2-[2-((4-(4-Cyanophenoxy)phenyl)methylene)hydrazinyl]-4-arylthiazole derivatives (1–10)
A mixture of compound A (0.001 mol) and aryl acyl bromide (0.001 mol) in ethanol (20 mL) was refluxed for 6 h. The reaction mixture was cooled and filtered. The product was crystallized from ethanol.
2-[2-((4-(4-Cyanophenoxy)phenyl)methylene)hydrazinyl]-4-phenylthiazole (1). Dark goldenrod powder. M.p. 214–215 °C. Yield: 74%. IR νmax (cm−1): 3452.58 (N-H stretching), 3089.96 (Aromatic C-H stretching), 2987.74, 2900.94, 2883.58 (Aliphatic C-H stretching), 2229.71 (C≡N stretching), 1625.99, 1591.27, 1490.97, 1444.68 (N-H bending, C=N and C=C stretching), 1409.96, 1361.74 (C-H bending), 1303.88, 1240.23, 1205.51, 1165.00, 1089.78, 1047.35, 1028.06 (C-N, C-O stretching and aromatic C-H in plane bending), 879.54, 864.11, 835.18, 773.46, 729.09, 715.59, 688.59, 648.08 (Aromatic C-H out of plane bending and C-S stretching). 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 7.08–7.41 (m, 8H), 7.72–7.86 (m, 6H), 8.06 (s, 1H), 12.01 (s, 1H). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 104.43 (CH), 106.20 (C), 119.17 (2CH), 119.36 (C), 121.11 (2CH), 126.24 (2CH), 128.62 (CH), 129.02 (2CH), 129.32 (2CH), 131.95 (C), 135.16 (C), 135.41 (2CH), 141.25 (CH), 150.90 (C), 156.03 (C), 161.31 (C), 168.90 (C). HRMS (ESI) (m/z): [M + H]+ 397.1121.
2-[2-((4-(4-Cyanophenoxy)phenyl)methylene)hydrazinyl]-4-(4-nitrophenyl)thiazole (2). Orange powder. M.p. 250–251 °C. Yield: 92%. IR νmax (cm−1): 3305.99 (N-H stretching), 3113.11, 3062.96 (Aromatic C-H stretching), 2987.74, 2927.94 (Aliphatic C-H stretching), 2229.71 (C≡N stretching), 1593.20, 1573.91, 1494.83 (N-H bending, C=N and C=C stretching), 1431.18, 1408.04, 1355.96 (C-H bending), 1338.60 (NO2 stretching), 1315.45, 1278.81, 1236.37, 1201.65, 1168.86, 1124.50, 1111.00, 1097.50, 1047.35, 1010.70 (C-N, C-O stretching and aromatic C-H in plane bending), 929.69, 879.54, 858.32, 837.11, 815.89, 715.59 (Aromatic C-H out of plane bending). 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 7.17 (d, J = 9.2 Hz, 2H), 7.21 (d, J = 9.2 Hz, 2H), 7.73–7.77 (m, 3H), 7.87 (d, J = 9.2 Hz, 2H), 8.09 (s, 1H), 8.11 (d, J = 9.2 Hz, 2H), 8.28 (d, J = 9.2 Hz, 2H), 12.33 (s, 1H). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 106.23 (C), 109.31 (CH), 119.18 (2CH), 119.35 (C), 121.10 (2CH), 124.81 (2CH), 127.02 (2CH), 129.07 (2CH), 131.82 (C), 135.41 (2CH), 141.34 (C), 141.54 (CH), 146.90 (C), 149.27 (C), 156.13 (C), 161.28 (C), 169.32 (C). HRMS (ESI) (m/z): [M + H]+ 442.0969.
2-[2-((4-(4-Cyanophenoxy)phenyl)methylene)hydrazinyl]-4-(4-fluorophenyl)thiazole (3). Cinnamon-colored powder. M.p. 204–205 °C. Yield: 77%. IR νmax (cm−1): 3419.79 (N-H stretching), 3057.17 (Aromatic C-H stretching), 2914.44 (Aliphatic C-H stretching), 2225.85 (C≡N stretching), 1622.13, 1593.20, 1568.13, 1558.48, 1494.83, 1456.26 (N-H bending, C=N and C=C stretching), 1411.89 (C-H bending), 1290.38, 1238.30, 1203.58, 1166.93, 1105.21, 1083.99, 1037.70, 1014.56 (C-N, C-O stretching and aromatic C-H in plane bending), 881.47, 833.25, 804.32, 794.67, 759.95, 746.45, 715.59, 692.44 (Aromatic C-H out of plane bending and C-S stretching). 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 7.10–7.34 (m, 8H), 7.71–7.77 (m, 2H), 7.83–7.91 (m, 3H), 8.08 (s, 1H), 12.31 (s, 1H). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 104.18 (CH), 106.19 (C), 116.13 (d, J = 21.3 Hz, 2CH), 119.15 (2CH), 119.34 (C), 121.10 (2CH), 128.21 (d, J = 7.6 Hz, 2CH), 129.01 (2CH), 131.91 (2C), 135.40 (2CH), 141.28 (CH), 149.91 (C), 156.03 (C), 161.28 (C), 163.52 (C), 168.98 (C). HRMS (ESI) (m/z): [M + H]+ 415.1047.
