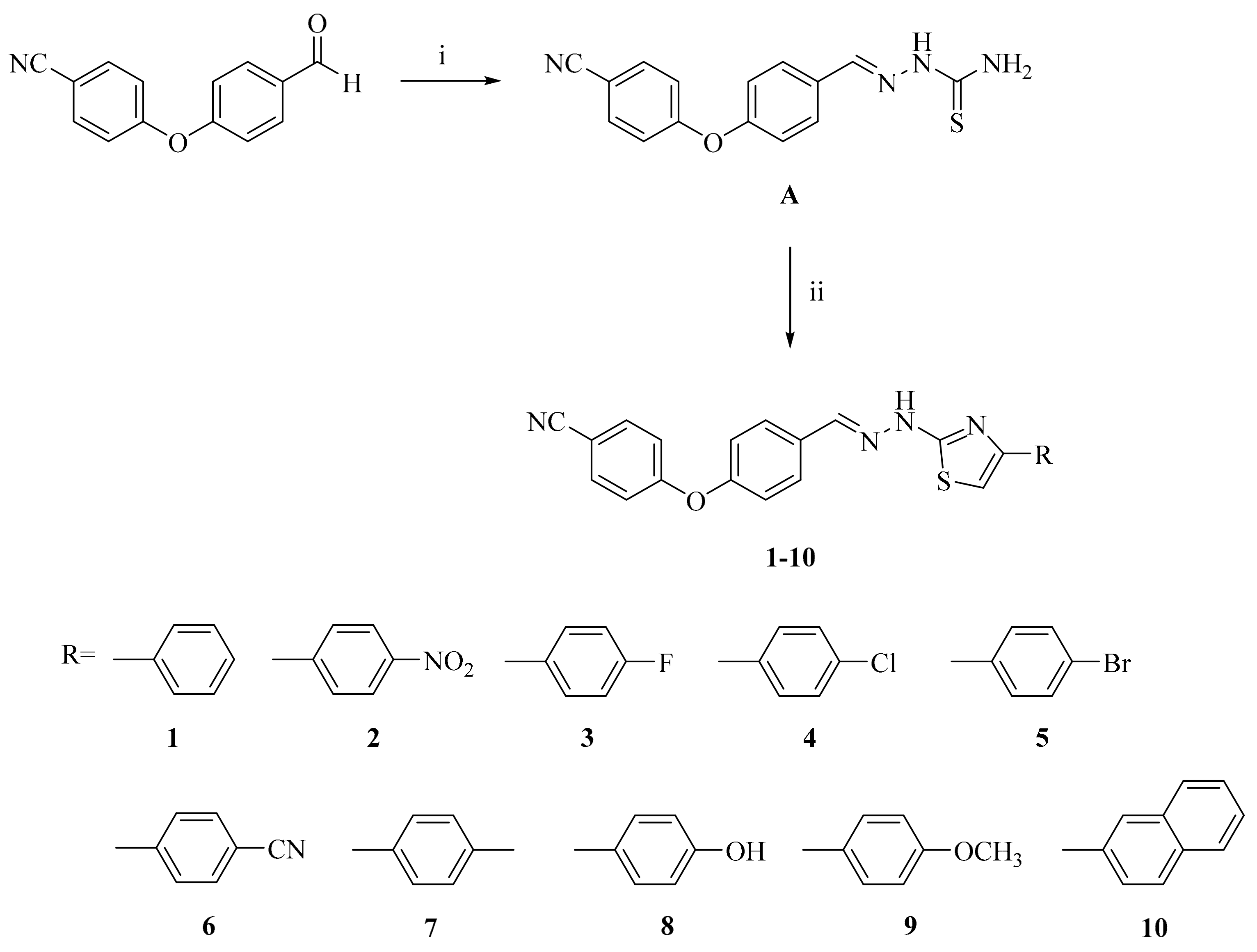
2-[2-((4-(4-Cyanophenoxy)phenyl)methylene)hydrazinyl]-4-arylthiazole derivatives (1–10)
3.3. Biochemistry
3.3.1. Cell Culture and Drug Treatment
3.3.2. MTT Assay
3.3.3. Flow Cytometric Analyses of Apoptosis
3.3.4. Determination of Akt Inhibition
3.3.5. Statistical Analyses
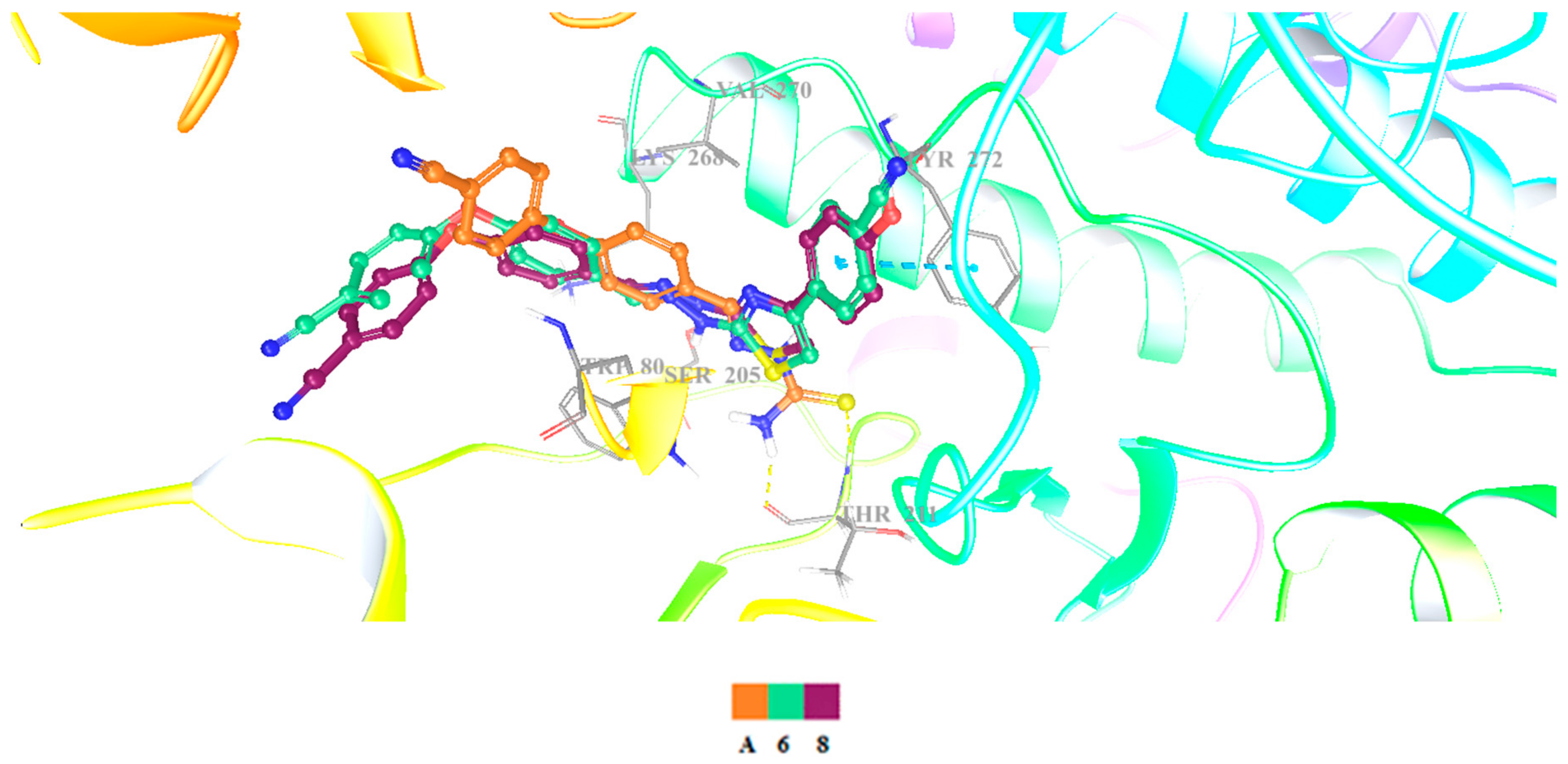
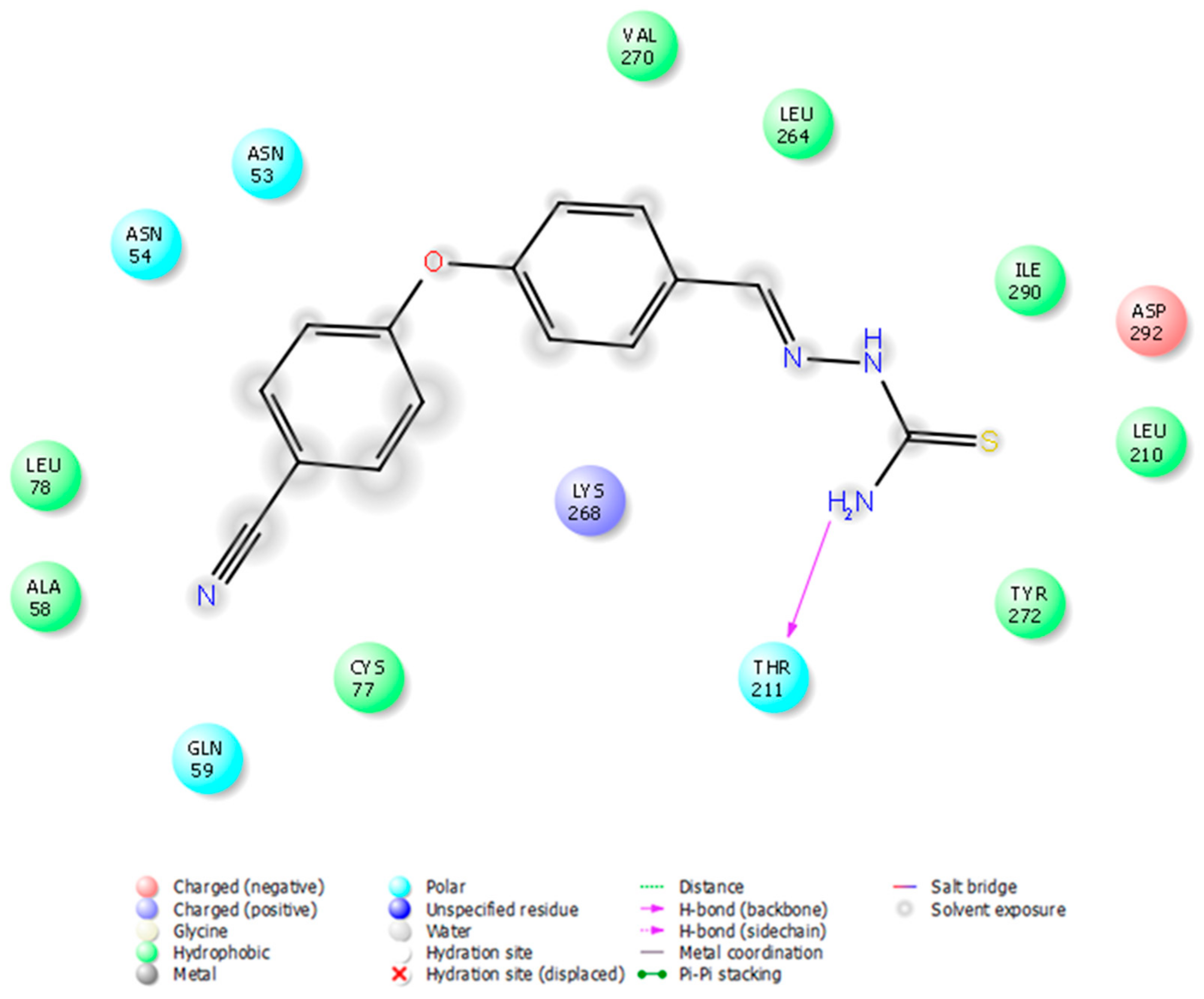
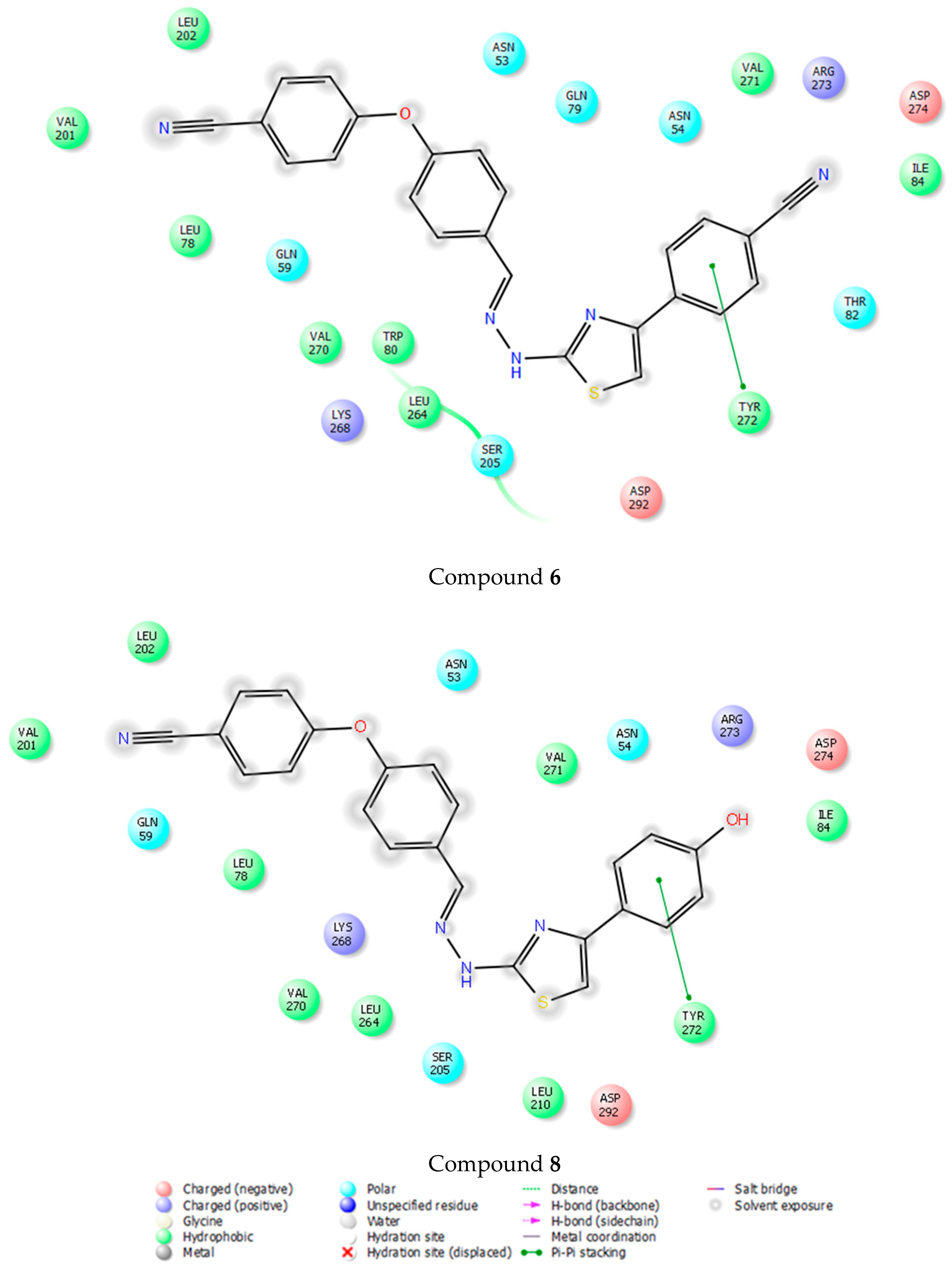
3.4. Molecular Docking Studies
3.5. Molinspiration Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Xue, Y.; Hou, S.; Ji, H.; Han, X. Evolution from Genetics to Phenotype: Reinterpretation of NSCLC Plasticity, Heterogeneity, and Drug Resistance. Protein Cell 2017, 8, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhu, T.; Gao, Y.-F.; Zheng, W.; Wang, C.-J.; Xiao, L.; Huang, M.-S.; Yin, J.-Y.; Zhou, H.-H.; Liu, Z.-Q. Targeting DNA Damage Response in the Radio(Chemo)therapy of Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2016, 17, 839. [Google Scholar] [CrossRef] [PubMed]
- Ansari, J.; Shackelford, R.E.; El-Osta, H. Epigenetics in Non-Small Cell Lung Cancer: From Basics to Therapeutics. Transl. Lung Cancer Res. 2016, 5, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Gyoba, J.; Shan, S.; Roa, W.; Bédard, E.L.R. Diagnosing Lung Cancers through Examination of Micro-RNA Biomarkers in Blood, Plasma, Serum and Sputum: A Review and Summary of Current Literature. Int. J. Mol. Sci. 2016, 17, 494. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Cai, J.; Jiang, C. Recent Advances in Targeted Therapy for Glioma. Curr. Med. Chem. 2017, 24, 1365–1381. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.A.; Hosni-Ahmed, A.; Jones, T.S.; Patil, R.; Pfeffer, L.M.; Miller, D.D. Novel Approaches to Glioma Drug Design and Drug Screening. Expert Opin. Drug Discov. 2013, 8, 1135–1151. [Google Scholar] [CrossRef] [PubMed]
