Neuroprotective Effects of Mitochondria-Targeted Plastoquinone in a Rat Model of Neonatal Hypoxic–Ischemic Brain Injury


 , ,
, , 
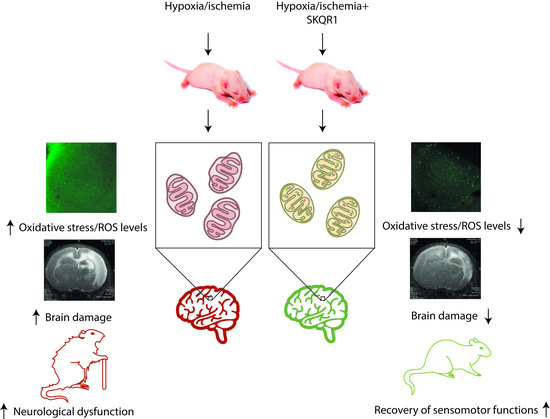
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
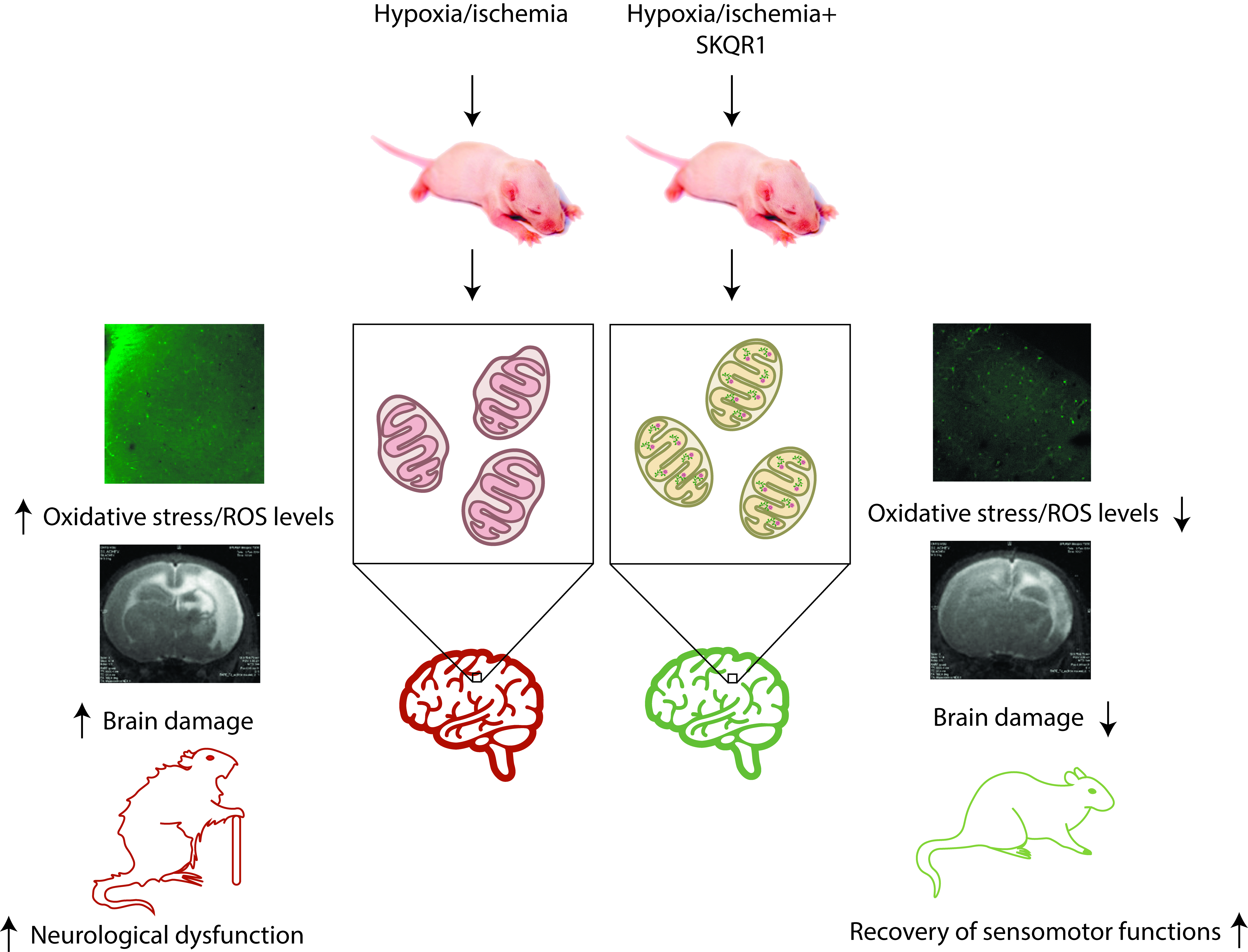
2.1. Hypoxia-Ischemia Induces Mitochondrial Dysfunction and Oxidative Stress
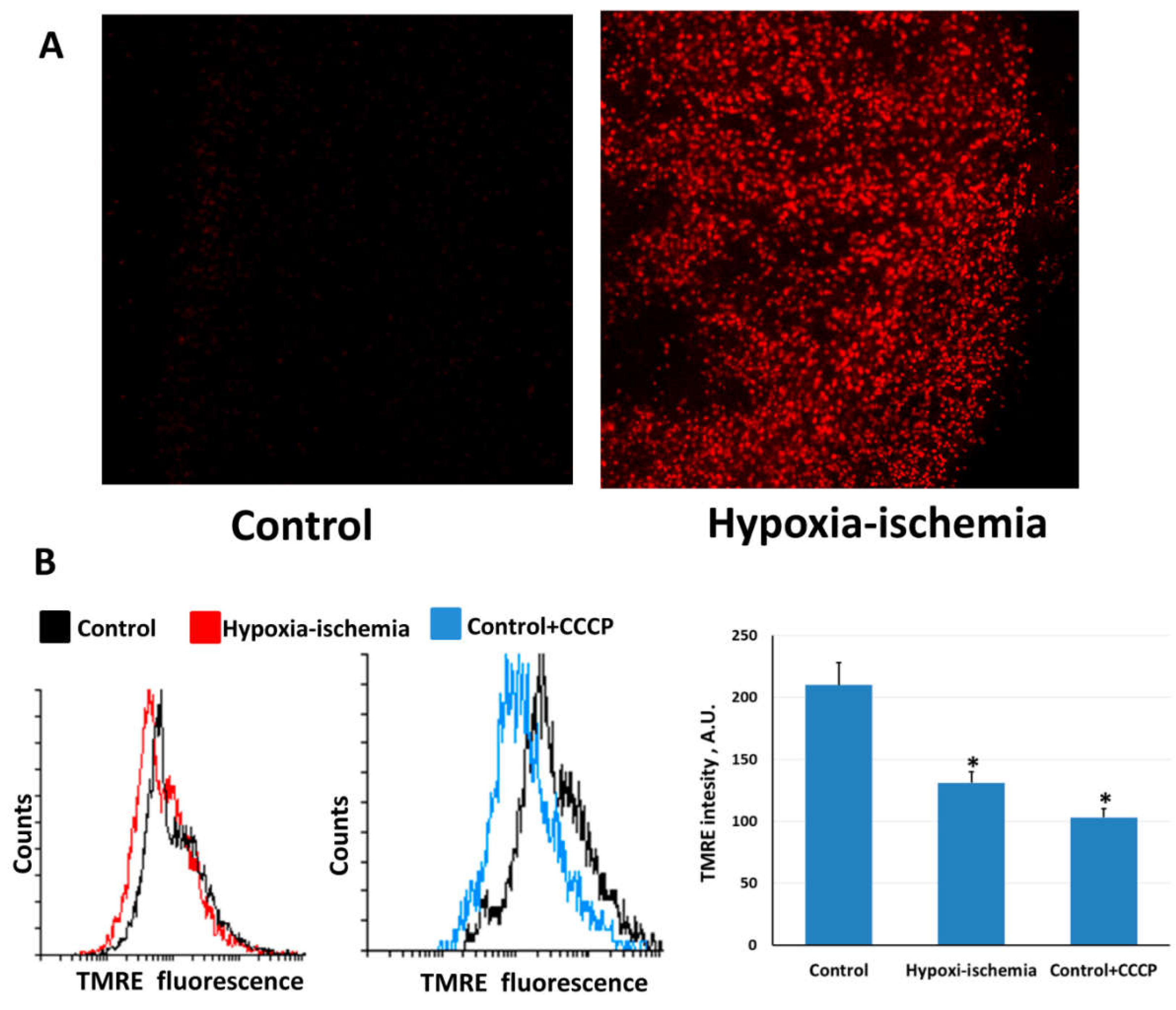
2.2. Mitochondria-Targeted Antioxidant SkQR1 Diminishes Oxidative Stress in Brain after Hypoxia–Ischemia
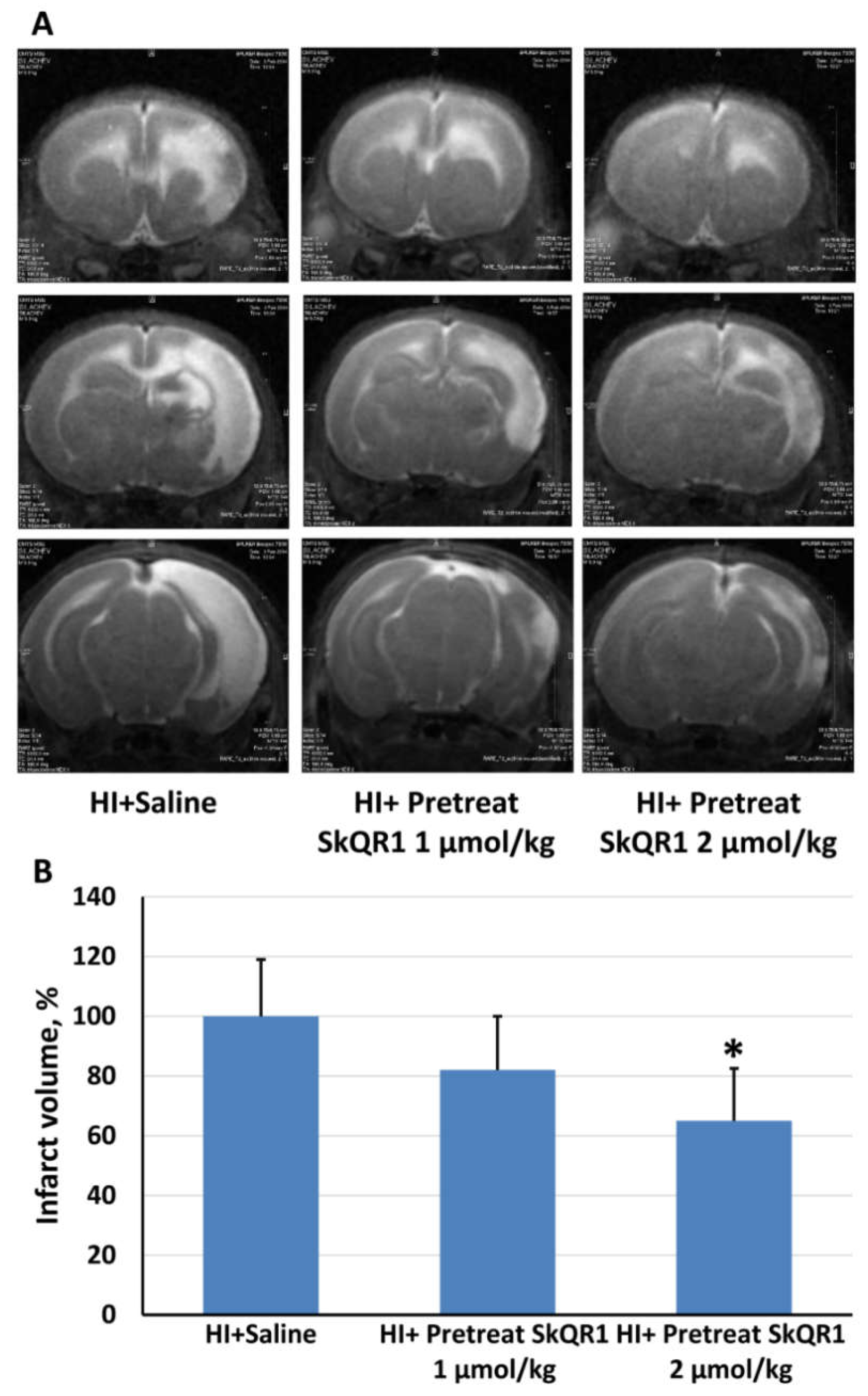
2.3. Effects of Pretreatment with Mitochondria-Targeted Antioxidant SkQR1 on Infarct Size
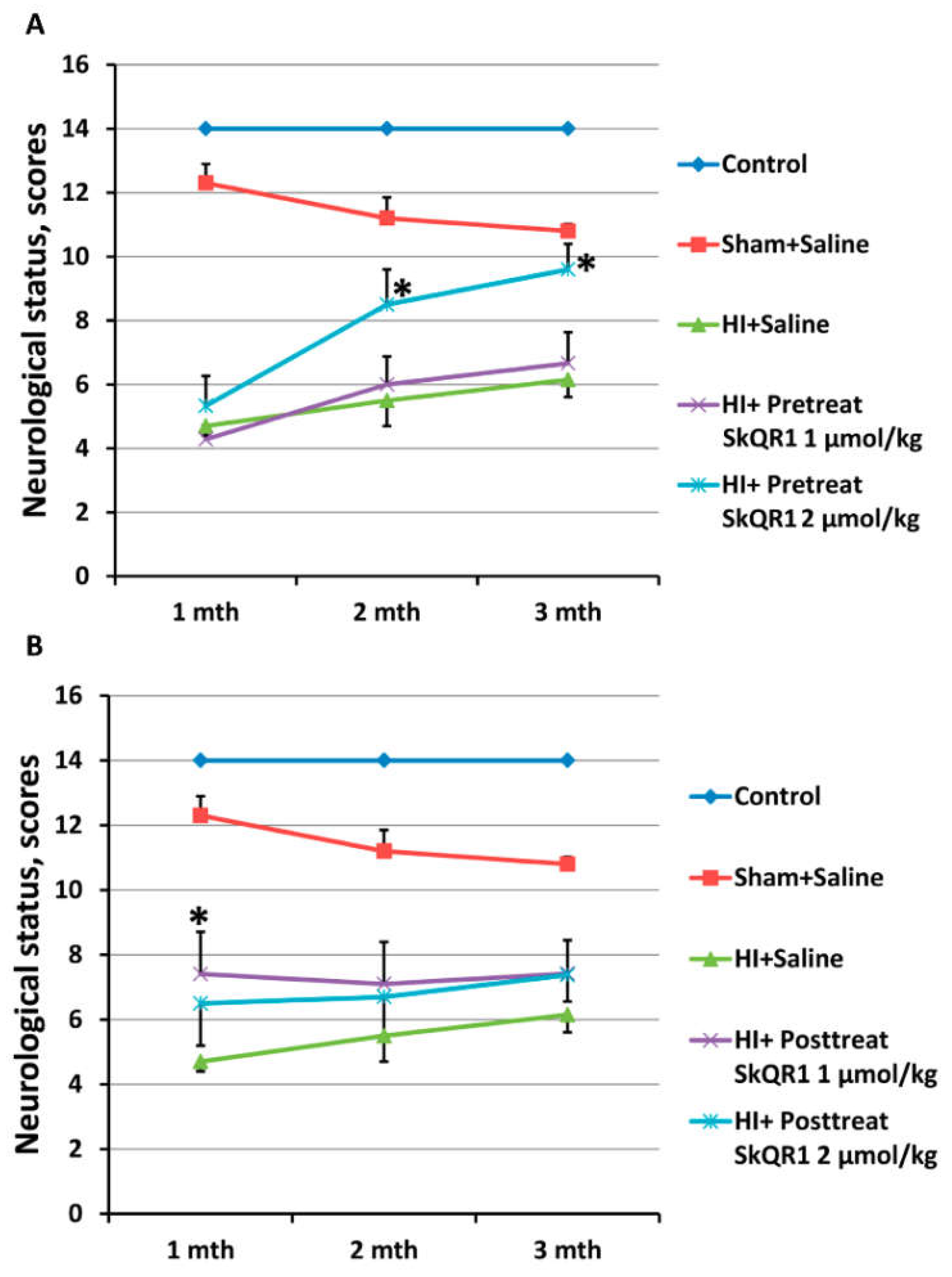
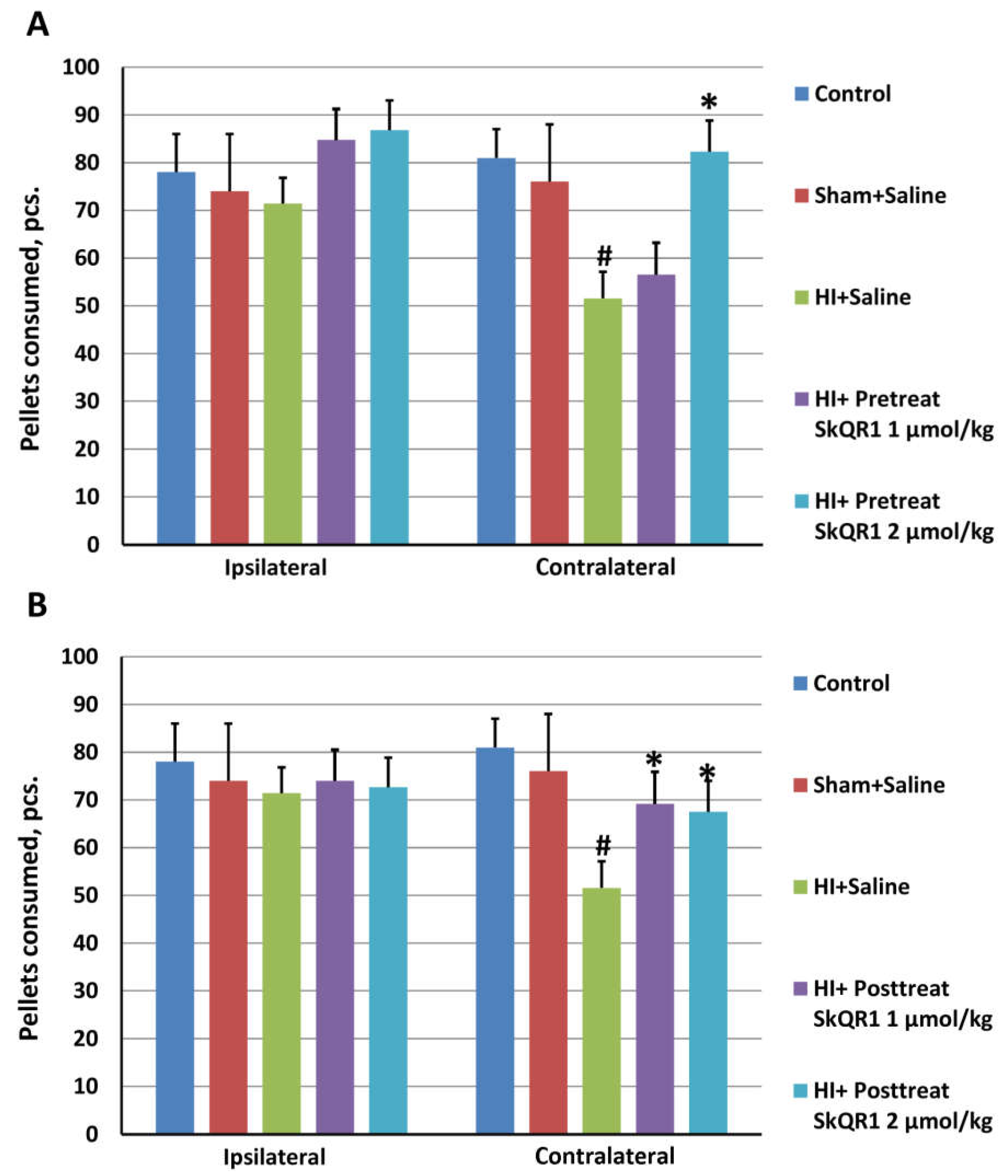
2.4. Effects of Mitochondria-Targeted Antioxidant SkQR1 Pre- and Post-Treatment on Sensorimotor Deficits in HI Rats
3. Discussion
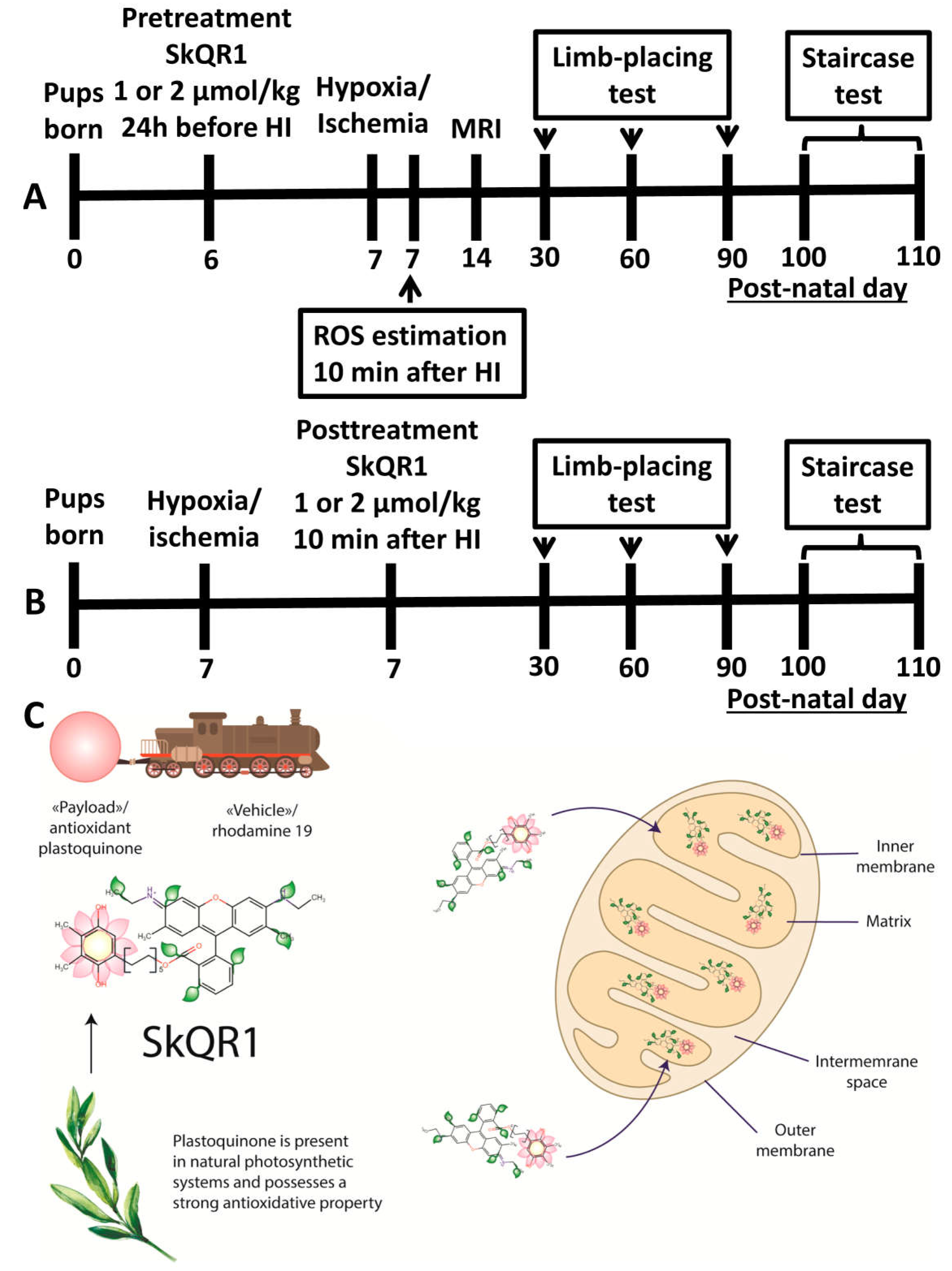
4. Materials and Methods
4.1. Use of Animals
4.2. Induction of Neonatal HI Animal Model and Treatment with SkQR1
4.3. MRI Studies of the Brain Damage