2-[2-((4-(4-Cyanophenoxy)phenyl)methylene)hydrazinyl]-4-(4-chlorophenyl)thiazole (4). Goldenrod powder. M.p. 227–228 °C. Yield: 83%. IR νmax (cm−1): 3402.43 (N-H stretching), 3055.24 (Aromatic C-H stretching), 2912.51, 2858.51 (Aliphatic C-H stretching), 2225.85 (C≡N stretching), 1614.42, 1606.70, 1593.20, 1492.90 (N-H bending, C=N and C=C stretching), 1244.09, 1201.65, 1166.93, 1093.64, 1012.63 (C-N, C-O stretching and aromatic C-H in plane bending), 879.54, 829.39, 759.95, 746.45 (Aromatic C-H out of plane bending). 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 7.17 (d, J = 9.2 Hz, 2H), 7.21 (d, J = 8.8 Hz, 2H), 7.41 (s, 1H), 7.47 (d, J = 8.4 Hz, 2H), 7.76 (d, J = 8.8 Hz, 2H), 7.88 (d, J = 8.8 Hz, 4H), 8.09 (s, 1H), 12.31 (s, 1H). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 105.23 (CH), 106.19 (C), 119.15 (2CH), 119.34 (C), 121.10 (2CH), 127.93 (2CH), 129.02 (2CH), 129.31 (2CH), 131.89 (C), 132.66 (C), 134.06 (C), 135.40 (2CH), 141.33 (CH), 149.78 (C), 156.04 (C), 161.28 (C), 169.02 (C). HRMS (ESI) (m/z): [M + H]+ 431.0751.
2-[2-((4-(4-Cyanophenoxy)phenyl)methylene)hydrazinyl]-4-(4-bromophenyl)thiazole (5). Dark beige powder. M.p. 217–218 °C. Yield: 88%. IR νmax (cm−1): 3284.77 (N-H stretching), 3116.97, 3047.53 (Aromatic C-H stretching), 2227.78 (C≡N stretching), 1593.20, 1571.99, 1556.55, 1494.83, 1473.62 (N-H bending, C=N and C=C stretching), 1415.75, 1396.46, 1352.10 (C-H bending), 1284.59, 1244.09, 1203.58, 1166.93, 1114.86, 1097.50, 1072.42, 1045.42, 1006.84 (C-N, C-O stretching and aromatic C-H in plane bending), 954.76, 933.55, 908.47, 881.47, 854.47, 833.25, 825.53, 729.09, 696.30, 686.66, 663.51, 632.65 (Aromatic C-H out of plane bending and C-S stretching). 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 7.17 (d, J = 9.2 Hz, 2H), 7.20 (d, J = 9.2 Hz, 2H), 7.41 (s, 1H), 7.61 (d, J = 8.4 Hz, 2H), 7.76 (d, J = 9.2 Hz, 2H), 7.81 (d, J = 8.4 Hz, 2H), 7.87 (d, J = 8.8 Hz, 2H), 8.08 (s, 1H), 12.31 (s, 1H). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 105.30 (CH), 106.19 (C), 119.14 (2CH), 119.34 (C), 121.09 (2CH), 121.24 (C), 128.23 (2CH), 129.01 (2CH), 131.90 (C), 132.21 (2CH), 134.46 (C), 135.39 (2CH), 141.27 (CH), 149.91 (C), 156.03 (C), 161.28 (C), 169.03 (C). HRMS (ESI) (m/z): [M + 2H]+ 477.0225.
2-[2-((4-(4-Cyanophenoxy)phenyl)methylene)hydrazinyl]-4-(4-cyanophenyl)thiazole (6). Metallic gold powder. M.p. 232–233 °C. Yield: 90%. IR νmax (cm−1): 3329.14 (N-H stretching), 3122.75, 3095.75, 3045.60 (Aromatic C-H stretching), 2978.09 (Aliphatic C-H stretching), 2218.14 (C≡N stretching), 1604.77, 1593.20, 1560.41, 1492.90, 1483.26 (N-H bending, C=N and C=C stretching), 1409.96, 1354.03 (C-H bending), 1284.59, 1238.30, 1193.94, 1168.86, 1122.57, 1101.35, 1049.28, 1014.56 (C-N, C-O stretching and aromatic C-H in plane bending), 908.47, 881.47, 867.97, 839.03, 815.89, 794.67, 769.60, 736.81, 715.59, 696.30, 657.73, 642.30, 605.65, 543.93 (Aromatic C-H out of plane bending and C-S stretching). 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 7.17 (d, J = 9.2 Hz, 2H), 7.21 (d, J = 8.8 Hz, 2H), 7.65 (s, 1H), 7.76 (d, J = 8.8 Hz, 2H), 7.87 (d, J = 9.2 Hz, 4H), 8.04 (d, J = 8.8 Hz, 2H), 8.09 (s, 1H), 12.29 (brs, 1H). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 106.22 (C), 108.27 (CH), 110.29 (C), 119.16 (2CH), 119.35 (C), 119.68 (C), 121.10 (2CH), 126.80 (2CH), 129.05 (2CH), 131.86 (C), 133.38 (2CH), 135.40 (2CH), 139.43 (C), 141.46 (CH), 149.53 (C), 156.08 (C), 161.28 (C), 169.22 (C). HRMS (ESI) (m/z): [M + H]+ 422.1092.
