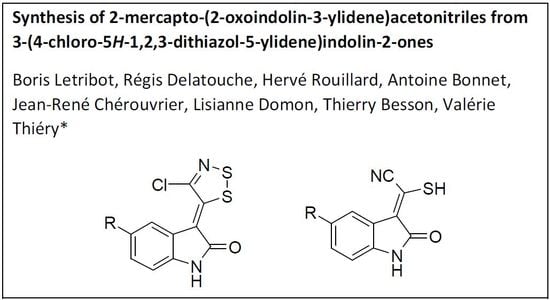
Synthesis of 2-Mercapto-(2-Oxoindolin-3-Ylidene)Acetonitriles from 3-(4-Chloro-5H-1,2,3-Dithiazol-5-Ylidene)Indolin-2-ones


 and
and
Abstract
:
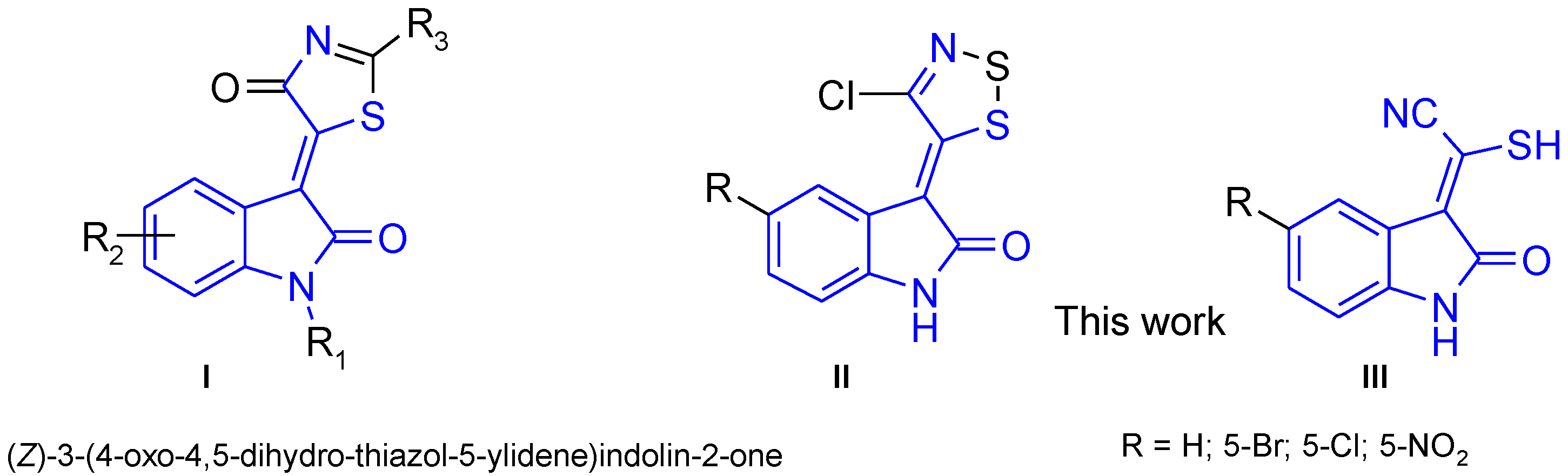
1. Introduction
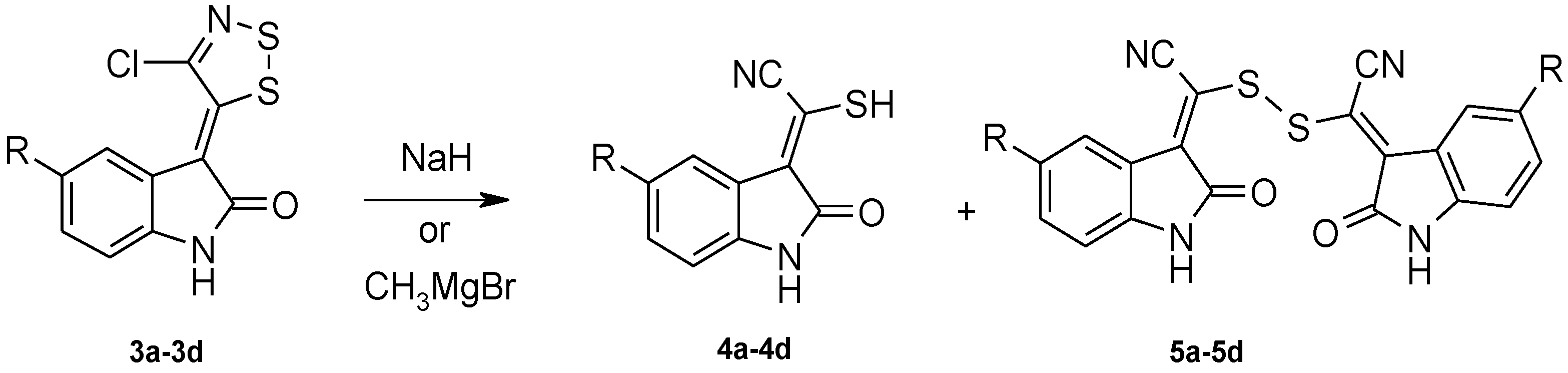
2. Results and Discussion
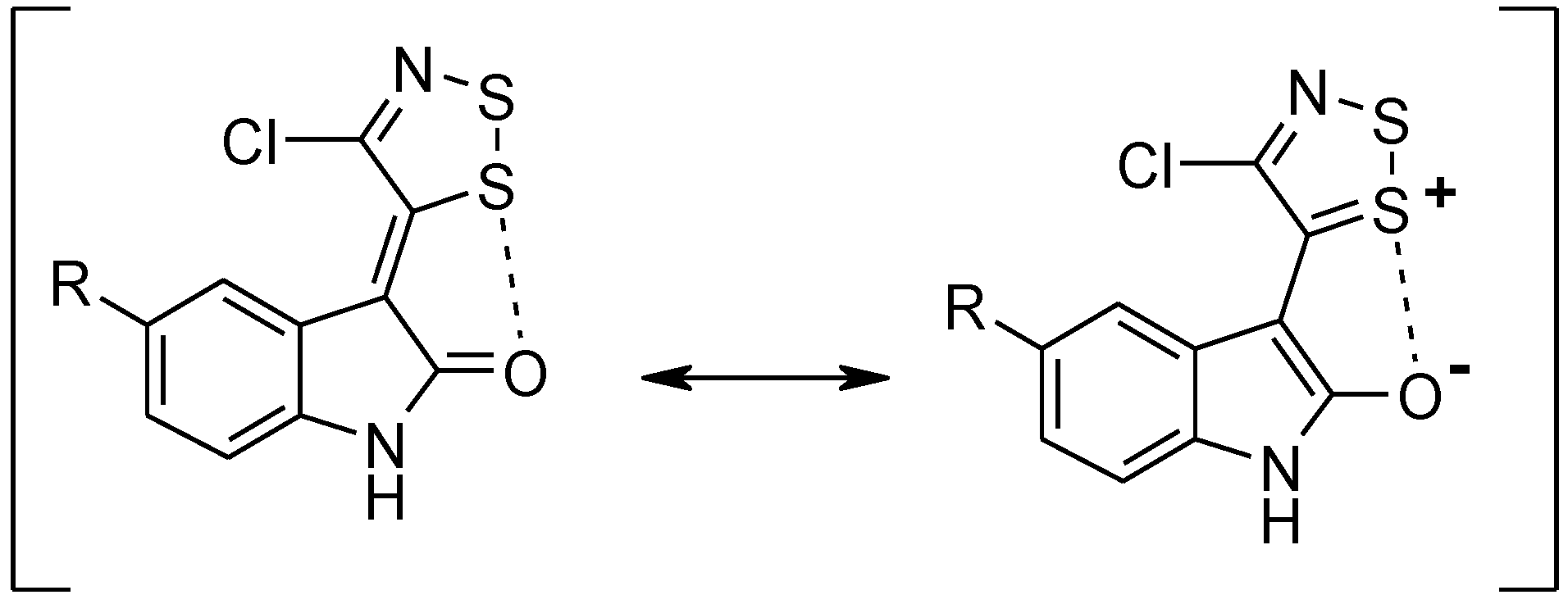
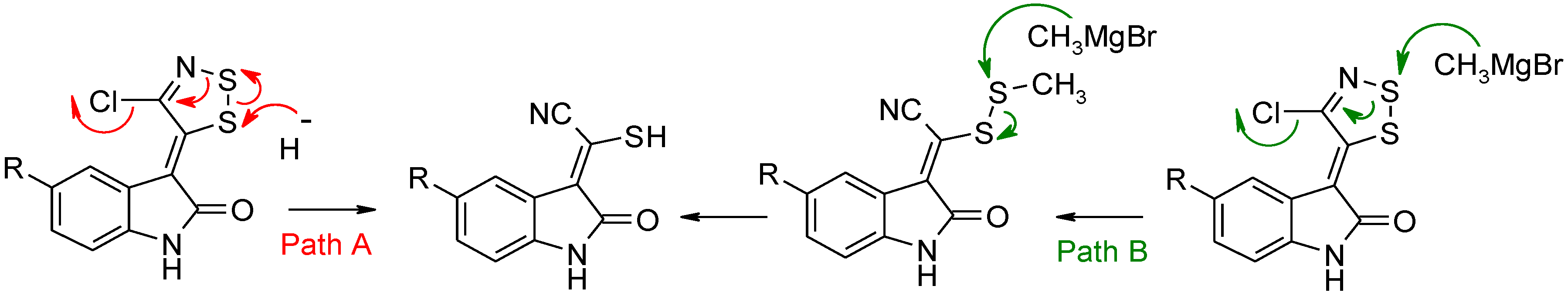
Dithiazole Ring Opening
3. Materials and Methods
3.1. General Information
3.2. Synthesis
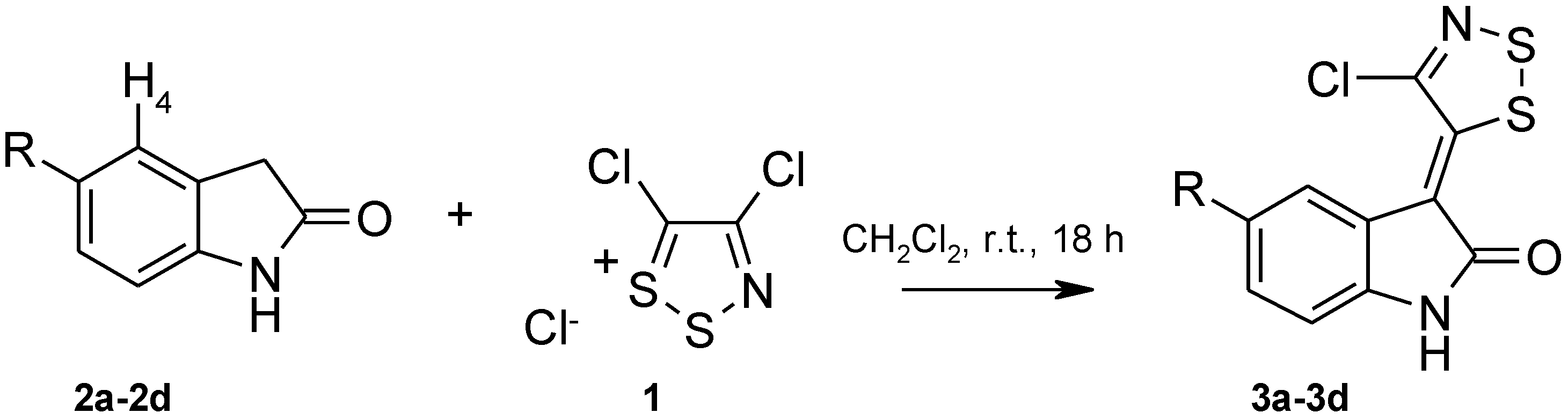
3.2.1. General Procedure for the Synthesis of 3-(4-Chloro-5H-1,2,3-Dithiazol-5-Ylidene)Indolin-2-one Derivatives (Table 2, 3a–d).
3.2.2. General Procedure for the Synthesis of 2-Mercapto-2-(2-Oxoindolin-3-Ylidene)Acetonitrile Derivatives (Table 3, 4a–4d)
Path A with NaH
Path B with CH3MgBr
3.2.3. General Procedure for the Synthesis of 2-(2-Oxoindolin-3-Ylidene)Acetonitrile Derivatives (Table 4, 6a–d)
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Weniger, B.; Jiang, Y.; Anton, R.; Bastida, J.; Varea, T.; Quirion, J.-C. Oxindole Alkaloids from Neolaugeria Resinosa. Phytochemistry 1993, 32, 1587–1590. [Google Scholar] [CrossRef]
- Hosoe, T.; Nozawa, K.; Kawahara, N.; Fukushima, K.; Nishimura, K.; Miyaji, M.; Kawai, K. Isolation of a New Potent Cytotoxic Pigment along with Indigotin from the Pathogenic Basidiomycetous Fungus Schizophyllum commune. Mycopathologia 1999, 146, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Tang, C.-P.; Ke, C.-Q.; Li, X.-Q.; Liu, J.; Gan, L.-S.; Weiss, H.-C.; Gesing, E.-R.; Ye, Y. Constituents of Trigonostemon chinensis. J. Nat. Prod. 2009, 73, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Millemaggi, A.; Perry, A.; Whitwood, A.C.; Taylor, R.J.K. Telescoped Enolate Arylation/HWE Procedure for the Preparation of 3-Alkenyl-Oxindoles: The First Synthesis of Soulieotine. Eur. J. Org. Chem. 2009, 18, 2947–2952. [Google Scholar] [CrossRef]
- Uehata, K.; Kimura, N.; Hasegawa, K.; Arai, S.; Nishida, M.; Hosoe, T.; Kawai, K.; Nishida, A. Total Synthesis of Schizocommunin and Revision of Its Structure. J. Nat. Prod. 2013, 76, 2034–2039. [Google Scholar] [CrossRef] [PubMed]