- Nitulescu, G.M.; Margina, D.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Saloustros, E.; Fenga, C.; Spandidos, D.Α.; Libra, M.; Tsatsakis, A.M. Akt inhibitors in cancer treatment: The Long Journey from Drug Discovery to Clinical Use (Review). Int. J. Oncol. 2016, 48, 869–885. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.K.; Bordoloi, D.; Monisha, J.; Padmavathi, G.; Kotoky, J.; Golla, R.; Kunnumakkara, A.B. Specific Targeting of Akt Kinase Isoforms: Taking the Precise Path for Prevention and Treatment of Cancer. Curr. Drug Targets 2017, 18, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Cassinelli, G.; Zuco, V.; Gatti, L.; Lanzi, C.; Zaffaroni, N.; Colombo, D.; Perego, P. Targeting the Akt Kinase to Modulate Survival, Invasiveness and Drug Resistance of Cancer Cells. Curr. Med. Chem. 2013, 20, 1923–1945. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.K.; Du-Cuny, L.; Chen, L.; Meuillet, E.J.; Mash, E.A.; Powis, G.; Zhang, S. Recent Development of Anticancer Therapeutics Targeting Akt. Recent Pat. Anti-Cancer Drug Discov. 2011, 6, 146–159. [Google Scholar] [CrossRef]
- Ayati, A.; Emami, S.; Asadipour, A.; Shafiee, A.; Foroumadi, A. Recent Applications of 1,3-Thiazole Core Structure in the Identification of New Lead Compounds and Drug Discovery. Eur. J. Med. Chem. 2015, 97, 699–718. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Sikdar, P.; Bairagi, M. Recent Developments of 2-Aminothiazoles in Medicinal Chemistry. Eur. J. Med. Chem. 2016, 109, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Chhabriaa, M.T.; Patel, S.; Modi, P.; Brahmkshatriya, P.S. Thiazole: A Review on Chemistry, Synthesis and Therapeutic Importance of its Derivatives. Curr. Top. Med. Chem. 2016, 16, 2841–2862. [Google Scholar] [CrossRef]
- Rouf, A.; Tanyeli, C. Bioactive Thiazole and Benzothiazole Derivatives. Eur. J. Med. Chem. 2015, 97, 911–927. [Google Scholar] [CrossRef] [PubMed]
