1. Introduction
Glutamic acid and aspartic acid are the only proteinogenic amino acids with acidic side chains. Since their side chains are charged and hydrophilic they are frequently found on the surface of proteins. There they enhance the solubility of the protein and may also be involved in ionic protein–protein interactions. Due to their ability to act as proton donators and acceptors, glutamate and aspartate residues are also frequently found in active centres of enzymes [
1], where they may be involved in the catalytic reaction and/or substrate binding. In addition to their role as building blocks of proteins, both amino acids fulfil a number of functions in their free form. Glutamic acid is a key compound in metabolism since it links the citrate cycle with amino acid metabolism [
2]. Moreover, it is a precursor for a multitude of compounds including the amino acid glutamine and γ-aminobutyric acid (GABA), the chief inhibitory neurotransmitter in the mammalian nervous system [
3]. Glutamate itself is the principal excitatory neurotransmitter in the mammalian brain [
4], where it is involved in cognitive processes like learning and memory [
5]. Aspartate can also stimulate neuronal receptors but less efficiently than glutamate [
6,
7]. In addition, glutamate is crucial for detoxification of ammonia in the mammalian brain [
8] and for detoxification of xenobiotic compounds in many organisms [
9,
10]. In plants, glutamate has an essential role in amino acid anabolism. The α-amino group allows assimilation and dissimilation of ammonia and is the building block of all other amino acids. In plants GABA, arginine and proline are synthesised from glutamate [
11,
12]. Glutamate is also a precursor for synthesis of chlorophyll [
13].
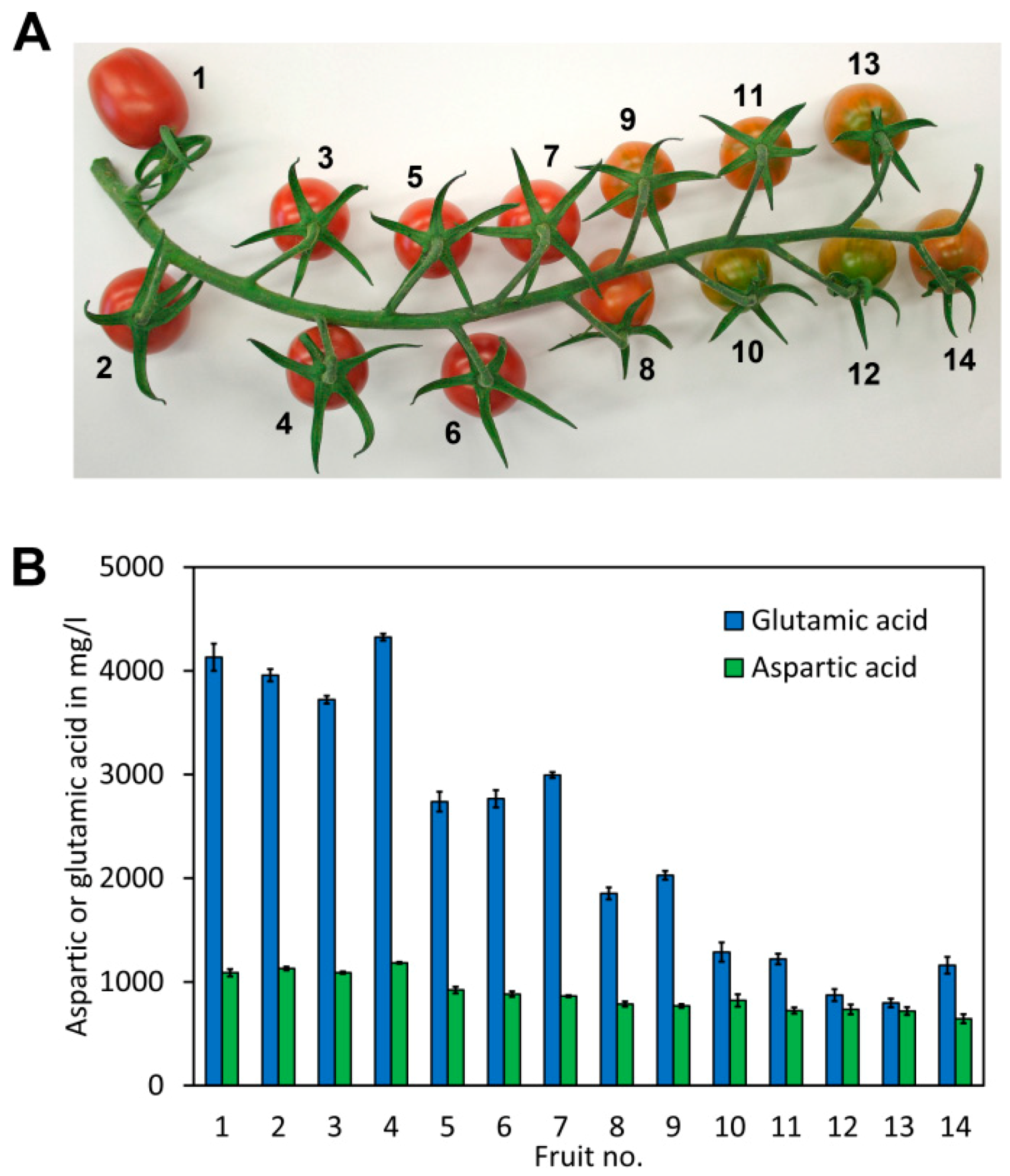
In a variety of vegetables and fruits such as tomatoes, green peas, mushrooms, and cabbage glutamic acid is found in considerable amounts. Ripe tomato fruits contain aspartate and glutamate in amounts of approximately 800 and 3000 mg/kg, respectively. During ripening of tomatoes, changes of aspartate and glutamate concentrations happen. In the case of glutamate, a red, fully ripened tomato contains a significantly higher concentration of glutamate than an immature green tomato. Aspartate is also reported to increase during tomato ripening, though at a much lower magnitude [
14]. Amongst the total free amino acids in ripened tomato fruit, glutamate represents approximately 55% of the relative molar concentration, making it the main free amino acid [
15].
Due to the importance of glutamate and aspartate in metabolism and development, a number of methods have been developed for their quantification. Both compounds can be quantified together with other amino acids by chromatographic methods. Classically, amino acids are separated in so-called amino acid analysers by cation exchange chromatography and post-column derivatisation with ninhydrin to give coloured compounds that are detected at 440 nm and 570 nm [