- Kaur, M.; Singh, M.; Chadha, N.; Silakari, O. Oxindole: A chemical prism carrying plethora of therapeutic benefits. Eur. J. Med. Chem. 2016, 123, 858–894. [Google Scholar] [CrossRef] [PubMed]
- Bort, A.; Quesada, S.; Ramos-Torres, Á.; Gargantilla, M.; Priego, E.M.; Raynal, S.; Lepifre, F.; Gasalla, J.M.; Rodriguez-Henche, N.; Castro, A.; et al. Identification of a novel 2-oxindole fluorinated derivative as in vivo antitumor agent for prostate cancer acting via AMPK activation. Sci. Rep. 2018, 8, 4370. [Google Scholar] [CrossRef] [PubMed]
- Riham, F.G. Facile synthesis of simple 2-oxindole-based compounds with promising antiproliferative activity. Future Med. Chem. 2018, 10, 269–282. [Google Scholar] [CrossRef]
- Davis, H.J.; Kavanagh, M.E.; Balan, T.; Abell, C.; Coyne, A.G. Spirooxindoles as novel 3D-fragment scaffolds: Synthesis and screening against CYP121 from M. tuberculosis. Bioorg. Med. Chem. Lett. 2016, 26, 3735–3740. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.-W.; Wang, J.; Xiao, J.-A.; Li, J.; Xiang, H.-Y.; Chen, X.-Q.; Yang, H. l-Pyroglutamic sulphonamide as hydrogen-bonding organocatalyst: Enantioselective Diels-Alder cyclization to construct carbazolespirooxindoles. J. Org. Chem. 2017, 82, 6441–6449. [Google Scholar] [CrossRef] [PubMed]
- Jayakmar, S.; Louven, K.; Strohmann, C.; Kumar, K. A tunable and enantioselective hetero-Diels–Alder reaction provides access to distinct piperidinoyl spirooxindoles. Angew. Chem. Int. Ed. 2017, 56, 15945–15949. [Google Scholar] [CrossRef] [PubMed]
- Morales-Ríos, M.S.; García-Velgara, M.; Cervantes-Cuevas, H.; Alvarez-Cisneros, C.; Joseph-Nathan, P. Push–pull and pull–push effects in isatylidenes. Magn. Reson. Chem. 2000, 38, 172–176. [Google Scholar] [CrossRef]
- Li, G.; Feng, X.; Du, H. Palladium-catalyzed asymmetric allylic amination of racemic butadiene monoxide with isatin derivatives. Org. Biomol. Chem. 2015, 13, 5826–5830. [Google Scholar] [CrossRef] [PubMed]
- Millemaggi, A.; Taylor, R.J.K. 3-Alkenyl-oxindoles: Natural products, pharmaceuticals, and recent synthetic advances in tandem/telescoped approaches. Eur. J. Org. Chem. 2010, 4527–4547. [Google Scholar] [CrossRef]