- Popsavin, M.; Kojić, V.; Spaić, S.; Svirčev, M.; Bogdanović, G.; Jakimov, D.; Aleksić, L.; Popsavin, V. 2-Substituted Thiazole-4-carboxamide Derivatives as Tiazofurin Mimics: Synthesis and In Vitro Antitumour Activity. Tetrahedron 2014, 70, 2343–2350. [Google Scholar] [CrossRef]
- Păunescu, E.; Clavel, C.M.; Nowak-Sliwinska, P.; Griffioen, A.W.; Dyson, P.J. Improved Angiostatic Activity of Dasatinib by Modulation with Hydrophobic Chains. ACS Med. Chem. Lett. 2015, 6, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Morigi, R.; Locatelli, A.; Leoni, A.; Rambaldi, M. Recent Patents on Thiazole Derivatives Endowed with Antitumor Activity. Recent Pat. Anti-Cancer Drug Discov. 2015, 10, 280–297. [Google Scholar] [CrossRef]
- Leoni, A.; Locatelli, A.; Morigi, R.; Rambaldi, M. Novel Thiazole Derivatives: A Patent Review (2008–2012; Part 1). Expert Opin. Ther. Pat. 2014, 24, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Zhang, Z.; Zhuang, X.; Luo, J.; Cao, X.; Li, H.; Tu, Z.; Lu, X.; Ren, X.; Ding, K. New Thiazole Carboxamides as Potent Inhibitors of Akt Kinases. Bioorg. Med. Chem. Lett. 2012, 22, 1208–1212. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Yang, F.; Chang, S.; Tang, J.; Qin, J.; Feng, G.-K.; Ding, K.; Zhu, X.-F. DC120, a Novel and Potent Inhibitor of AKT Kinase, Induces Tumor Cell Apoptosis and Suppresses Tumor Growth. Mol. Pharmacol. 2012, 82, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Mathew, B.; Suresh, J.; Ahsan, M.J.; Mathew, G.E.; Usman, D.; Subramanyan, P.N.S.; Safna, K.F.; Maddela, S. Hydrazones as A Privileged Structural Linker in Antitubercular Agents: A Review. Infect. Disord. Drug Targets 2015, 15, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Narang, R.; Narasimhan, B.; Sharma, S. A Review on Biological Activities and Chemical Synthesis of Hydrazide Derivatives. Curr. Med. Chem. 2012, 19, 569–612. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Narasimhan, B. Hydrazides/Hydrazones as Antimicrobial and Anticancer Agents in the New Millennium. Mini-Rev. Med. Chem. 2013, 13, 971–987. