Ultra-Liquid Chromatography Tandem Mass Spectrometry (UPLC-MS/MS)-Based Pharmacokinetics and Tissue Distribution Study of Koumine and the Detoxification Mechanism of Glycyrrhiza uralensis Fisch on Gelsemium elegans Benth.


Abstract
1. Introduction
2. Results
2.1. Method Development
2.2. Method Validation
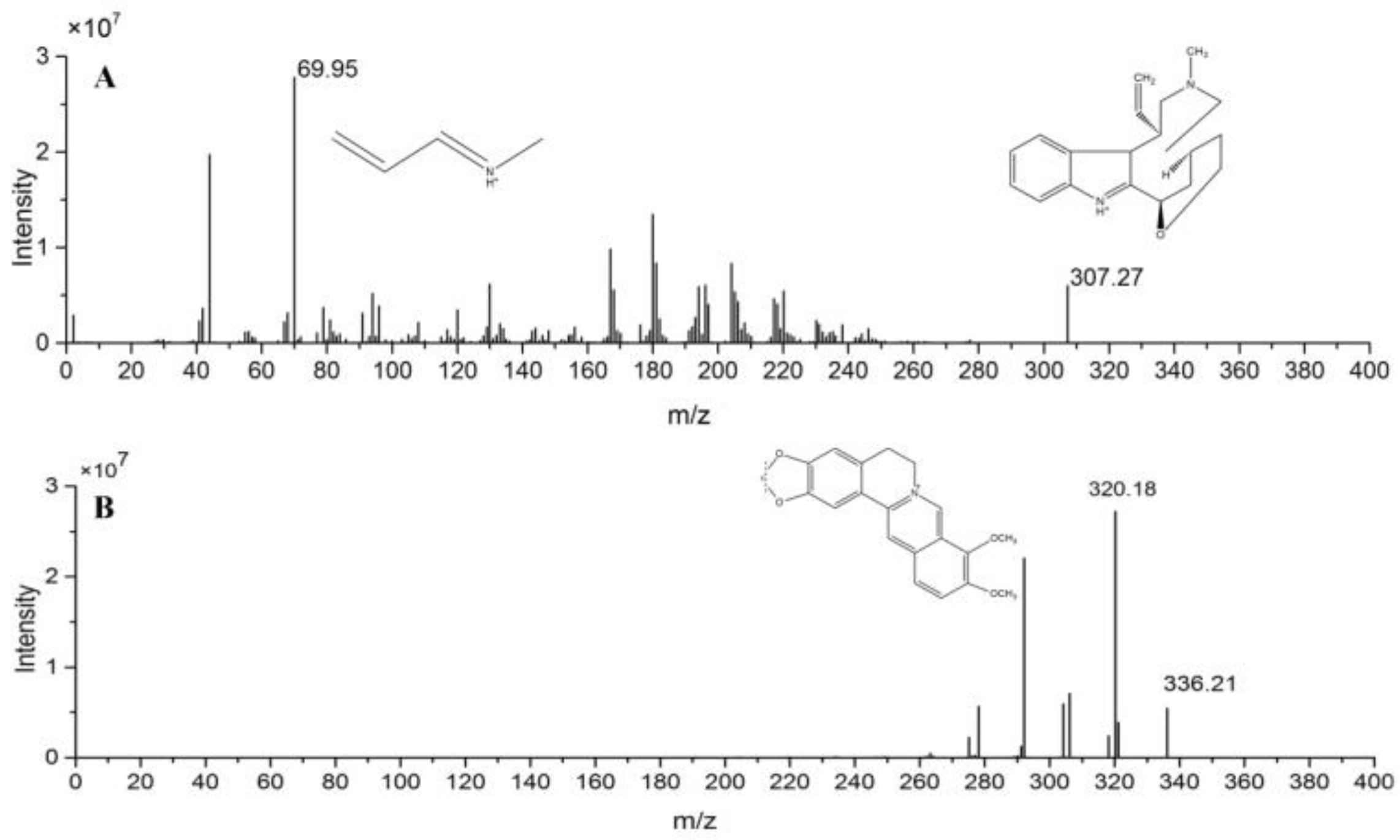
2.2.1. Specificity
2.2.2. Linearity and Lower Limit of Quantification LLOQ
2.2.3. Precision and Accuracy
2.2.4. Extraction Recovery and Matrix Effect
2.2.5. Stability
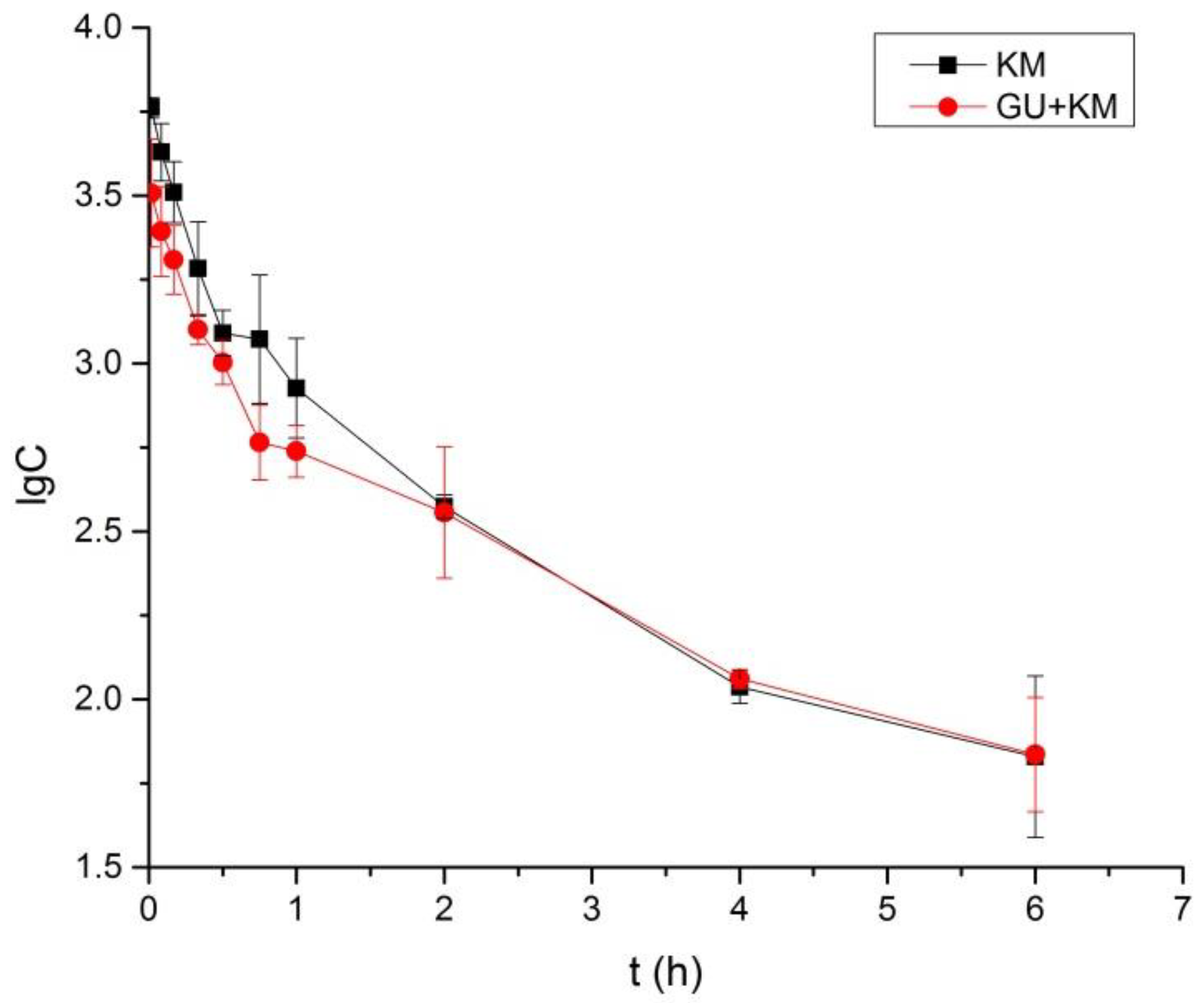
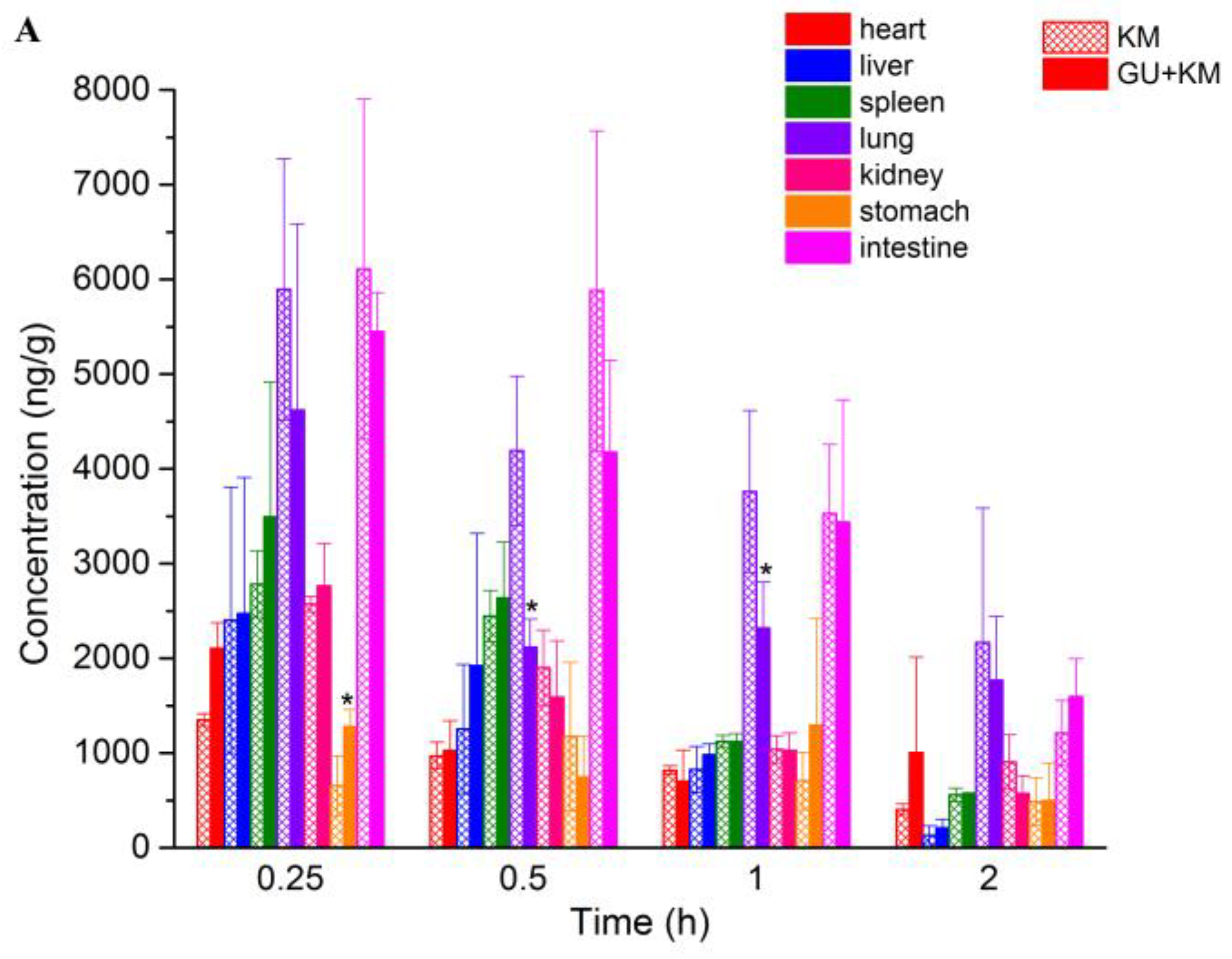
2.3. Pharmacokinetics Study and Tissue Distribution
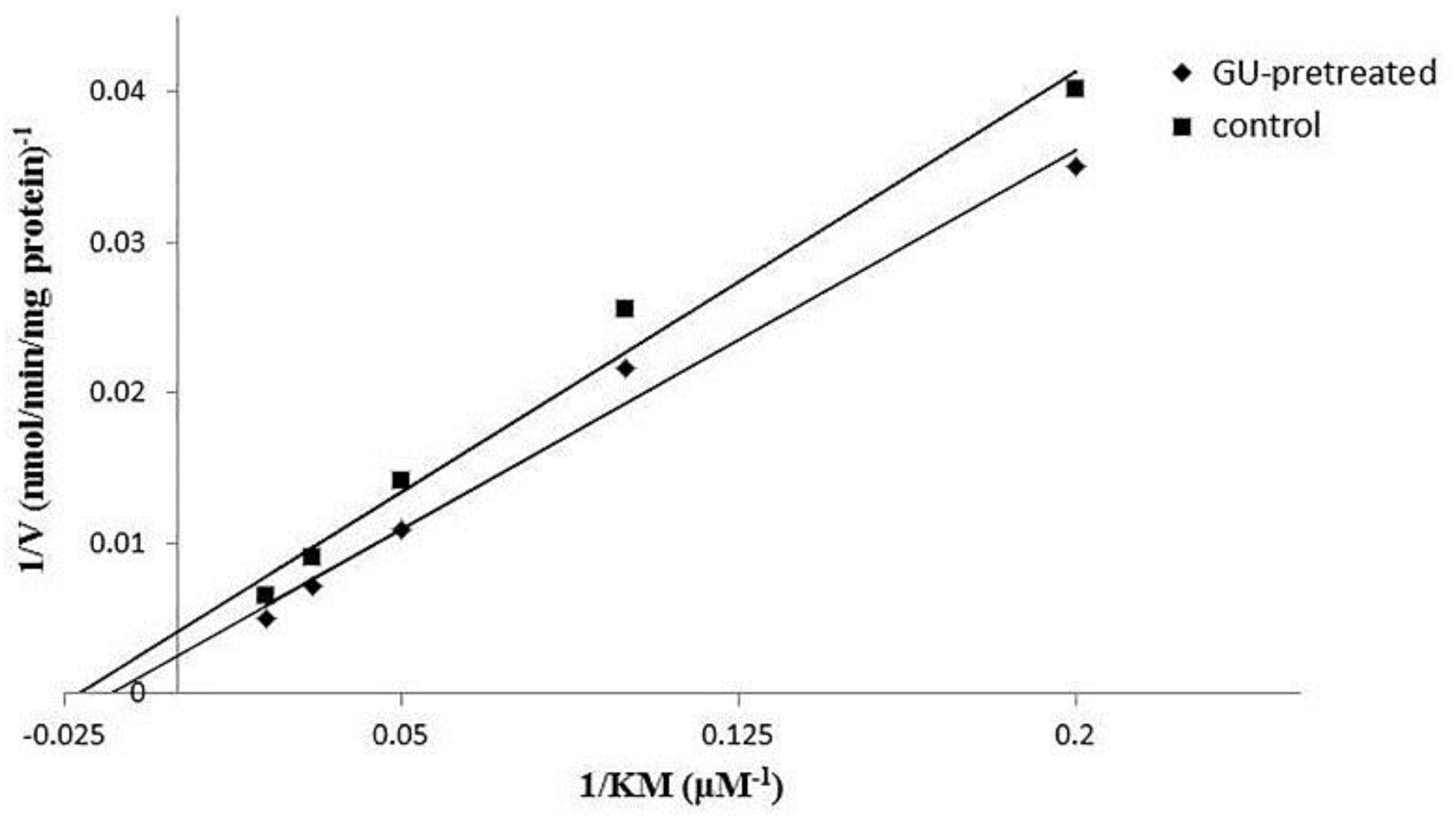
2.4. In Vitro Metabolism of Koumine in Rat Liver Microsomes
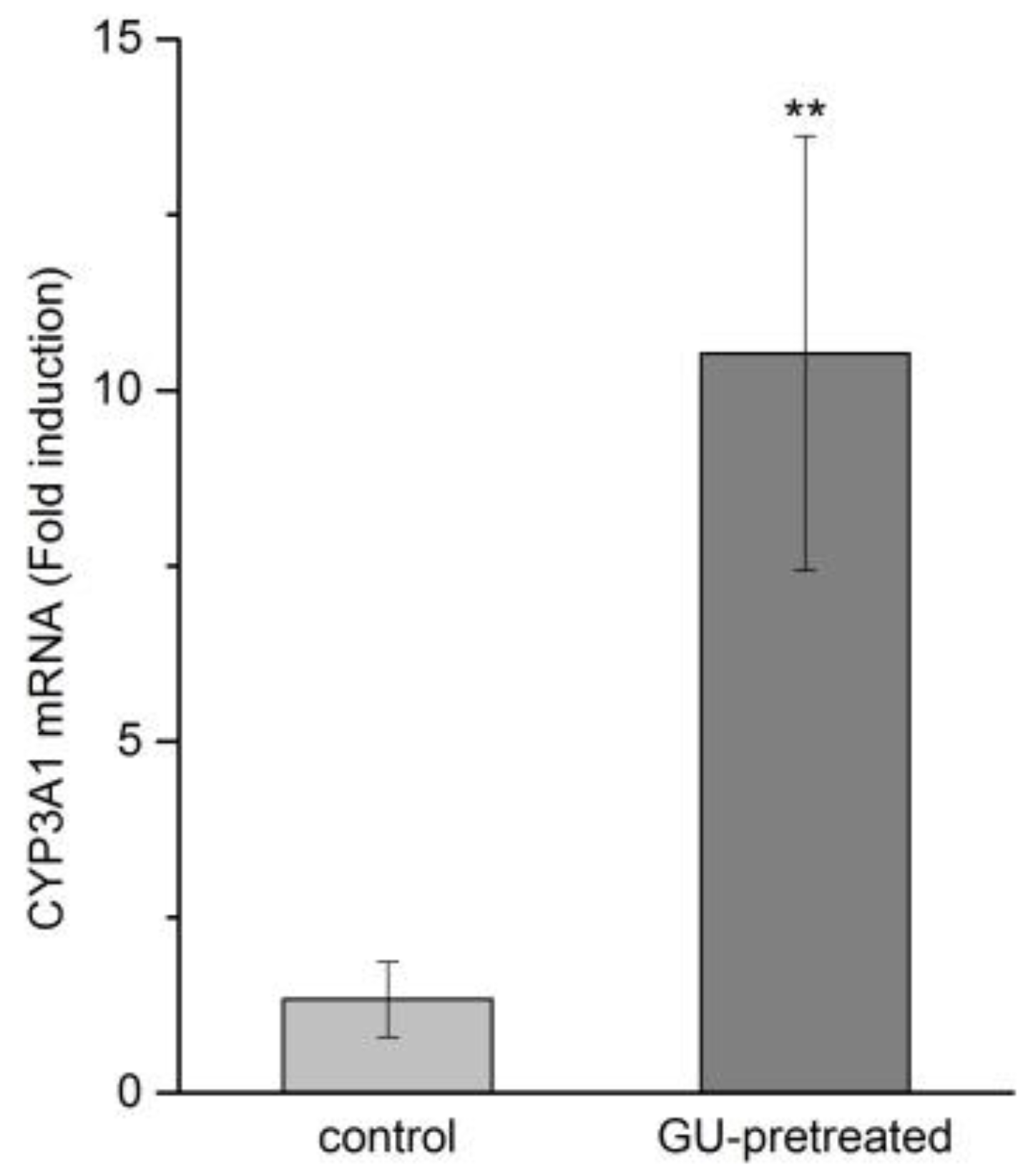
2.5. Effects of GU Treatment on CYP3A1 Expression by RT-qPCR
3. Experimental
3.1. Materials and Reagents
3.2. Animals
3.3. Preparation of Herbal Decoctions
3.4. Preparation of Calibration Standards and Quality Control (QC) Samples
3.5. Sample Preparation
3.6. Instruments and Analytical Conditions
3.7. Method Validation
3.8. In Vivo Pharmacokinetics and Tissue Distribution Study
3.9. Preparation of RLM
3.10. In Vitro Metabolism of Koumine in RLM