- Lubkoll, J.; Millemaggi, A.; Perry, A.; Taylor, R.J.K. Tandem Horner–Wadsworth–Emmons/Heck procedures for the preparation of 3-alkenyl-oxindoles: The synthesis of Semaxanib and GW441756. Tetrahedron 2010, 66, 6606–6612. [Google Scholar] [CrossRef]
- Miura, T.; Toyoshima, T.; Ito, Y.; Murakami, M. Synthesis of stereodefined 3-alkylideneoxindoles by palladium-catalyzed reactions of 2-(alkynyl)aryl isocyanates with thiols and alcohols. Chem. Lett. 2009, 38, 1174–1175. [Google Scholar] [CrossRef]
- Tang, S.; Yu, Q.-F.; Peng, P.; Li, J.-H.; Zhong, P.; Tang, R.-Y. Palladium-catalyzed carbonylative annulation reaction of 2-(1-alkynyl)benzenamines: Selective synthesis of 3-(halomethylene)indolin-2-ones. Org. Lett. 2007, 9, 3413–3416. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Okada, T.; Nagasawa, H. Oxindole Synthesis by Palladium-catalysed aromatic C–H alkenylation. Chem. Commun. 2010, 46, 2462–2464. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.-H.; Zhang, X.-C.; Wei, Y.; Shi, M. Chemoselective reduction of isatin-derived electron-deficient alkenes using alkylphosphanes as reduction reagents. Eur. J. Org. Chem. 2011, 14, 2668–2672. [Google Scholar] [CrossRef]
- Rassu, G.; Zambrano, V.; Pinna, L.; Curti, C.; Battistini, L.; Sartori, A.; Pelosi, G.; Zanardi, F.; Casiraghi, G. Direct regio-, diastereo-, and enantioselective vinylogous Michael Addition of prochiral 3-alkylideneoxindoles to nitroolefins. Adv. Synth. Catal. 2013, 355, 1881–1886. [Google Scholar] [CrossRef]
- Yang, X.-H.; Li, K.; Song, R.-J.; Li, J.-H. Room-temperature Palladium-catalyzed intramolecular oxidative aminocarbonylation of vinylic C(sp2)–H Bonds with amines and CO. Eur. J. Org. Chem. 2014, 3, 616–623. [Google Scholar] [CrossRef]
- Amol, P.J.; Amjad, A.; Ravi, P.S. Vinylogous nucleophilic substitution of the hydroxy group in diarylmethanols with 3-propenyl-2-silyloxyindoles: Towards the synthesis of α-alkylidene-δ-diaryl-2-oxindoles. Adv. Synth. Catal. 2017, 359, 1508–1510. [Google Scholar] [CrossRef]
- Craig, R.; Sorrentino, E.; Connon, S.J. Enantioselective alkylation of 2-oxindoles catalyzed by a bifunctional phase-transfer catalyst: Synthesis of (−)-Debromoflustramine B. Chem. Eur. J. 2018, 24, 4528–4531. [Google Scholar] [CrossRef] [PubMed]