4.4. The Staircase Test
4.5. Limb-Placing Test
4.6. ROS Determination in Brain Slices
4.7. Confocal Microscopy
4.8. Assessment of Changes in Mitochondrial Transmembrane Potential
4.9. Statistics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, L.; Oza, S.; Hogan, D.; Perin, J.; Rudan, I.; Lawn, J.E.; Cousens, S.; Mathers, C.; Black, R.E. Global, regional, and national causes of child mortality in 2000–13, with projections to inform post-2015 priorities: An updated systematic analysis. Lancet 2015, 385, 430–440. [Google Scholar] [CrossRef]
- Tagin, M.; Abdel-Hady, H.; ur Rahman, S.; Azzopardi, D.V.; Gunn, A.J. Neuroprotection for Perinatal Hypoxic Ischemic Encephalopathy in Low- and Middle-Income Countries. J. Pediatr. 2015, 167, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.A.; Brandon, D.H. Hypoxic Ischemic Encephalopathy: Pathophysiology and Experimental Treatments. Newborn Infant Nurs. Rev. 2011, 11, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Zhu, P.; Fujino, M.; Zhuang, J.; Guo, H.; Sheikh, I.; Zhao, L.; Li, X.-K. Oxidative Stress in Hypoxic-Ischemic Encephalopathy: Molecular Mechanisms and Therapeutic Strategies. Int. J. Mol. Sci. 2016, 17, 2078. [Google Scholar] [CrossRef] [PubMed]
- Silachev, D.N.; Plotnikov, E.Y.; Pevzner, I.B.; Zorova, L.D.; Babenko, V.A.; Zorov, S.D.; Popkov, V.A.; Jankauskas, S.S.; Zinchenko, V.P.; Sukhikh, G.T.; et al. The mitochondrion as a key regulator of ischaemic tolerance and injury. Heart Lung Circ. 2014, 23, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Ferriero, D.M. Neonatal brain injury. N. Engl. J. Med. 2004, 351, 1985–1995. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.H.; Han, S.W.; Lee, S.K. Free radicals as triggers of brain edema formation after stroke. Free Radic. Biol. Med. 2005, 39, 51–70. [Google Scholar] [CrossRef] [PubMed]
- Slemmer, J.E.; Shacka, J.J.; Sweeney, M.I.; Weber, J.T. Antioxidants and free radical scavengers for the treatment of stroke, traumatic brain injury and aging. Curr. Med. Chem. 2008, 15, 404–414. [Google Scholar] [PubMed]