3.11. RNA Isolation, cDNA Synthesis and Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR) Analysis of CYP3A1 mRNA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jin, G.L.; Su, Y.P.; Liu, M.; Xu, Y.; Yang, J.; Liao, K.J.; Yu, C.X. Medicinal plants of the genus Gelsemium (Gelsemiaceae, Gentianales)—A review of their phytochemistry, pharmacology, toxicology and traditional use. J. Ethnopharmacol. 2014, 152, 33–52. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.H.; Liang, Z.E.; Wu, J.; Yi, J.E.; Chen, X.J.; Sun, Z.L. A Potential Mechanism for the Anti-Apoptotic Property of Koumine Involving Mitochondrial Pathway in LPS-Mediated RAW 264.7 Macrophages. Molecules 2016, 21, 1317. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Matias, F.B.; Wu, J.; Liang, Z.; Sun, Z. Koumine Attenuates Lipopolysaccaride-Stimulated Inflammation in RAW264.7 Macrophages, Coincidentally Associated with Inhibition of NF-kappaB, ERK and p38 Pathways. Int. J. Mol. Sci. 2016, 17, 430. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Zhong, Z.F.; Xin, Z.M.; Hong, L.H.; Su, Y.P.; Yu, C.X. Koumine exhibits anxiolytic properties without inducing adverse neurological effects on functional observation battery, open-field and Vogel conflict tests in rodents. J. Nat. Med. 2017, 71, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Karwacki, Z.; Niewiadomski, S.; Rzaska, M.; Witkowska, M. The effect of bispectral index monitoring on anaesthetic requirements in target-controlled infusion for lumbar microdiscectomy. Anaesthesiol. Intensive