- Petrini, M.; Chiurchiù, E.; Rossi, F.V.; Palmieri, A. Oxidative conversion of sulfonyl indoles into 3-alkylidene-2-oxindoles under flow chemical conditions. Synthesis 2018, 50, 371–376. [Google Scholar] [CrossRef]
- Beauchard, A.; Ferandin, Y.; Frère, S.; Lozach, O.; Blairvacq, M.; Meijer, L.; Thiéry, V.; Besson, T. Synthesis of novel 5-substituted Indirubins as protein kinases inhibitors. Bioorg. Med. Chem. 2006, 14, 6434–6443. [Google Scholar] [CrossRef] [PubMed]
- Havrylyuk, D.; Zimenkovsky, B.; Vasylenko, O.; Gzella, A.; Lesyk, R. Synthesis of new 4-thiazolidinone-, pyrazoline-, and Isatin-based conjugates with promising antitumor Activity. J. Med. Chem. 2012, 55, 8630–8641. [Google Scholar] [CrossRef] [PubMed]
- Erben, F.; Michalik, D.; Feist, H.; Kleeblatt, D.; Hein, M.; Matin, A.; Iqbal, J.; Langer, P. Synthesis and antiproliferative activity of (Z)-1-Glycosyl-3-(5-oxo-2-thioxoimidazolidin-4-ylidene)indolin-2-ones and (Z)-3-(2-glycosylsulfanyl-4-oxo-4,5-dihydro-thiazol-5-ylidene)indolin-2-Ones. RSC Adv. 2014, 4, 10879–10893. [Google Scholar] [CrossRef]
- Appel, R.; Janssen, H.; Siray, M.; Knoch, F. Synthese und reaktionen des 4,5-dichlor-1,2,3-dithiazolium-chlorids. Eur. J. Inorg. Chem. 1985, 118, 1632–1643. [Google Scholar] [CrossRef]
- Rees, C.W. Polysulfur-nitrogen heterocyclic chemistry. J. Heterocycl. Chem. 1992, 29, 639–651. [Google Scholar] [CrossRef]
- Rakitin, O.A. Comprehensive Heterocyclic Chemistry II I; Katritzky, A.R., Ramsden, C.A., Scriven, E.F.V., Taylor, R.J.K., Eds.; Elsevier: Oxford, UK, 2008; Volume 6, p. 1. [Google Scholar]
- Konstantinova, L.S.; Rakitin, O. Synthesis and properties of 1,2,3-dithiazoles. Russ. Chem. Rev. 2008, 77, 521–546. [Google Scholar] [CrossRef]
- Foucourt, A.; Chosson, E.; Besson, T. 4,5-Dichloro-1,2,3-dithiazol-1-ium chloride. e-EROS 2009. [Google Scholar] [CrossRef]
- Koutentis, P.A. The preparation and characterization of 5-substituted-4-chloro-1,2,3-dithiazolium salts and their conversion into 4-substituted-3-chloro-1,2,5-thiadiazoles. Molecules 2005, 10, 346–359. [Google Scholar] [CrossRef] [PubMed]
- Cuadro, A.M.; Alvarez-Buila, J. 4,5-Dichloro-1,2,3-dithiazolium chloride (Appel salt). Reactions with N-nucleophiles. Tetrahedron 1994, 50, 10037–10046. [Google Scholar] [CrossRef]