- Margaill, I.; Plotkine, M.; Lerouet, D. Antioxidant strategies in the treatment of stroke. Free Radic. Biol. Med. 2005, 39, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Kamata, H.; Hirata, H. Redox regulation of cellular signalling. Cell. Signal. 1999, 11, 1–14. [Google Scholar] [CrossRef]
- Owuor, E.D.; Kong, A.-N.T. Antioxidants and oxidants regulated signal transduction pathways. Biochem. Pharmacol. 2002, 64, 765–770. [Google Scholar] [CrossRef]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [PubMed]
- Ten, V.S.; Starkov, A. Hypoxic-ischemic injury in the developing brain: The role of reactive oxygen species originating in mitochondria. Neurol. Res. Int. 2012, 2012, 542976. [Google Scholar] [CrossRef] [PubMed]
- Burns, R.J.; Smith, R.A.; Murphy, M.P. Synthesis and characterization of thiobutyltriphenylphosphonium bromide, a novel thiol reagent targeted to the mitochondrial matrix. Arch. Biochem. Biophys. 1995, 322, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Porteous, C.M.; Coulter, C.V.; Murphy, M.P. Selective targeting of an antioxidant to mitochondria. Eur. J. Biochem. 1999, 263, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Antonenko, Y.N.; Avetisyan, A.V.; Bakeeva, L.E.; Chernyak, B.V.; Chertkov, V.A.; Domnina, L.V.; Ivanova, O.Y.; Izyumov, D.S.; Khailova, L.S.; Klishin, S.S.; et al. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 1. Cationic plastoquinone derivatives: Synthesis and in vitro studies. Biochem. Mosc. 2008, 73, 1273–1287. [Google Scholar] [CrossRef]
- Silachev, D.N.; Plotnikov, E.Y.; Zorova, L.D.; Pevzner, I.B.; Sumbatyan, N.V.; Korshunova, G.A.; Gulyaev, M.V.; Pirogov, Y.A.; Skulachev, V.P.; Zorov, D.B. Neuroprotective Effects of Mitochondria-Targeted Plastoquinone and Thymoquinone in a Rat Model of Brain Ischemia/Reperfusion Injury. Molecules 2015, 20, 14487–14503. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P.; Anisimov, V.N.; Antonenko, Y.N.; Bakeeva, L.E.; Chernyak, B.V.; Erichev, V.P.; Filenko, O.F.; Kalinina, N.I.; Kapelko, V.I.; Kolosova, N.G.; et al. An attempt to prevent senescence: A mitochondrial approach. Biochim. Biophys. Acta 2009, 1787, 437–461. [Google Scholar] [CrossRef] [PubMed]