Ther. 2014, 46, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Rujjanawate, C.; Kanjanapothi, D.; Panthong, A. Pharmacological effect and toxicity of alkaloids from Gelsemium elegans Benth. J. Ethnopharmacol. 2003, 89, 91–95. [Google Scholar] [CrossRef]
- Gong, H.; Zhang, B.K.; Yan, M.; Fang, P.F.; Li, H.D.; Hu, C.P.; Yang, Y.; Cao, P.; Jiang, P.; Fan, X.R. A protective mechanism of licorice (Glycyrrhiza uralensis): Isoliquiritigenin stimulates detoxification system via Nrf2 activation. J. Ethnopharmacol. 2015, 162, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Xiong, B.J.; Xu, Y.; Jin, G.L.; Liu, M.; Yang, J.; Yu, C.X. Analgesic effects and pharmacologic mechanisms of the Gelsemium alkaloid koumine on a rat model of postoperative pain. Sci. Rep. 2017, 7, 14269. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Lei, L.; Fang, F.; Yang, S.; Wang, J. Inhibitory effects of koumine on splenocyte proliferation and humoral immune response in mice. Pharmacol. Clin. Chin. Mater. Med. 1999, 15, 10–12. [Google Scholar]
- Wang, L.; Wen, Y.; Meng, F. Simultaneous determination of gelsemine and koumine in rat plasma by UPLC-MS/MS and application to pharmacokinetic study after oral administration of Gelsemium elegans Benth extract. Biomed. Chromatogr. 2018, 31, e4201. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Su, Y.P.; Yu, C.X.; Xu, Y.; Yang, J. Cytotoxic effects of alkaloidal compounds from Gelsemium elegans Benth on the tumor cells of digestive system in vitro. Strait Pharm. J. 2010, 22, 197–200. [Google Scholar]