- Rakitin, O.A.; Rees, C.W.; Vlasova, O.G. 1,2,4-Thiadiazole 4-oxides. Chem. Commun. 1996, 11, 1273–1274. [Google Scholar] [CrossRef]
- Koyioni, M.; Manoli, M.; Manolis, M.J.; Koutentis, P.A. Reinvestigating the reaction of 1H-pyrazol-5-amines with 4,5-dichloro-1,2,3-dithiazolium chloride: A route to pyrazolo[3,4-c]isothiazoles and pyrazolo[3,4-d]thiazoles. J. Org. Chem. 2014, 79, 4025–4037. [Google Scholar] [CrossRef] [PubMed]
- Baraldi, P.G.; Pavani, M.G.; del Carmen Nunez, M.; Brigidi, P.; Vitali, B.; Gambari, R.; Romagnoli, R. Antimicrobial and antitumor activity of N-heteroimmine-1,2,3-dithiazoles and their transformation in triazolo-, imidazo-, and pyrazolopirimidines. Bioorg. Med. Chem. 2002, 10, 449–456. [Google Scholar] [CrossRef]
- Mirallai, S.I.; Manos, M.J.; Koutentis, P.A. One-Step Conversion of 2-amino-N′-arylbenzamidines into 3-aryl-4-imino-3,4-dihydroquinazoline-2-carbonitriles using 4,5-dichloro-1,2,3-dithiazolium chloride. J. Org. Chem. 2013, 78, 9906–9913. [Google Scholar] [CrossRef] [PubMed]
- Konstantinova, L.S.; Bol’shakov, O.I.; Obruchnikova, N.V.; Laborie, H.; Tanga, A.; Sopéna, V.; Lanneluc, I.; Picot, L.; Sablé, S.; Thiéry, V. One-Pot Synthesis of 5-phenylimino, 5-thieno or 5-oxo-1,2,3-dithiazoles and evaluation of their antimicrobial and antitumor activity. Bioorg. Med. Chem. Lett. 2009, 19, 136–141. [Google Scholar] [CrossRef] [PubMed]
- De Fatima Pereira, M.; Sarghe, E.; Rouillard, H.; Domon, L.; Chérouvrier, J.-R.; Thiéry, V. Synthesis of novel heterocyclic fused pyrimidin-4-one derivatives from imino-1,2,3-dithiazoles. Arkivoc 2017, 335–345. [Google Scholar] [CrossRef]
- Emayan, K.; English, R.F.; Koutentis, P.A.; Rees, C.W. New routes to benzothiophenes, isothiazoles and 1,2,3-dithiazoles. J. Chem. Soc. Perkin Trans. 1 1997, 22, 3345–3349. [Google Scholar] [CrossRef]
- Koutentis, P.A.; Rees, C.W. Reactions of tetracyanoethylene oxide with 1,2,3-dithiazoles. J. Chem. Soc. Perkin Trans. 1 1998, 2505–2509. [Google Scholar] [CrossRef]
- Christoforou, I.C.; Koutentis, P.A.; Rees, C.W. Reactions of 1,2,3-dithiazoles with halogenated malononitriles. J. Chem. Soc. Perkin Trans. 1 2002, 1236–1241. [Google Scholar] [CrossRef]