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.; Kaufmann, D.; Pojarová, M.; Müller, C.; Pfaller, T.; Kühne, S.; Bednarski, P.J.; von Angerer, E. Aroyl Hydrazones of 2-Phenylindole-3-carbaldehydes as Novel Antimitotic Agents. Bioorg. Med. Chem. 2008, 16, 6436–6447. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Fan, C.-D.; Zhao, B.-X.; Zhao, J.; Shin, D.-S.; Miao, J.-Y. Synthesis and Structure‒Activity Relationships of Novel 1-Arylmethyl-3-aryl-1H-pyrazole-5-carbohydrazide Hydrazone Derivatives as Potential Agents against A549 Lung Cancer Cells. Eur. J. Med. Chem. 2008, 43, 2347–2353. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.S.; Lee, D.-U. Synthesis, Biological Evaluation, Drug-likeness, and In Silico Screening of Novel Benzylidene-hydrazone Analogues as Small Molecule Anticancer Agents. Arch. Pharm. Res. 2016, 39, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Şenkardeş, S.; Kaushik-Basu, N.; Durmaz, İ.; Manvar, D.; Basu, A.; Atalay, R.; Küçükgüzel, Ş.G. Synthesis of Novel Diflunisal Hydrazide‒hydrazones as Anti-hepatitis C Virus Agents and Hepatocellular Carcinoma Inhibitors. Eur. J. Med. Chem. 2016, 108, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.S.; Choi, S.-U.; Lee, D.-U. Synthesis, Anticancer, and Docking Studies of Salicyl-hydrazone Analogues: A Novel Series of Small Potent Tropomyosin Receptor Kinase A Inhibitors. Bioorg. Med. Chem. 2017, 25, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, F.; Bizzarri, B.; Maccioni, E.; Secci, D.; Bolasco, A.; Chimenti, P.; Fioravanti, R.; Granese, A.; Carradori, S.; Tosi, F.; et al. A Novel Histone Acetyltransferase Inhibitor Modulating Gcn5 Network: Cyclopentylidene-[4-(4′-chlorophenyl)thiazol-2-yl)hydrazone. J. Med. Chem. 2009, 52, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Secci, D.; Carradori, S.; Bizzarri, B.; Bolasco, A.; Ballario, P.; Patramani, Z.; Fragapane, P.; Vernarecci, S.; Canzonetta, C.; Filetici, P. Synthesis of a Novel Series of Thiazole-Based Histone Acetyltransferase Inhibitors. Bioorg. Med. Chem. 2014, 22, 1680–1689. [Google Scholar] [CrossRef] [PubMed]