- Hu, Y.; Wang, Z.; Huang, X.; Xia, B.; Tang, L.; Zheng, Z.; Ye, L. Oxidative metabolism of koumine is mainly catalyzed by microsomal CYP3A4/3A5. Xenobiotica 2017, 47, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
Sample Availability: Samples of the compounds the analytes are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Regression Equations | R2 | Linear Range (ng/mL) | LLOQ (ng/mL) |
|---|---|---|---|---|
| Plasma | Y = 0.129 × 10−3X − 0.0021 | 0.9967 | 10–5000 | 10 |
| Heart | Y = 2.76 × 10−3X − 0.3078 | 0.9904 | 25–5000 | 25 |
| Liver | Y = 2.76 × 10−3X − 0.3595 | 0.9978 | 25–5000 | 25 |
| Spleen | Y = 3.01 × 10−3X − 0.0547 | 0.9943 | 25–5000 | 25 |
| Lung | Y = 1.58 × 10−3X + 0.1471 | 0.9975 | 25–5000 | 25 |
| Kidney | Y = 2.80 × 10−3X − 0.1467 | 0.9964 | 25–5000 | 25 |
| Stomach | Y = 1.52 × 10−3X + 0.1601 | 0.9968 | 25–5000 | 25 |
| Intestine | Y = 1.52 × 10−3X + 0.05300 | 0.9979 | 25–5000 | 25 |
| Rat Liver Microsome | Y = 0.0959 × 10−3X + 0.1471 | 0.9910 | 50–50000 | 50 |
| Samples | Analyte Concentration | Accuracy (%) | Intra-Day Precsion (%) | Inter-Day Precsion (%) |
|---|---|---|---|---|
| (ng/mL) | ||||
| Plasma | 10 | −4.0 | 7.8 | 9.6 |
| 25 | −4.3 | 10 | 11 | |
| 300 | 3.2 | 9.7 | 8.5 | |