16]. For more sensitive and rapid quantification a number of high-performance liquid chromatography (HPLC) techniques, mainly with pre-column derivatisation, were reported. A widely applied reagent is phenyl isothiocyanate [
17,
18,
19], which is also part of Edman chemistry for protein sequencing [
20]. For highly sensitive quantification of amino acids, derivatisation with o-phthaldialdehyde (OPA) and a thiole like 2-mercaptoethanol [
21,
22,
23] or more recently 3-mercaptopropionic acid [
24] and fluorescence detection is frequently applied. Disadvantages of o-phthaldialdehyde derivatisation are the low stability of the derivatives and that it does not react with proline, making a second derivatisation step with fluorenylmethyloxycarbonyl (FMOC) necessary [
25]. In contrast, 6-aminoquinoline-
N-hydroxy-succinimidyl carbamate (AQC) reacts rapidly with both primary and secondary amines [
26] to highly fluorescent derivatives. Disadvantages of AQC are that the emission wavelength depends on the water content of the eluent, that in solutions with a high water content significant fluorescence quenching is observed [
26], and that the reagent is very expensive. More recently, derivatisation with AQC has been applied for quantification of amino acids by liquid chromatography-mass spectrometry (LC-MS) [
27] and liquid chromatography-tandem mass spectrometry (LC-MS/MS) in plant material [
28]. In these methods the AQC tag is used to enhance binding of the amino acids to the reversed phase column and for improvement of detection. Underivatised amino acids can also be analysed by LC-MS/MS [
29,
30]. LC-MS/MS-based methods are often extremely sensitive and highly selective. However, major drawbacks are frequently observed matrix effects caused by ion suppression [
31], which can be particularly significant for methods with limited separation of the amino acids from the matrix [
30]. These effects can be compensated by using stable isotope labelled standards. However, such standards are very costly and also the equipment required for LC-MS/MS is very expensive and requires specially trained personnel. In addition, the high sensitivity offered by LC-MS/MS is not an advantage for analysis of amino acids in tomatoes since they contain free amino acids in the mg/L to g/L range, which makes several hundred-fold dilution necessary and may thereby even introduce dilution errors. In addition, for tomato breeding and fruit quality assessment only the glutamate and aspartate contents are relevant since these two amino acids have, in contrast to other amino acids, a significant impact on the taste [
32,
33].