- Łączkowski, K.Z.; Misiura, K.; Świtalska, M.; Wietrzyk, J.; Baranowska-Łączkowska, A.; Fernandez, B.; Paneth, A.; Plech, T. Synthesis and In Vitro Antiproliferative Activity of Thiazole-Based Nitrogen Mustards: The Hydrogen Bonding Interaction between Model Systems and Nucleobases. Anti-Cancer Agents Med. Chem. 2014, 14, 1271–1281. [Google Scholar] [CrossRef]
- Altıntop, M.D.; Özdemir, A.; Ilgın, S.; Atlı, Ö. Synthesis and Biological Evaluation of New Pyrazole-Based Thiazolyl Hydrazone Derivatives as Potential Anticancer Agents. Lett. Drug Des. Discov. 2014, 11, 833–839. [Google Scholar] [CrossRef]
- Altıntop, M.D.; Özdemir, A.; Turan-Zitouni, G.; Ilgın, S.; Atlı, Ö.; Demirci, F.; Kaplancıklı, Z.A. Synthesis and In Vitro Evaluation of New Nitro-Substituted Thiazolyl Hydrazone Derivatives as Anticandidal and Anticancer Agents. Molecules 2014, 19, 14809–14820. [Google Scholar] [CrossRef] [PubMed]
- Kaplancıklı, Z.A.; Sever, B.; Altıntop, M.D.; Atlı, Ö.; Baysal, M.; Özdemir, A. Synthesis and Evaluation of New Thiazolyl Hydrazone Derivatives as Potential Anticancer Agents. Lett. Drug Des. Discov. 2017, 14, 672–677. [Google Scholar] [CrossRef]
- Ashwell, M.A.; Lapierre, J.M.; Brassard, C.; Bresciano, K.; Bull, C.; Cornell-Kennon, S.; Eathiraj, S.; France, D.S.; Hall, T.; Hill, J.; et al. Discovery and Optimization of a Series of 3-(3-Phenyl-3H-imidazo[4,5-b]pyridin-2-yl)pyridin-2-amines: Orally Bioavailable, Selective, and Potent ATP-Independent Akt Inhibitors. J. Med. Chem. 2012, 55, 5291–5310. [Google Scholar] [CrossRef] [PubMed]
- Van Den Driessche, G.; Fourches, D. Adverse Drug Reactions Triggered by the Common HLA-B*57:01 Variant: A Molecular Docking Study. J. Cheminform. 2017, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Molinspiration Cheminformatics on the Web. Available online: http://www.molinspiration.com (accessed on 31 December 2017).