| 4000 | 3.9 | 9.0 | 9.7 | |
| Heart | 25 | 4.2 | 8.1 | 10 |
| 100 | 3.9 | 12 | 9.1 | |
| 600 | −3.1 | 10 | 7.6 | |
| 4000 | 2.5 | 11 | 10 | |
| Liver | 25 | 3.6 | 8.3 | 9.2 |
| 100 | 4.5 | 11 | 11 | |
| 600 | 3.9 | 8.5 | 9.0 | |
| 4000 | −3.5 | 9.5 | 8.8 | |
| Spleen | 25 | −4.1 | 8.6 | 10 |
| 100 | 2.9 | 12 | 8.7 | |
| 600 | −2.4 | 10 | 9.2 | |
| 4000 | 3.4 | 9.7 | 7.6 | |
| Lung | 25 | 3.3 | 9.8 | 10 |
| 100 | 3.1 | 12 | 9.4 | |
| 600 | −2.6 | 7.2 | 10 | |
| 4000 | 3.2 | 10.0 | 6.6 | |
| Kidney | 25 | −4.6 | 11 | 11 |
| 100 | 4.5 | 9.9 | 8.8 | |
| 600 | −3.8 | 10 | 7.8 | |
| 4000 | 2.2 | 7.4 | 9.5 | |
| Stomach | 25 | 4.3 | 10.2 | 8.4 |
| 100 | 3.0 | 10 | 9.8 | |
| 600 | 2.5 | 5.1 | 7.0 | |
| 4000 | 3.3 | 8.3 | 7.5 | |
| Intestine | 25 | 3.7 | 10 | 8.8 |
| 100 | 3.8 | 7.6 | 11 | |
| 600 | 2.2 | 6.9 | 9.6 | |
| 4000 | −3.0 | 9.2 | 7.1 | |
| Rat Liver Microsome | 50 | 2.9 | 10 | 9.2 |
| 250 | 3.0 | 6.2 | 7.2 | |
| 2000 | 2.6 | 7.7 | 8.1 | |
| 20000 | 2.2 | 7.4 | 7.2 |
| Samples | Spiked Concentration (ng/mL) | Matrix Effect | Extraction Recovery | ||
|---|---|---|---|---|---|
| Mean (%) | RSD (%) | Mean (%) | RSD (%) | ||
| Plasma | 25 | 92.4 | 9.4 | 81.9 | 8.4 |
| 300 | 96.5 | 6.3 | 88.0 | 9.8 | |
| 4000 | 102.5 | 8.6 | 82.9 | 7.4 | |
| Heart | 100 | 88.9 | 10.9 | 85.0 | 9.7 |
| 600 | 93.5 | 7.9 | 87.4 | 8.8 | |
| 4000 | 87.0 | 8.7 | 88.6 | 8.0 | |