2-[2-((4-(4-Cyanophenoxy)phenyl)methylene)hydrazinyl]-4-(4-methylphenyl)thiazole (7). Pale yellow powder. M.p. 228–229 °C. Yield: 74%. IR νmax (cm−1): 3412.08 (N-H stretching), 3051.39 (Aromatic C-H stretching), 2918.30, 2864.29 (Aliphatic C-H stretching), 2223.92 (C≡N stretching), 1593.20, 1564.27, 1494.83 (N-H bending, C=N and C=C stretching), 1435.04 (C-H bending), 1246.02, 1201.65, 1166.93, 1085.92, 1035.77, 1014.56 (C-N, C-O stretching and aromatic C-H in plane bending), 879.54, 856.39, 831.32, 819.75, 759.95, 746.45, 715.59, 694.37 (Aromatic C-H out of plane bending and C-S stretching). 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 2.33 (s, 3H), 7.05–7.26 (m, 7H), 7.73–7.89 (m, 6H), 8.12 (s, 1H), 12.27 (s, 1H). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 21.50 (CH3), 103.61 (CH), 106.20 (C), 119.16 (2CH), 119.34 (C), 121.07 (2CH), 126.25 (2CH), 129.09 (2CH), 129.88 (2CH), 131.82 (C), 132.10 (C), 135.39 (2CH), 137.75 (C), 141.74 (CH), 150.27 (C), 156.09 (C), 161.25 (C), 168.85 (C). HRMS (ESI) (m/z): [M + H]+ 411.1300.
2-[2-((4-(4-Cyanophenoxy)phenyl)methylene)hydrazinyl]-4-(4-hydroxyphenyl)thiazole (8). Brown powder. M.p. 249–250 °C. Yield: 73%. IR νmax (cm−1): 3444.87, 3361.93 (N-H stretching), 3165.19, 3115.04, 3037.89 (Aromatic C-H stretching), 2877.79 (Aliphatic C-H stretching), 2223.92 (C≡N stretching), 1624.06, 1593.20, 1512.19, 1492.90 (N-H bending, C=N and C=C stretching), 1435.04, 1371.39 (C-H bending), 1249.87, 1203.58, 1184.29, 1166.93, 1078.21, 1041.56, 1012.63 (C-N, C-O stretching and aromatic C-H in plane bending), 956.69, 879.54, 852.54, 829.39, 802.39, 736.81, 702.09, 651.94, 632.65, 599.86 (Aromatic C-H out of plane bending and C-S stretching). 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 6.79 (d, J = 8.8 Hz, 2H), 7.05 (s, 1H), 7.15 (dd, J = 16.0, 8.4 Hz, 4H), 7.61 (d, J = 8.0 Hz, 2H), 7.74 (d, J = 8.8 Hz, 2H), 7.83 (d, J = 8.4 Hz, 2H), 8.13-8.14 (m, 1H), 9.21 (s, 1H), 12.30 (s, 1H). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 101.76 (CH), 106.24 (C), 116.08 (2CH), 119.21 (2CH), 119.34 (C), 121.04 (2CH), 125.33 (C), 127.91 (2CH), 129.29 (2CH), 131.56 (C), 135.40 (2CH), 143.30 (CH), 149.50 (C), 156.32 (C), 158.18 (C), 161.18 (C), 168.82 (C). HRMS (ESI) (m/z): [M + H]+ 413.1096.
2-[2-((4-(4-Cyanophenoxy)phenyl)methylene)hydrazinyl]-4-(4-methoxyphenyl)thiazole (9). Mustard-colored powder. M.p. 210–211 °C. Yield: 75%. IR νmax (cm−1): 3151.69, 3061.03 (Aromatic C-H stretching), 2958.80, 2835.36 (Aliphatic C-H stretching), 2223.92 (C≡N stretching), 1593.20, 1562.34, 1494.83 (N-H bending, C=N and C=C stretching), 1435.04, 1413.82, 1365.60 (C-H bending), 1288.45, 1244.09, 1201.65, 1166.93, 1085.92, 1028.06, 1014.56 (C-N, C-O stretching and aromatic C-H in plane bending), 879.54, 858.32, 833.25, 748.38, 711.73, 692.44 (Aromatic C-H out of plane bending and C-S stretching). 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 3.80 (s, 3H), 6.83 (d, J = 8.8 Hz, 1H), 6.99 (d, J = 8.4 Hz, 2H), 7.12–7.27 (m, 4H), 7.74–7.89 (m, 6H), 8.14 (s, 1H), 12.30 (s, 1H). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 55.06 (CH3), 101.67 (CH), 105.45 (C), 113.91 (2CH), 118.39 (2CH), 118.57 (C), 120.28 (2CH), 126.93 (2CH), 128.35 (2CH), 129.34 (C), 131.02 (C), 134.62 (2CH), 141.46 (CH), 149.27 (C), 155.80 (C), 159.08 (C), 160.60 (C), 168.03 (C). HRMS (ESI) (m/z): [M + H]+ 427.1254.