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties that Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Gabr, M.T.; El-Gohary, N.S.; El-Bendary, E.R.; El-Kerdawy, M.M.; Ni, N. Synthesis, In Vitro Antitumor Activity and Molecular Modeling Studies of A New Series of Benzothiazole Schiff Bases. Chin. Chem. Lett. 2016, 27, 380–386. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 16, 55–63. [Google Scholar] [CrossRef]
- Altıntop, M.D.; Temel, H.E.; Sever, B.; Akalın Çiftçi, G.; Kaplancıklı, Z.A. Synthesis and Evaluation of New Benzodioxole-Based Thiosemicarbazone Derivatives as Potential Antitumor Agents. Molecules 2016, 21, 1598. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1–10 are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µg/mL) | ||
|---|---|---|---|
| A549 Cell Line | C6 Cell Line | NIH/3T3 Cell Line | |
| A | 92.5 ± 3.54 | 26.33 ± 1.53 | 225 ± 18.03 |
| 1 | 410 ± 14.14 | >500 | 196.67 ± 20.82 |
| 2 | 100 ± 20 | >500 | 71.67 ± 24.66 |
| 3 | 193.33 ± 15.28 | >500 | 170 ± 36.06 |
| 4 | 206.67 ± 23.09 | >500 | 180 ± 10 |
| 5 | 198.33 ± 10.41 | >500 | 163.33 ± 5.77 |
| 6 | 12 ± 1.73 | 3.83 ± 0.76 | >500 |
| 7 | 270 ± 95.39 | 16 ± 5.66 | 185 ± 5 |
| 8 | 106.67 ± 5.77 | 5.83 ± 0.76 | 226.67 ± 11.55 |
| 9 | 233.33 ± 57.74 | >500 | 433.33 ± 28.87 |
| 10 | >500 | >500 | >500 |
| Cisplatin | 17.33 ± 2.08 | 12.67 ± 3.06 | ND |
| Groups | Early Apoptotic Cells% | Late Apoptotic Cells% | Viable Cells% |
|---|---|---|---|
| Control (untreated) | 0.9 | 0.3 | 94.0 |