- Kalogirou, A.S.; Koutentis, P.A. The reaction of 4,5-dichloro-1,2,3-dithiazolium chloride with dimethylsulfonium dicyanomethylide: An improved synthesis of (4-chloro-1,2,3-dithiazolylidene)-malononitrile. Tetrahedron 2009, 65, 6850–6854. [Google Scholar] [CrossRef]
- D’hooge, B.; Dehaen, W. Stereoisomerism in the adducts of Appel salt with activated methylenes. Bull. Soc. Chim. Belg. 1997, 106, 729–730. [Google Scholar]
- Clarke, D.; Emayan, K.; Rees, C.W. New synthesis of isothiazoles from primary enamines. J. Chem. Soc. Perkin Trans. 1 1998, 1, 77–81. [Google Scholar] [CrossRef]
- Kim, K. Synthesis and Reactions of 1,2,3-Dithiazoles. Sulfur Rep. 1998, 21, 147–210. [Google Scholar] [CrossRef]
- Chang, Y.-G.; Cho, H.S.; Kim, K. Novel synthesis and reactions of 5,7-dialkyl-4,6-dioxo-4,5,6,7-tetrahydro-isothiazolo[3,4-d]pyrimidine-3-carbonitriles and 6-methyl-4-oxo-4H-1-aza-5-oxa-2-thiaindene-3-carbonitrile. Org. Lett. 2003, 5, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Jeon, M.-K.; Kim, K. Synthesis of new 5-alkylidene-4-chloro-5H-1,2,3-dithiazoles and their stereochemistry. Tetrahedron 1999, 55, 9651–9667. [Google Scholar] [CrossRef]
- Kalogirou, A.S.; Koutentis, P.A. The degradation of 4,5-dichloro-1,2,3-dithiazolium chloride in wet solvents. Tetrahedron 2009, 65, 6859–6862. [Google Scholar] [CrossRef]
- Besson, T.; Rees, C.W. New route to 4-alkoxyquinazoline-2-carbonitriles. J. Chem. Soc. Perkin Trans. 1 1996, 23, 2857–2860. [Google Scholar] [CrossRef]
- Michaelidou, S.S.; Koutentis, P.A. The Conversion of 2-(4-chloro-5H-1,2,3-dithiazolylideneamino)benzonitriles into 3-aminoindole-2-carbonitriles using triphenylphosphine. Tetrahedron 2009, 65, 8428–8433. [Google Scholar] [CrossRef]
- Bénéteau, V.; Besson, T.; Guillard, J.; Léonce, S.; Pfeiffer, B. Synthesis and in vitro antitumour evaluation of benzothiazole-2-carbonitrile derivatives. Eur. J. Med. Chem. 1999, 34, 1053–1060. [Google Scholar] [CrossRef]
- Rees, C.W.; Roe, D.G.; Thiéry, V. Imidazoquinolinethiones from quinolines: A new molecular rearrangement. Chem. Commun. 1996, 24, 2775–2776. [Google Scholar] [CrossRef]