| Liver | 100 | 94.3 | 9.2 | 90.1 | 8.6 |
| 600 | 94.5 | 8.9 | 86.3 | 7.9 | |
| 4000 | 87.7 | 10.0 | 81.2 | 8.3 | |
| Spleen | 100 | 101.2 | 9.6 | 80.2 | 7.0 |
| 600 | 105.1 | 7.3 | 79.6 | 8.5 | |
| 4000 | 94.3 | 9.0 | 82.0 | 9.7 | |
| Lung | 100 | 88.0 | 8.0 | 81.4 | 6.7 |
| 600 | 90.1 | 8.5 | 78.6 | 7.9 | |
| 4000 | 84.3 | 9.7 | 84.2 | 8.5 | |
| Kidney | 100 | 104.6 | 9.4 | 87.6 | 9.2 |
| 600 | 92.6 | 7.4 | 80.0 | 9.0 | |
| 4000 | 103.1 | 9.1 | 87.1 | 7.2 | |
| Stomach | 100 | 98.6 | 8.2 | 84.3 | 6.4 |
| 600 | 87.4 | 9.1 | 83.1 | 7.4 | |
| 4000 | 100.2 | 7.6 | 87.1 | 8.2 | |
| Intestine | 100 | 98.5 | 10.0 | 79.0 | 9.7 |
| 600 | 87.3 | 8.9 | 82.0 | 7.3 | |
| 4000 | 95.3 | 7.5 | 85.3 | 8.5 | |
| Rat Liver Microsome | 250 | 91.5 | 7.8 | 81.3 | 8.0 |
| 2000 | 91.0 | 8.3 | 87.1 | 8.2 | |
| 20000 | 90.3 | 8.6 | 82.3 | 7.7 | |
| Samples | Spiked Concentration | Stability (% RE) | |||
|---|---|---|---|---|---|
| (ng/mL) | Three Freeze-Thaw | Short-Term | Long-Term | Post-Preparative | |
| Plasma | 25 | 4.3 | −4.7 | 3.8 | 4.7 |
| 300 | −3.2 | 2.3 | 4.7 | −4.4 | |
| 4000 | 4.4 | 4.7 | −4.9 | 2.5 | |
| Heart | 100 | −6.2 | 3.0 | 4.0 | 4.4 |
| 600 | −5.3 | 4.8 | 3.0 | −3.9 | |
| 4000 | 2.4 | −4.4 | 3.2 | 4.7 | |
| Liver | 100 | −4.7 | 3.6 | 4.1 | −4.0 |
| 600 | 2.2 | −4.3 | 3.9 | 4.2 | |
| 4000 | 3.3 | 4.2 | 3.1 | 4.8 | |
| Spleen | 100 | −3.2 | 2.7 | -3.1 | 3.8 |
| 600 | 4.5 | 3.5 | 2.7 | −4.1 | |