| Compound A treated cells | 7.6 | 45.9 | 24.2 |
| Compound 6 treated cells | 2.3 | 36.5 | 14.4 |
| Compound 8 treated cells | 2.1 | 38.7 | 18.5 |
| Cisplatin treated cells | 2.6 | 63.5 | 14.1 |
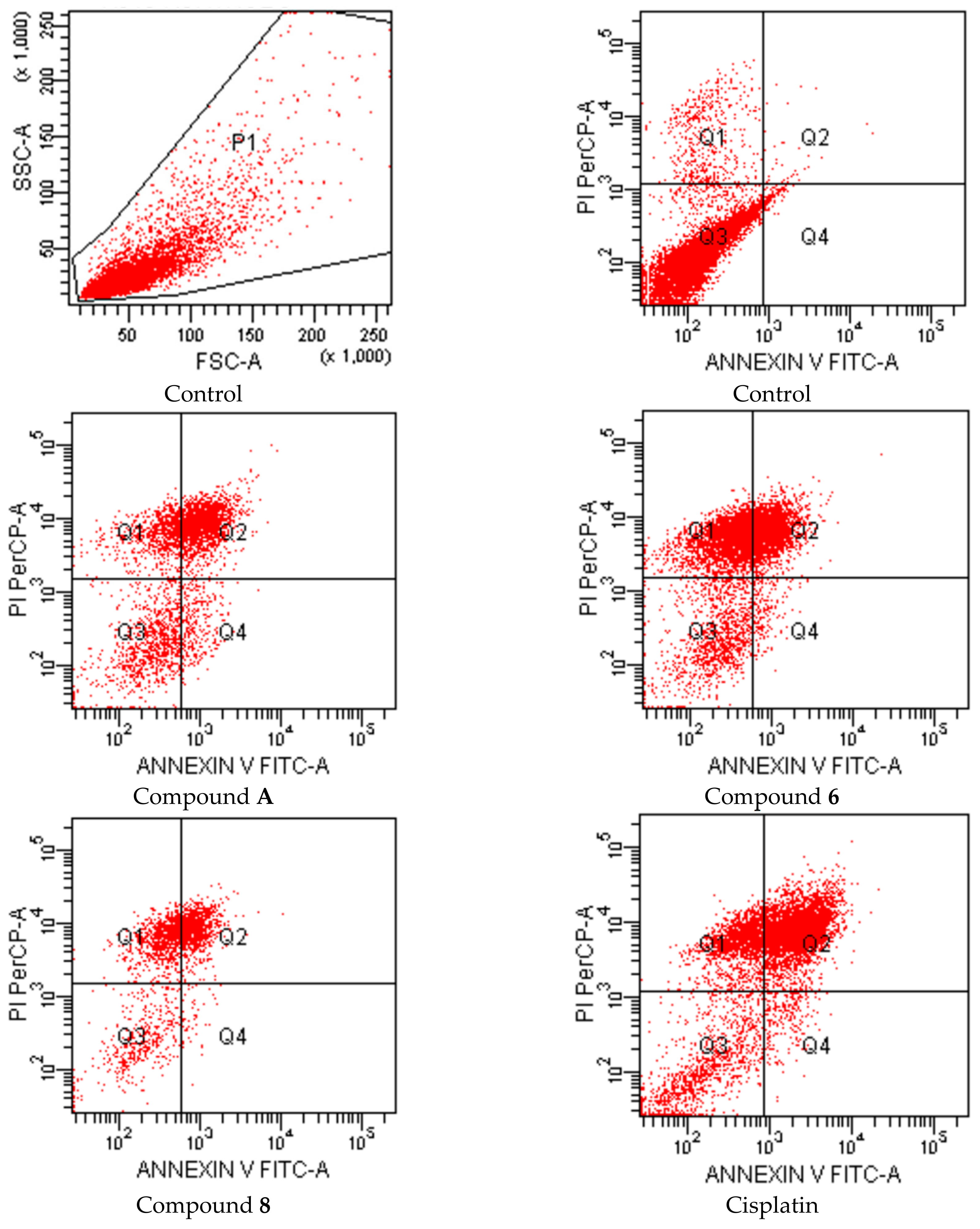
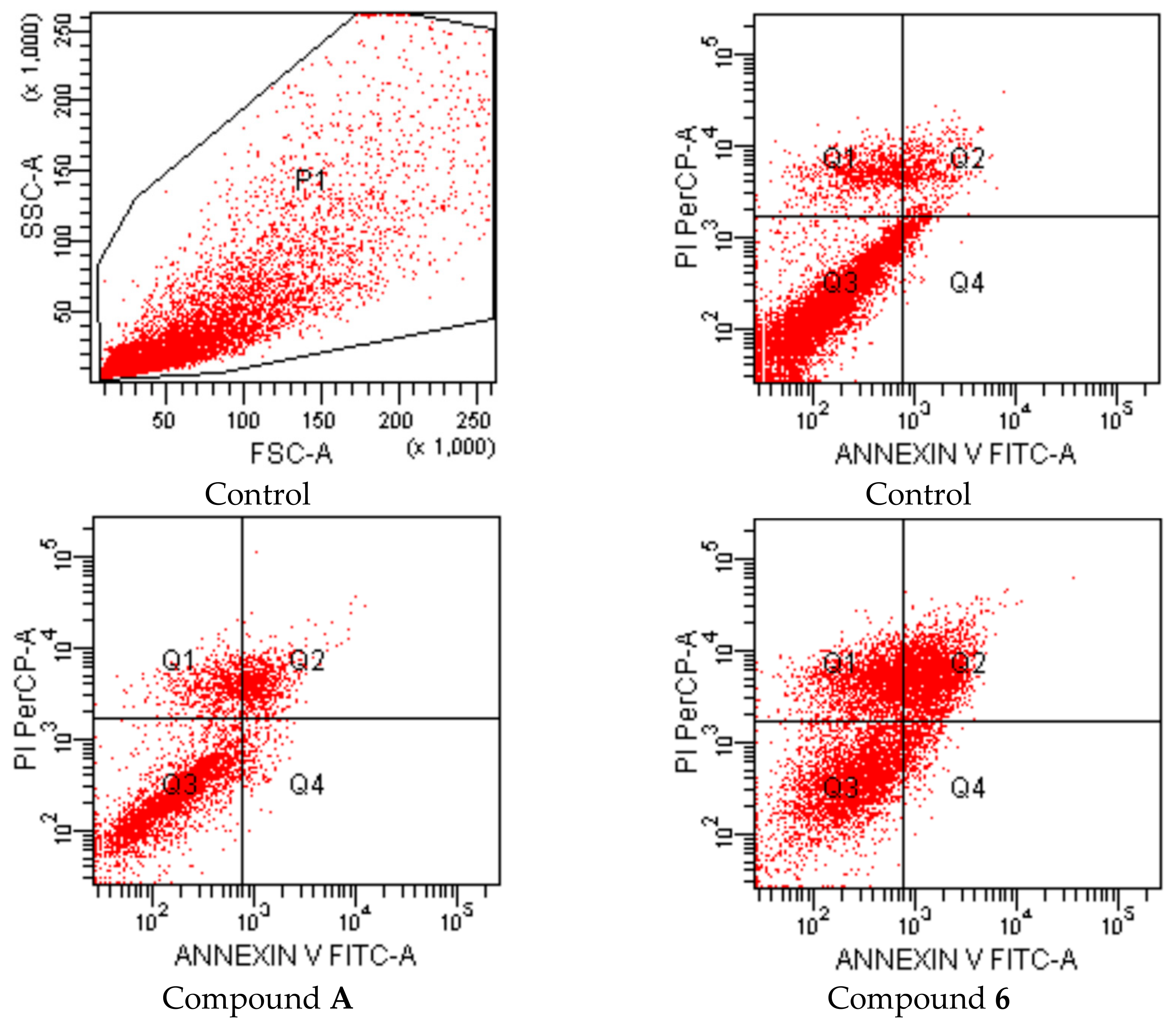
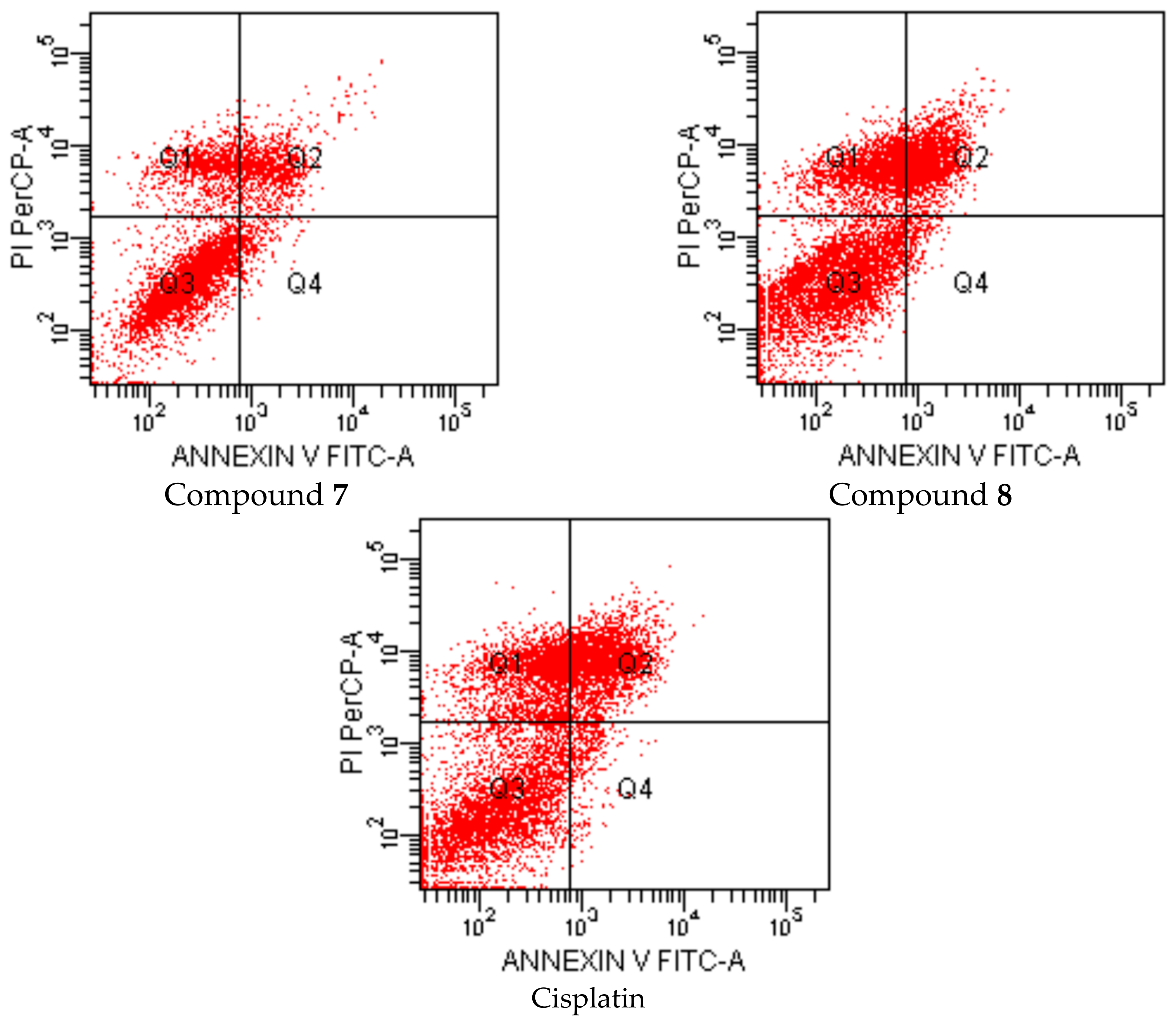
| Groups | Early Apoptotic Cells% | Late Apoptotic Cells% | Viable Cells% |
|---|---|---|---|
| Control (untreated) | 2.5 | 5.5 | 82.7 |
| Compound A treated cells | 3.2 | 13.1 | 70.2 |
| Compound 6 treated cells | 4.6 | 28.2 | 42.2 |
| Compound 7 treated cells | 3.2 | 14.3 | 69.2 |
| Compound 8 treated cells | 1.9 | 23.8 | 49.7 |
| Cisplatin treated cells | 3.3 | 25.5 | 47.6 |
| Compound | Inhibition % | |
|---|---|---|
| A549 Cell Line | C6 Cell Line | |
| A | 68.08 ± 2.48 | 25.23 ± 8.62 |
| 6 | 45.77 ± 10.58 | 71.66 ± 4.09 |
| 7 | - | 70.42 ± 10.37 |
| 8 | 57.37 ± 17.30 | 24.99 ± 0.52 |
| Cisplatin | 31.01 ± 3.18 | 77.25 ± 5.75 |
| Compound | 4EJN | ||
|---|---|---|---|
| Docking Score | Glide Gscore | Glide Emodel | |
| A | −6.54 | −6.54 | −55.88 |
| 6 | −5.08 | −7.00 | −71.40 |
| 8 | −5.45 | −6.81 | −69.00 |
| Compound | Molecular Properties a | |||||||
|---|---|---|---|---|---|---|---|---|
| MW | logP | TPSA | nrotb | HBA | HBD | Volume | Violation | |
| A | 296.36 | 3.39 | 83.44 | 5 | 5 | 3 | 256.11 | 0 |
| 1 | 396.48 | 5.47 | 70.31 | 6 | 5 | 1 | 346.33 | 1 |
| 2 | 441.47 | 5.42 | 116.13 | 7 | 8 | 1 | 369.67 | 1 |