- Besson, T.; Guillard, J.; Rees, C.W.; Thiéry, V. New syntheses of aryl isothiocyanates. J. Chem. Soc. Perkin Trans. 1 1998, 5, 889–892. [Google Scholar] [CrossRef]
- Logé, C.; Testard, A.; Thiéry, V.; Lozach, O.; Blairvacq, M.; Robert, J.-M.; Meijer, L.; Besson, T. Novel 9-oxo-thiazolo[5,4-f]quinazoline-2-carbonitrile derivatives as dual cyclin-dependent kinase 1 (CDK1)/glycogen synthase kinase-3 (GSK-3) inhibitors: Synthesis, biological evaluation and molecular modeling studies. Eur. J. Med. Chem. 2008, 43, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Kalogirou, A.S.; Christoforou, I.C.; Ioannidou, H.A.; Manos, M.J.; Koutentis, P.A. Ring transformation of (4-chloro-5H-1,2,3-dithiazol-5-ylidene)acetonitriles to 3-haloisothiazole-5-carbonitriles. RSC Adv. 2014, 4, 7735–7748. [Google Scholar] [CrossRef]
- Christoforou, I.C.; Kalogirou, A.S.; Koutentis, P.A. The preparation of dicyano-1,3,4-thiadiazole and tricyanothiazole via 1,2,3-dithiazole chemistry. Tetrahedron 2009, 65, 9967–9972. [Google Scholar] [CrossRef]
- Besson, T.; Guillaumet, G.; Lamazzi, C.; Rees, C.W.; Thiéry, V. N-(Cyanothioformyl)indoline; a new indoline ring forming reaction. J. Chem. Soc. Perkin Trans. 1 1998, 24, 4057–4060. [Google Scholar] [CrossRef]
- Long, D.R.; Richards, C.G.; Ross, M.S.F. The stereochemistry of 2-oxoindolin-3-ylidene derivatives. J. Heterocycl. Chem. 1978, 15, 633–636. [Google Scholar] [CrossRef]
- Osman, F.H.; El-Samahy, F.A. On the reaction of Isatin with cyanomethylene(triphenyl)-phosphorane. A nucleophilic attack of alkyl phosphites on the carbon–carbon double bond of (E)-oxindolylideneacetonitrile. Tetrahedron 2000, 56, 1863–1871. [Google Scholar] [CrossRef]
- Chen, M.; Gan, L.; Lin, S.; Wang, X.; Li, L.; Li, Y.; Zhu, C.; Wang, Y.; Jiang, B.; Jiang, J.; et al. Alkaloids from the root of Isatis Indigotica. J. Nat. Prod. 2012, 75, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 3 and 4 are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Starting Material | R | Dithiazole 3 | Yield (%) a | Chemical Shift of H4 in ppm b |
|---|---|---|---|---|
| 2a | H | 3a | 96 | 8.19 (7.20) c |
| 2b | 5-Br | 3b | 96 | 8.36 (7.36) |
| 2c | 5-Cl | 3c | 99 | 8.24 (7.20) |
| 2d | 5-NO2 | 3d | 85 | 9.13 (8.07) |
| Dithiazole | R | Mercapto | Yield % | Yield % | Dimer | Yield % | Yield % |