Underivatised amino acids can also be detected electrochemically [
34,
35,
36]. Nevertheless, amino acids are often derivatised to improve sensitivity or enhance separation on reverse phase columns [
37,
38]. Although electrochemical detection of amino acids offers high sensitivity this technique is comparatively little applied since the use of electrochemical detectors is not very widespread.
For specific quantification of glutamate enzyme assays are available where glutamate is oxidised by glutamate oxidase to α-ketoglutarate and hydrogen peroxide, the latter reacts subsequently by catalysis of horse radish peroxidase with an artificial substrate to a fluorescent compound [
39]. Alternatively, glutamate dehydrogenase can be used to reduce NAD (oxidised β-nicotinamide adenine dinucleotide) to NADH (reduced β-nicotinamide adenine dinucleotide), which can be either directly measured at 340 nm [
40] or used for reduction of a formazan to a blue dye [
41]. Similar enzymatic assays have also been developed for quantification of aspartic acid [
42,
43]. Enzymatic assays for both glutamate and aspartate have been commercialised as test kits allowing convenient quantification of these metabolites. However, disadvantages are the limited shelf-life of the reagents, sensitivity to inhibitors, and the relatively high costs per assay.
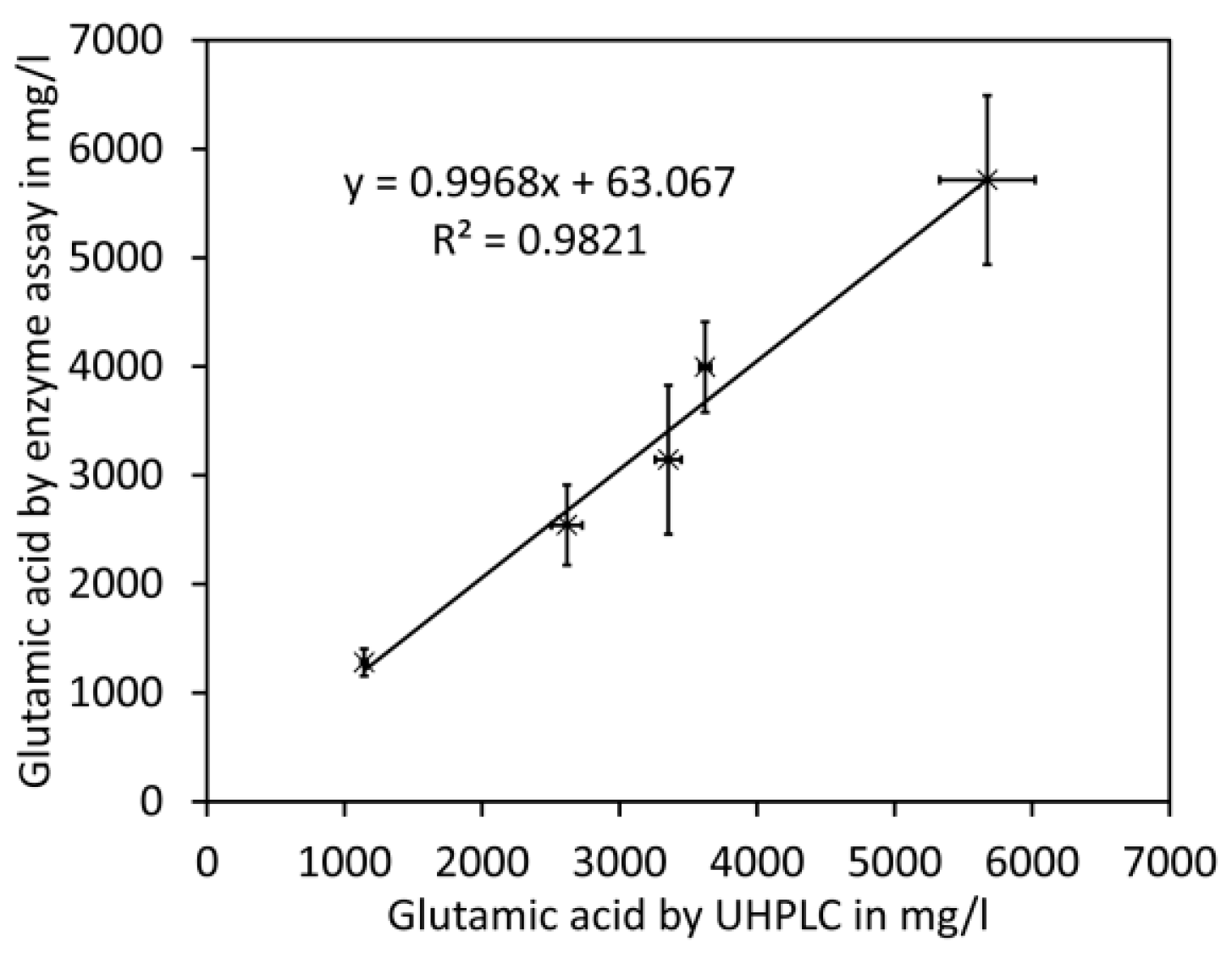
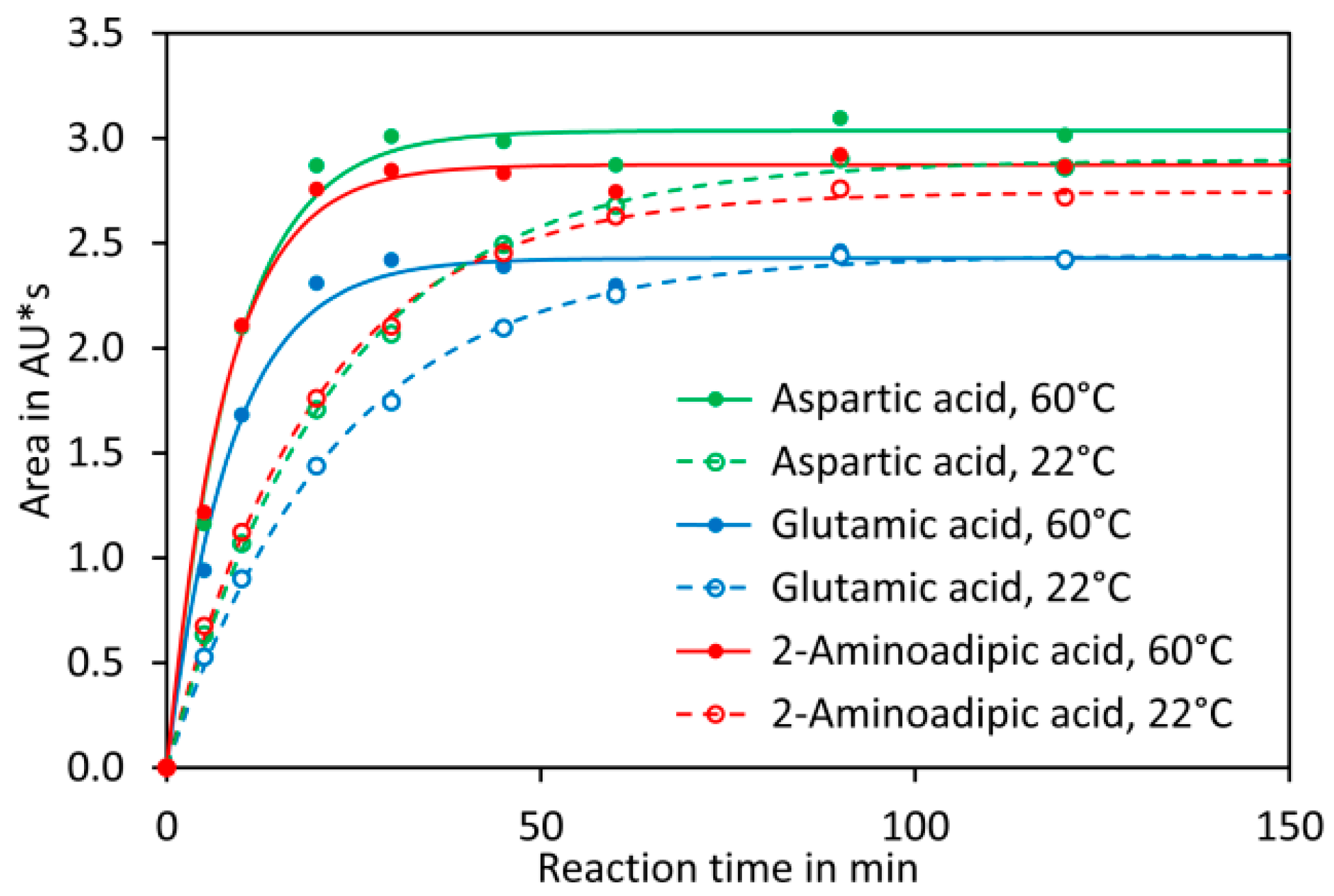
Here we present a quick and high-throughput ultra-high performance liquid chromatography (UHPLC) method for simultaneous quantification of glutamate and aspartate in tomato fruit by pre-column derivatisation with 2,4-dinitro-1-fluorobenzene (DNFB) and UV detection at 363 nm. DNFB was originally introduced by Frederick Sanger for labelling the N-terminal amino acid of proteins and peptides [
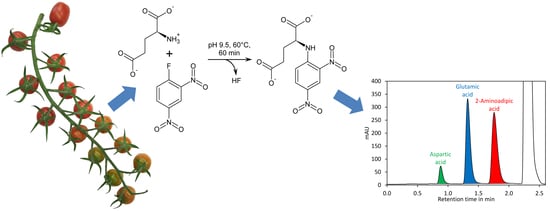
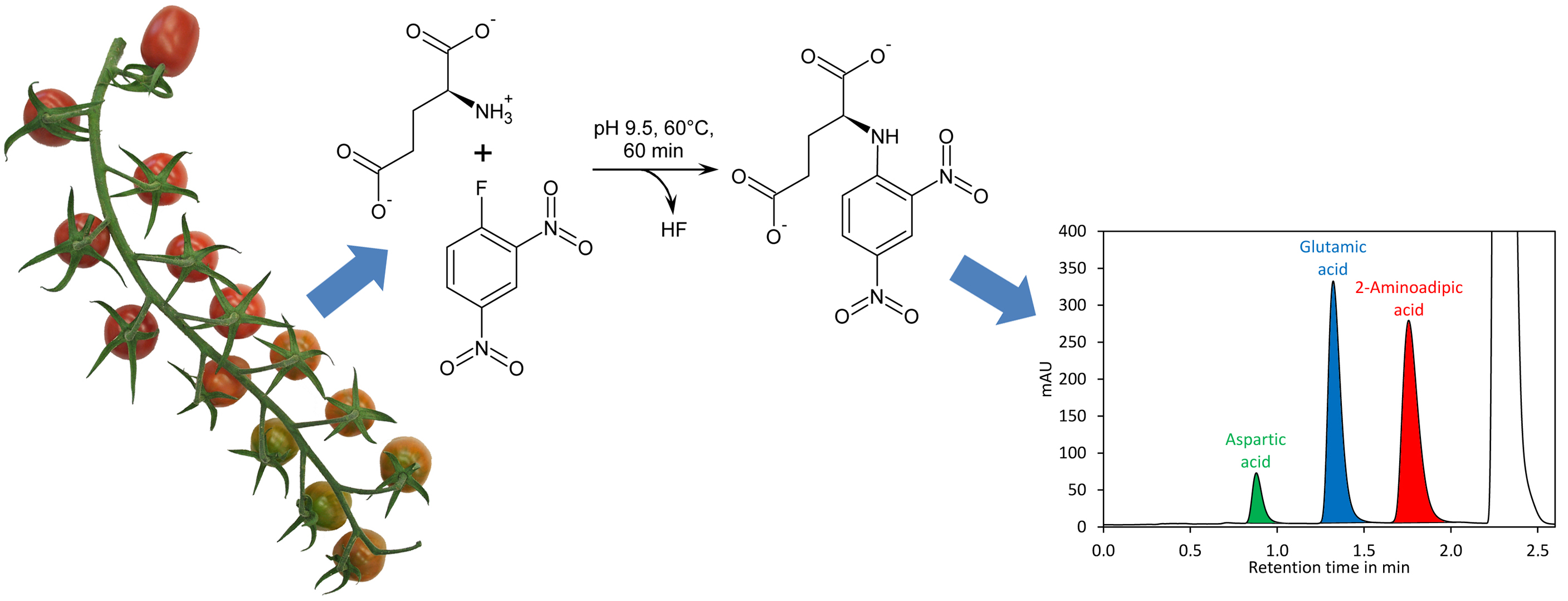
44]. The derivatisation of amino acids with DNFB is unique since an amine bond is formed (
Figure 1A), which can even resist harsh conditions required for hydrolysis of proteins (incubation in 6 M hydrochloric acid at 110 °C for 24 h). In addition, the amine nitrogen in the obtained derivative has, due to the electron capturing properties of the 2,4-dinitrobenzene moiety, an extremely low nucleophilicity and thus double derivatisation is not observed. DNFB has been used for labelling of free amino acids and subsequent quantification of the derivatives by HPLC with UV detection, but the formation of 2,4-dinitrophenol from the reaction of excess reagent with water (
Figure 1A) made this method complicated [
45]. Recently, a method for quantification of glutamate by pre-column derivatisation with 2,4-dinitrofluorobenzene and subsequent reversed-phase HPLC has been described. However, this method required removal of 2,4-dinitrophenol by extraction with diethyl ether, making sample preparation tedious [
46].