| 3 | 414.46 | 5.63 | 70.31 | 6 | 5 | 1 | 351.26 | 1 |
| 4 | 430.92 | 6.14 | 70.31 | 6 | 5 | 1 | 359.87 | 1 |
| 5 | 475.37 | 6.28 | 70.31 | 6 | 5 | 1 | 364.22 | 1 |
| 6 | 421.49 | 5.22 | 94.10 | 6 | 6 | 1 | 363.19 | 1 |
| 7 | 410.50 | 5.92 | 70.31 | 6 | 5 | 1 | 362.89 | 1 |
| 8 | 412.47 | 4.99 | 90.54 | 6 | 6 | 2 | 354.35 | 0 |
| 9 | 426.50 | 5.52 | 79.54 | 7 | 6 | 1 | 371.88 | 1 |
| 10 | 446.54 | 6.65 | 70.31 | 6 | 5 | 1 | 390.32 | 1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altıntop, M.D.; Sever, B.; Akalın Çiftçi, G.; Özdemir, A. Design, Synthesis, and Evaluation of a New Series of Thiazole-Based Anticancer Agents as Potent Akt Inhibitors. Molecules 2018, 23, 1318. https://doi.org/10.3390/molecules23061318
Altıntop MD, Sever B, Akalın Çiftçi G, Özdemir A. Design, Synthesis, and Evaluation of a New Series of Thiazole-Based Anticancer Agents as Potent Akt Inhibitors. Molecules. 2018; 23(6):1318. https://doi.org/10.3390/molecules23061318
Chicago/Turabian StyleAltıntop, Mehlika Dilek, Belgin Sever, Gülşen Akalın Çiftçi, and Ahmet Özdemir. 2018. "Design, Synthesis, and Evaluation of a New Series of Thiazole-Based Anticancer Agents as Potent Akt Inhibitors" Molecules 23, no. 6: 1318. https://doi.org/10.3390/molecules23061318
APA StyleAltıntop, M. D., Sever, B., Akalın Çiftçi, G., & Özdemir, A. (2018). Design, Synthesis, and Evaluation of a New Series of Thiazole-Based Anticancer Agents as Potent Akt Inhibitors. Molecules, 23(6), 1318. https://doi.org/10.3390/molecules23061318