2-[2-((4-(4-Cyanophenoxy)phenyl)methylene)hydrazinyl]-4-(naphthalen-2-yl)thiazole (10). Vanilla powder. M.p. 204–205 °C. Yield: 88% IR νmax (cm−1): 3381.21 (N-H stretching), 3118.90, 3059.10 (Aromatic C-H stretching), 2960.73, 2914.44, 2872.01 (Aliphatic C-H stretching), 2223.92 (C≡N stretching), 1620.21, 1595.13, 1566.20, 1492.90 (N-H bending, C=N and C=C stretching), 1440.83, 1361.74 (C-H bending), 1278.81, 1242.16, 1197.79, 1166.93, 1124.50, 1095.57, 1047.35, 1008.77 (C-N, C-O stretching and aromatic C-H in plane bending), 968.27, 958.62, 947.05, 918.12, 900.76, 866.04, 825.53, 802.39, 754.17, 713.66, 680.87, 650.01, 636.51, 609.51, 542.00 (Aromatic C-H out of plane bending and C-S stretching). 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 7.16–7.24 (m, 4H), 7.48–7.58 (m, 3H), 7.76–7.82 (m, 2H), 7.84–7.98 (m, 5H), 8.02 (m, 1H), 8.12 (s, 1H), 8.40 (s, 1H), 12.30 (s, 1H). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 