|---|---|---|---|---|---|---|---|
| Compound | NaH | CH3MgBr | NaH | CH3MgBr | |||
| 3a | H | 4a | 49 | 33 | 5a | 46 | 30 |
| 3b | 5-Br | 4b | 66 | 25 | 5b | 33 | 5 |
| 3c | 5-Cl | 4c | 68 | - | 5c | 30 | - |
| 3d | 5-NO2 | 4d | 88 | - | 5d | - | - |
| Mercapto | δ of H4 (in ppm) | Dimer | δ of H4 (in ppm) |
|---|---|---|---|
| acetonitrile | |||
| 4a | 8.99 | 5a | 8.10 |
| 4b | 9.23 | 5b | 9.08 |
| 4c | 9.55 | 5c | 8.86 |
| 4d | 9.91 | 5d | - |
| Compd. | R | Yield (%) a | δ ppm (Z) Isomer | δ ppm (Z) Isomer | δ ppm (E) Isomer | δ ppm (E) Isomer | % (E) Isomer b |
|---|---|---|---|---|---|---|---|
| 6 | H4 | H-vinyl | H4 | H-vinyl | |||
| 6a | H | 88 | 7.72 | 6.94 | 8.08 | 6.31 | 80 |
| 6b | 5-Br | 26 | 7.93 | 6.82 | 8.07 | 6.54 | 80 |
| 6c | 5-Cl | 27 | 7.81 | 6.83 | 7.93 | 6.55 | 86 |
| 6d | 5-NO2 | 56 | 8.61 | 7.34 | 8.72 | 6.80 | 83 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Letribot, B.; Delatouche, R.; Rouillard, H.; Bonnet, A.; Chérouvrier, J.-R.; Domon, L.; Besson, T.; Thiéry, V. Synthesis of 2-Mercapto-(2-Oxoindolin-3-Ylidene)Acetonitriles from 3-(4-Chloro-5H-1,2,3-Dithiazol-5-Ylidene)Indolin-2-ones. Molecules 2018, 23, 1390. https://doi.org/10.3390/molecules23061390
Letribot B, Delatouche R, Rouillard H, Bonnet A, Chérouvrier J-R, Domon L, Besson T, Thiéry V. Synthesis of 2-Mercapto-(2-Oxoindolin-3-Ylidene)Acetonitriles from 3-(4-Chloro-5H-1,2,3-Dithiazol-5-Ylidene)Indolin-2-ones. Molecules. 2018; 23(6):1390. https://doi.org/10.3390/molecules23061390
Chicago/Turabian StyleLetribot, Boris, Régis Delatouche, Hervé Rouillard, Antoine Bonnet, Jean-René Chérouvrier, Lisianne Domon, Thierry Besson, and Valérie Thiéry. 2018. "Synthesis of 2-Mercapto-(2-Oxoindolin-3-Ylidene)Acetonitriles from 3-(4-Chloro-5H-1,2,3-Dithiazol-5-Ylidene)Indolin-2-ones" Molecules 23, no. 6: 1390. https://doi.org/10.3390/molecules23061390
APA StyleLetribot, B., Delatouche, R., Rouillard, H., Bonnet, A., Chérouvrier, J. -R., Domon, L., Besson, T., & Thiéry, V. (2018). Synthesis of 2-Mercapto-(2-Oxoindolin-3-Ylidene)Acetonitriles from 3-(4-Chloro-5H-1,2,3-Dithiazol-5-Ylidene)Indolin-2-ones. Molecules, 23(6), 1390. https://doi.org/10.3390/molecules23061390