| 4000 | 3.0 | −4.0 | 4.3 | −2.2 | |
| Lung | 100 | −3.1 | 2.8 | 3.6 | 3.6 |
| 600 | 4.2 | 4.1 | 3.8 | 3.7 | |
| 4000 | 3.5 | 4.0 | −2.9 | 4.4 | |
| Kidney | 100 | 3.0 | −2.7 | 3.2 | 4.9 |
| 600 | −2.8 | 3.1 | 4.1 | −3.0 | |
| 4000 | 5.0 | 4.7 | 4.0 | 2.8 | |
| Stomach | 100 | 3.2 | −4.1 | 4.9 | 3.1 |
| 600 | −4.2 | 2.6 | 4.9 | 3.8 | |
| 4000 | 4.0 | −3.9 | −3.1 | −4.1 | |
| Intestine | 100 | 4.8 | 3.7 | 3.0 | 4.1 |
| 600 | 4.7 | −3.6 | 3.6 | 4.0 | |
| 4000 | −3.5 | 3.1 | 3.0 | 4.5 | |
| Rat liver microsome | 250 | 2.2 | 3.6 | 3.2 | 3.3 |
| 2000 | 3.1 | 2.7 | 2.3 | 3.6 | |
| 20000 | 3.0 | 2.6 | 3.7 | 3.0 | |
| Parameters | Unit | KM Group | GU + KM Group |
|---|---|---|---|
| AUC0–6 | μg/L * h | 3340 ± 410 | 2410 ± 130 * |
| AUC0–∞ | μg/L * h | 3400 ± 420 | 2600 ± 200 * |
| MRT0–t | h | 1.1 ± 0.1 | 1.3 ± 0.2 |
| MRT0–∞ | h | 1.2 ± 0.1 | 1.8 ± 0.4 |
| t1/2z | h | 1.2 ± 0.1 | 1.7 ± 0.3 * |
| CLZ | L/h/kg | 3.0 ± 0.4 | 3.9 ± 0.3 * |
| VZ | L/kg | 5.0 ± 0.4 | 9.2 ± 0.9 ** |
| Cmax | μg/L | 5790 ± 410 | 3600 ± 1260 * |
| Parameters | Unit | Control Group | GU-Pretreated Group |
|---|---|---|---|
| Km | μM | 39.8 ± 23.3 | 69.9 ± 22.4 |
| Vmax | nmol/min/mg protein | 193.8 ± 96.6 | 419.6 ± 121.7 * |