105.22 (CH), 106.21 (C), 119.17 (2CH), 119.37 (C), 121.12 (2CH), 124.63 (CH), 124.78 (CH), 126.71 (C), 127.14 (CH), 128.27 (CH), 128.80 (CH), 128.85 (CH), 129.02 (2CH), 131.98 (C), 132.73 (CH), 133.14 (C), 133.85 (C), 135.41 (2CH), 141.19 (CH), 151.10 (C), 156.03 (C), 161.31 (C), 168.98 (C). HRMS (ESI) (m/z): [M + H]+ 447.1317.






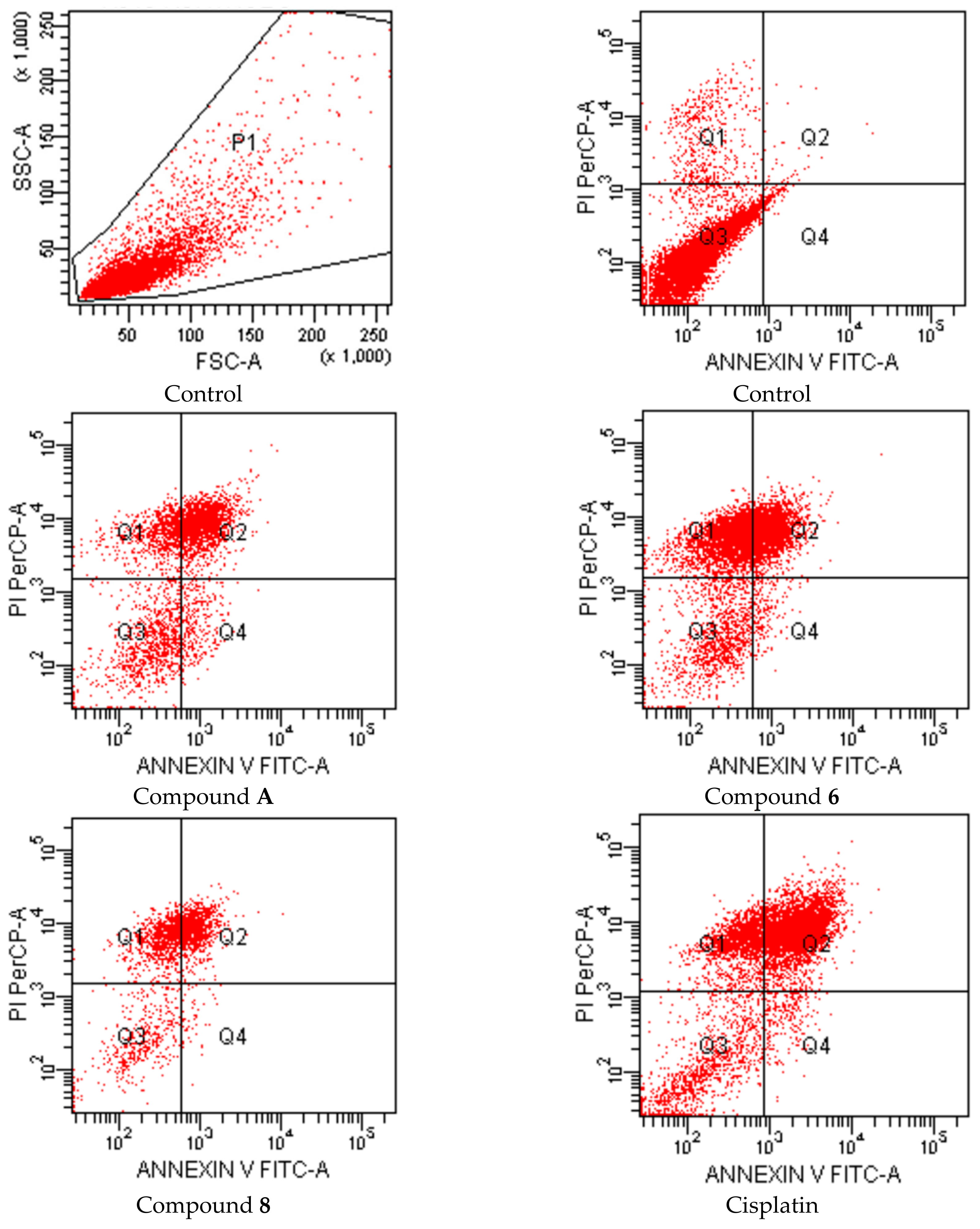
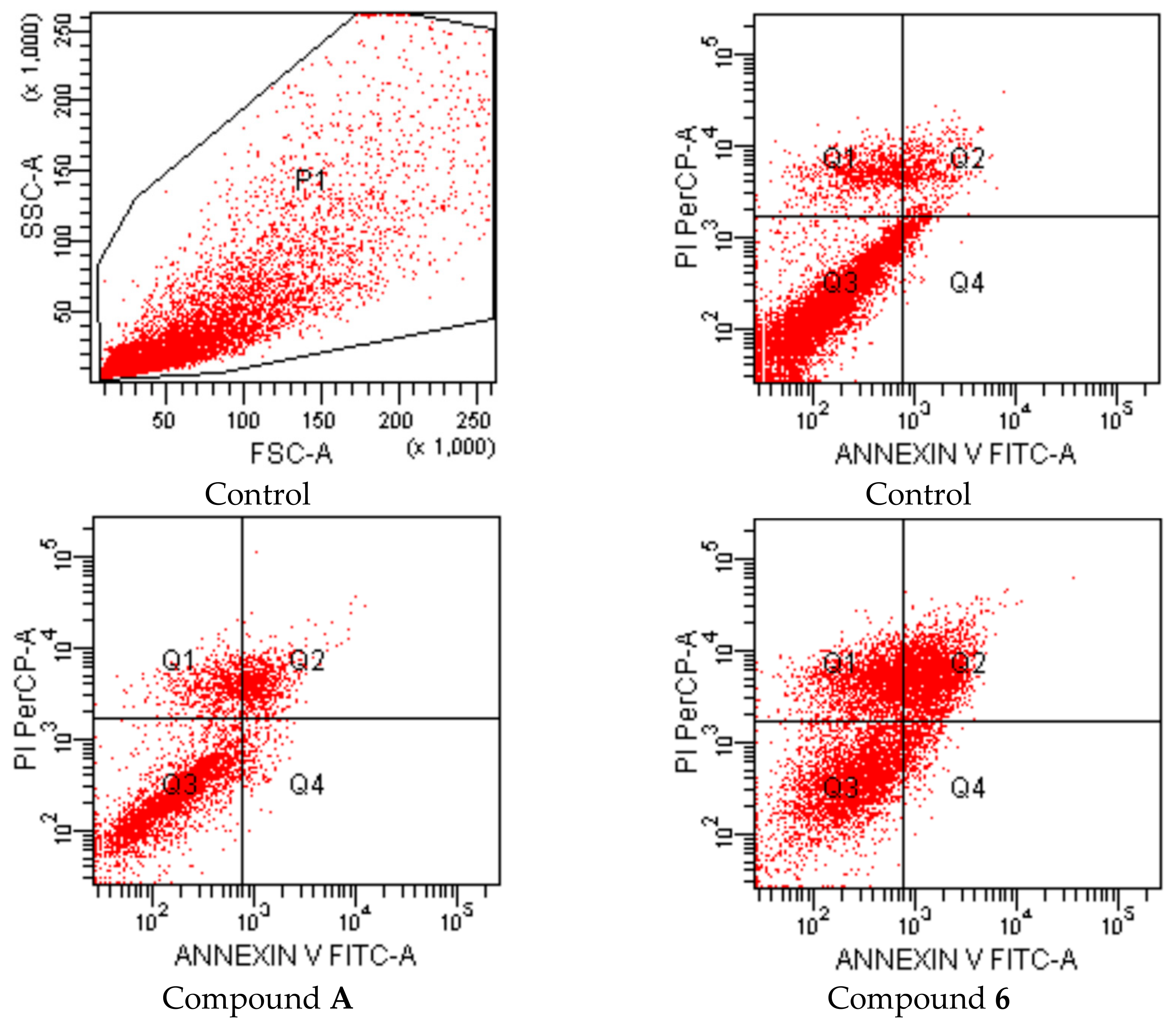
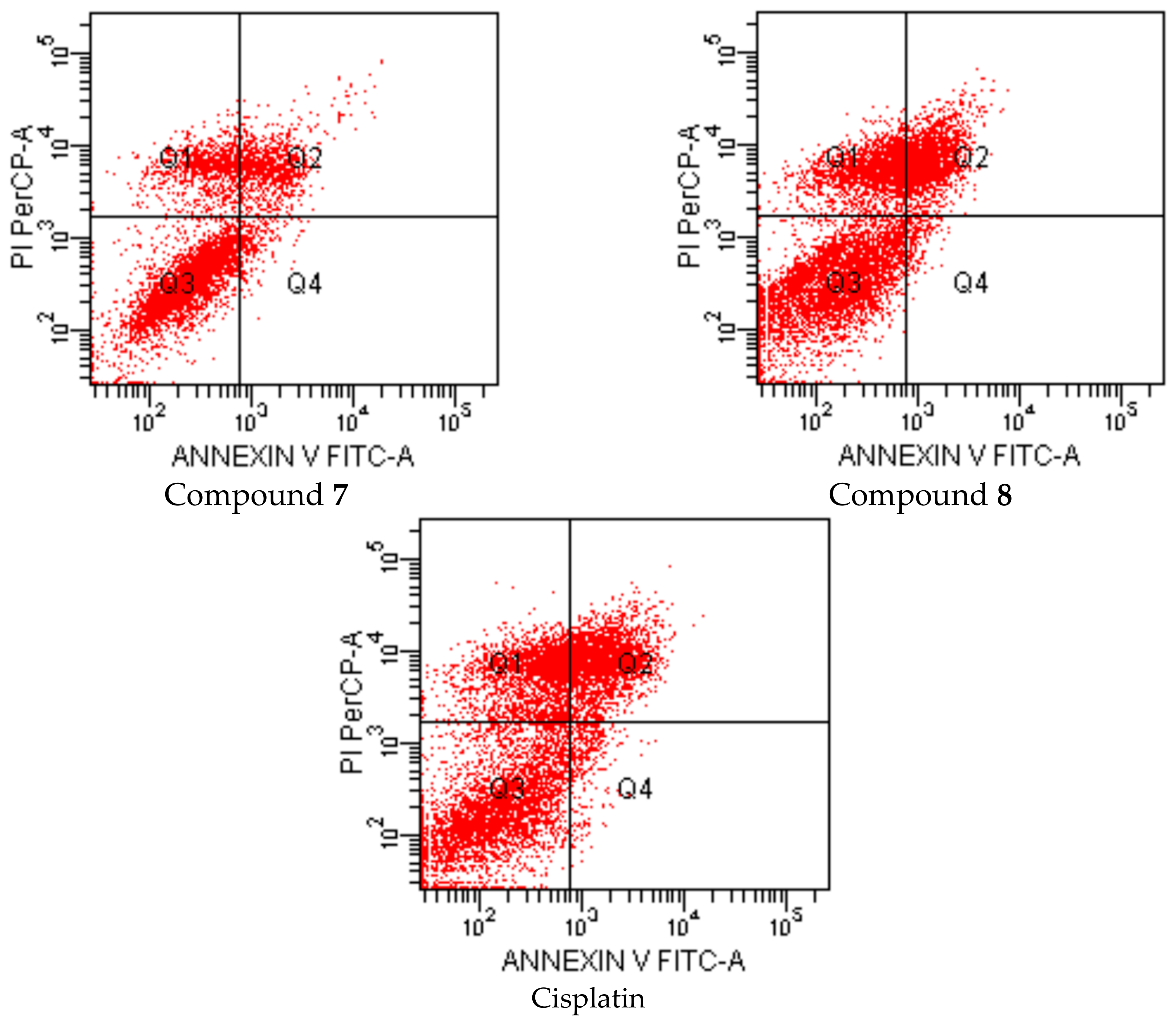
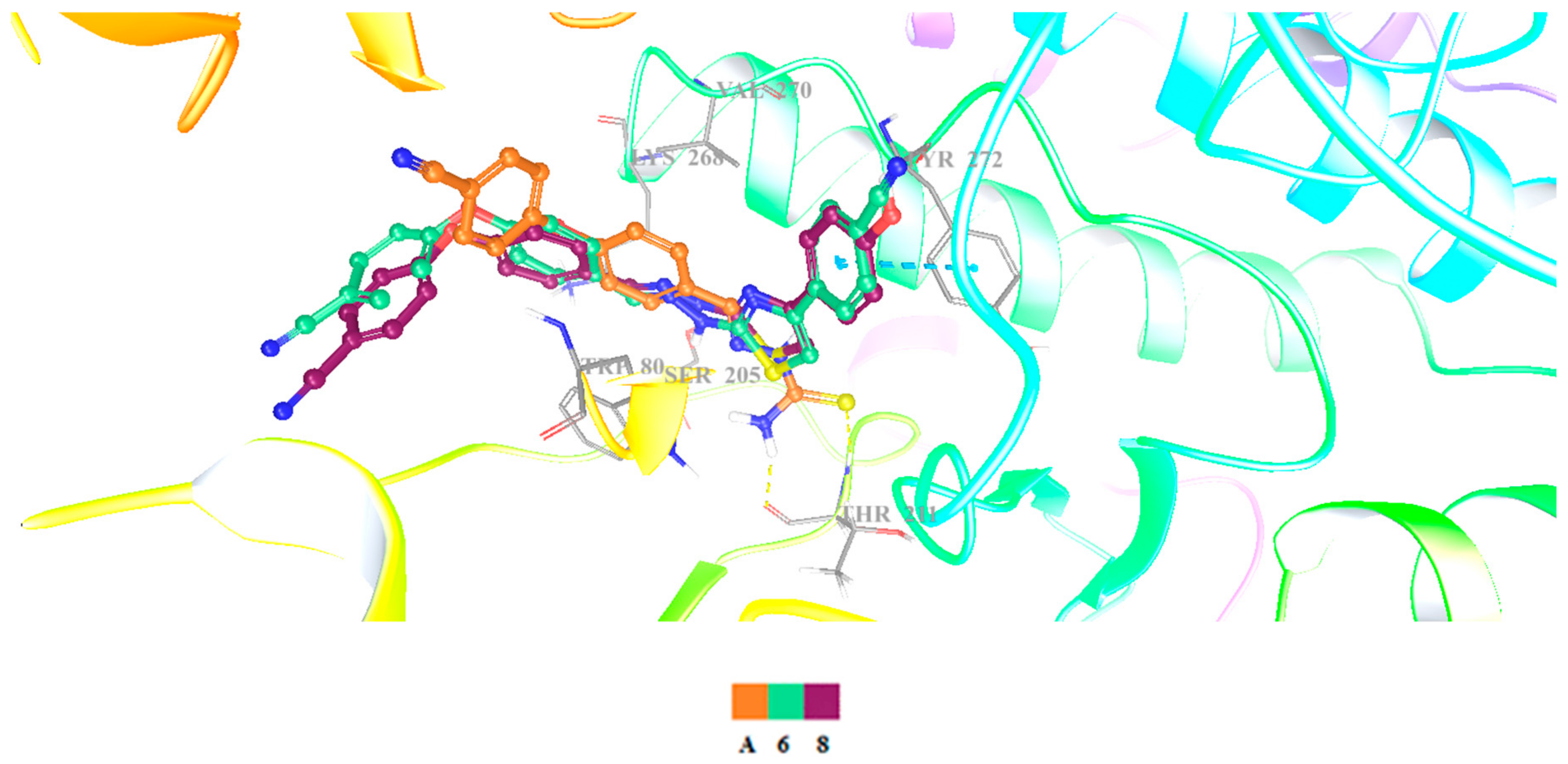
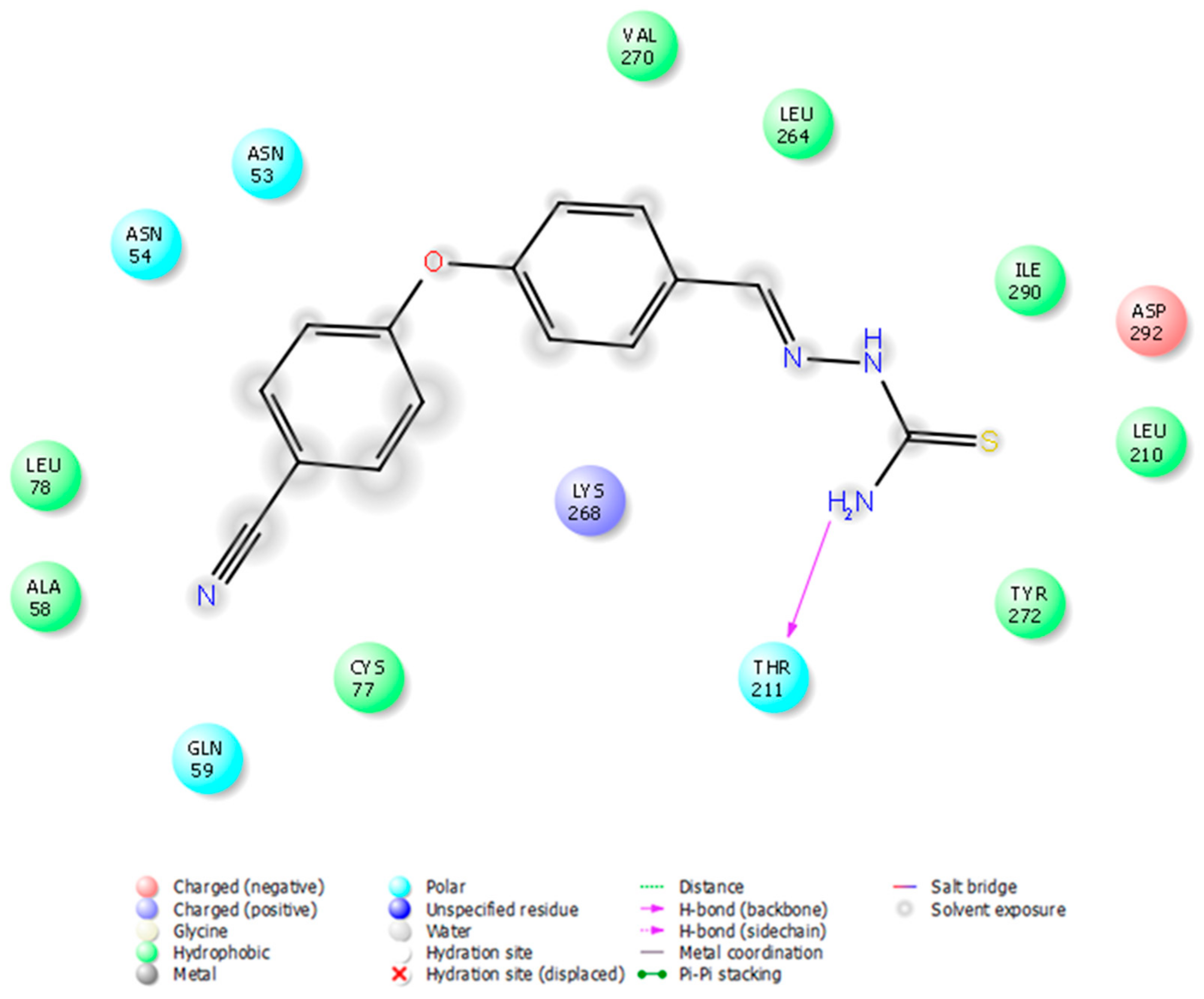
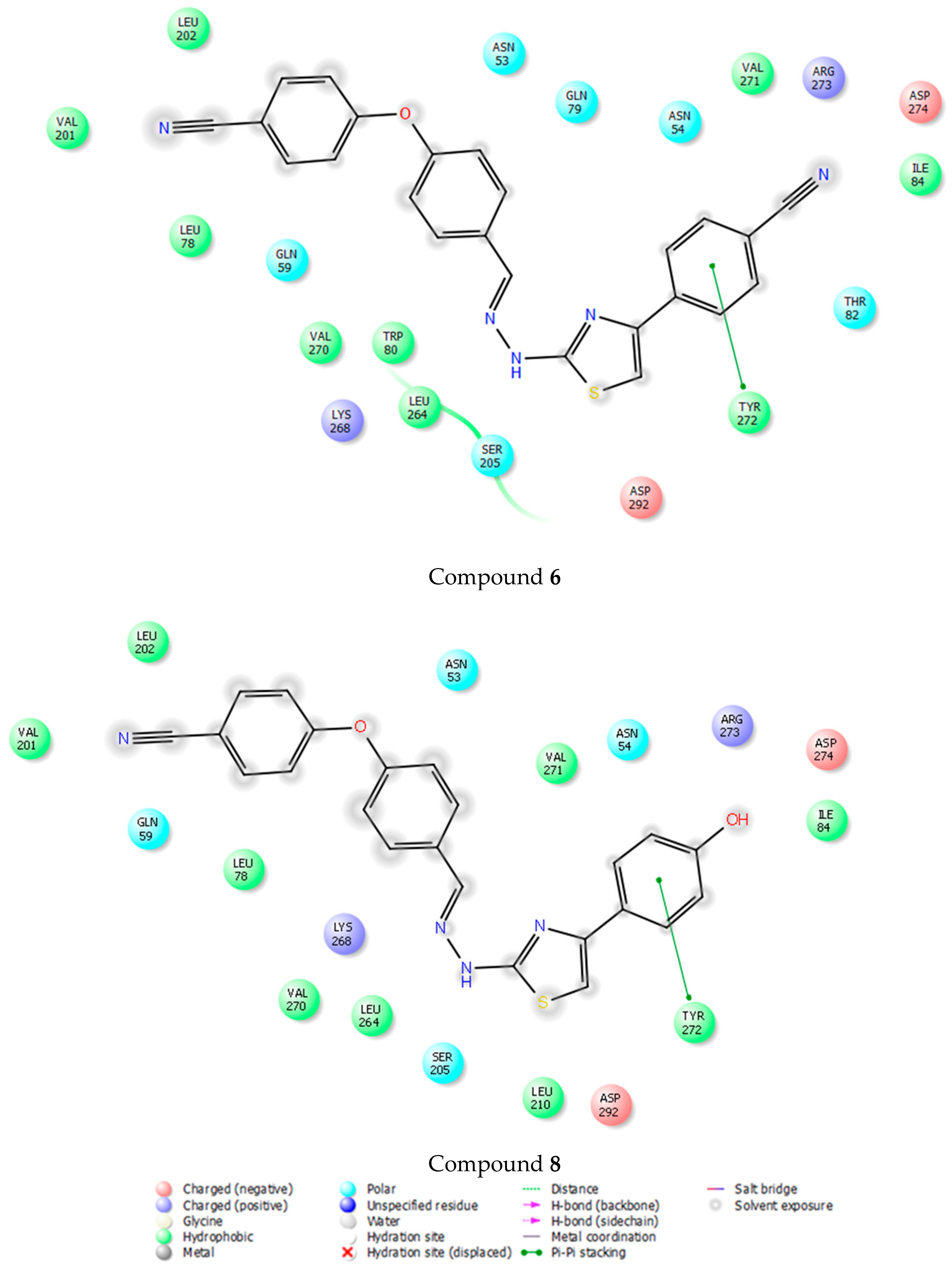

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}