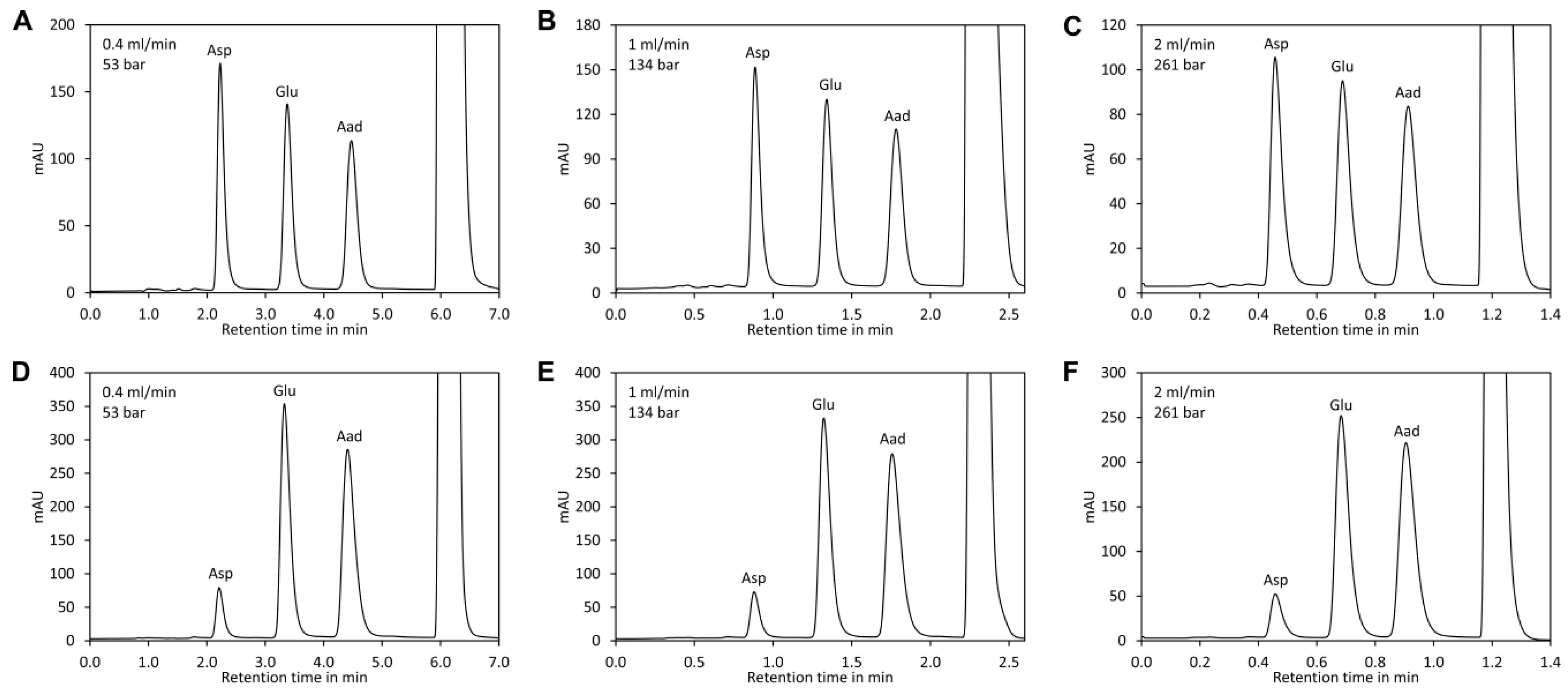
Here we provided an optimised protocol for derivatisation and chromatographic conditions that make removal of 2,4-dinitrophenol unnecessary and allow separation including column re-equilibration in 1.6 min. Importantly, the derivatisation reagent and the required buffers are inexpensive. Thus, the method is simple, rapid, and cost-efficient.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}