| CLint | μL/min/mg protein | 5.0 ± 0.4 | 6.0 ± 5.4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Sun, Q.; Zhao, N.; Wen, Y.-Q.; Song, Y.; Meng, F.-H. Ultra-Liquid Chromatography Tandem Mass Spectrometry (UPLC-MS/MS)-Based Pharmacokinetics and Tissue Distribution Study of Koumine and the Detoxification Mechanism of Glycyrrhiza uralensis Fisch on Gelsemium elegans Benth. Molecules 2018, 23, 1693. https://doi.org/10.3390/molecules23071693
Wang L, Sun Q, Zhao N, Wen Y-Q, Song Y, Meng F-H. Ultra-Liquid Chromatography Tandem Mass Spectrometry (UPLC-MS/MS)-Based Pharmacokinetics and Tissue Distribution Study of Koumine and the Detoxification Mechanism of Glycyrrhiza uralensis Fisch on Gelsemium elegans Benth. Molecules. 2018; 23(7):1693. https://doi.org/10.3390/molecules23071693
Chicago/Turabian StyleWang, Lin, Qi Sun, Nan Zhao, Yan-Qing Wen, Yang Song, and Fan-Hao Meng. 2018. "Ultra-Liquid Chromatography Tandem Mass Spectrometry (UPLC-MS/MS)-Based Pharmacokinetics and Tissue Distribution Study of Koumine and the Detoxification Mechanism of Glycyrrhiza uralensis Fisch on Gelsemium elegans Benth." Molecules 23, no. 7: 1693. https://doi.org/10.3390/molecules23071693
APA StyleWang, L., Sun, Q., Zhao, N., Wen, Y.-Q., Song, Y., & Meng, F.-H. (2018). Ultra-Liquid Chromatography Tandem Mass Spectrometry (UPLC-MS/MS)-Based Pharmacokinetics and Tissue Distribution Study of Koumine and the Detoxification Mechanism of Glycyrrhiza uralensis Fisch on Gelsemium elegans Benth. Molecules, 23(7), 1693. https://doi.org/10.3390/molecules23071693