- Korshunova, G.A.; Shishkina, A.V.; Skulachev, M.V. Design, Synthesis, and Some Aspects of the Biological Activity of Mitochondria-Targeted Antioxidants. Biochem. Mosc. 2017, 82, 760–777. [Google Scholar] [CrossRef] [PubMed]
- Silachev, D.N.; Isaev, N.K.; Pevzner, I.B.; Zorova, L.D.; Stelmashook, E.V.; Novikova, S.V.; Plotnikov, E.Y.; Skulachev, V.P.; Zorov, D.B. The Mitochondria-Targeted Antioxidants and Remote Kidney Preconditioning Ameliorate Brain Damage through Kidney-to-Brain Cross-Talk. PLoS ONE 2012, 7, e51553. [Google Scholar] [CrossRef] [PubMed]
- Isaev, N.K.; Novikova, S.V.; Stelmashook, E.V.; Barskov, I.V.; Silachev, D.N.; Khaspekov, L.G.; Skulachev, V.P.; Zorov, D.B. Mitochondria-targeted plastoquinone antioxidant SkQR1 decreases trauma-induced neurological deficit in rat. Biochem. Mosc. 2012, 77, 996–999. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, E.Y.; Silachev, D.N.; Chupyrkina, A.A.; Danshina, M.I.; Jankauskas, S.S.; Morosanova, M.A.; Stelmashook, E.V.; Vasileva, A.K.; Goryacheva, E.S.; Pirogov, Y.A.; et al. New-generation Skulachev ions exhibiting nephroprotective and neuroprotective properties. Biochem. Mosc. 2010, 75, 145–150. [Google Scholar] [CrossRef]
- Plotnikov, E.Y.; Chupyrkina, A.A.; Jankauskas, S.S.; Pevzner, I.B.; Silachev, D.N.; Skulachev, V.P.; Zorov, D.B. Mechanisms of nephroprotective effect of mitochondria-targeted antioxidants under rhabdomyolysis and ischemia/reperfusion. Biochim. Biophys. Acta 2011, 1812, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Jankauskas, S.S.; Andrianova, N.V.; Alieva, I.B.; Prusov, A.N.; Matsievsky, D.D.; Zorova, L.D.; Pevzner, I.B.; Savchenko, E.S.; Pirogov, Y.A.; Silachev, D.N.; et al. Dysfunction of Kidney Endothelium after Ischemia/Reperfusion and Its Prevention by Mitochondria-Targeted Antioxidant. Biochem. Mosc. 2016, 81, 1538–1548. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, E.Y.; Kazachenko, A.V.; Vyssokikh, M.Y.; Vasileva, A.K.; Tcvirkun, D.V.; Isaev, N.K.; Kirpatovsky, V.I.; Zorov, D.B. The role of mitochondria in oxidative and nitrosative stress during ischemia/reperfusion in the rat kidney. Kidney Int. 2007, 72, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, E.Y.; Morosanova, M.A.; Pevzner, I.B.; Zorova, L.D.; Manskikh, V.N.; Pulkova, N.V.; Galkina, S.I.; Skulachev, V.P.; Zorov, D.B. Protective effect of mitochondria-targeted antioxidants in an acute bacterial infection. Proc. Natl. Acad. Sci. USA 2013, 110, E3100–E3108. [Google Scholar] [CrossRef] [PubMed]
- Bakeeva, L.E.; Barskov, I.V.; Egorov, M.V.; Isaev, N.K.; Kapelko, V.I.; Kazachenko, A.V.; Kirpatovsky, V.I.; Kozlovsky, S.V.; Lakomkin, V.L.; Levina, S.B.; et al. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 2. Treatment of some ROS- and age-related diseases (heart arrhythmia, heart infarctions, kidney ischemia, and stroke). Biochem. Mosc. 2008, 73, 1288–1299. [Google Scholar] [CrossRef]
- Manskikh, V.N.; Gancharova, O.S.; Nikiforova, A.I.; Krasilshchikova, M.S.; Shabalina, I.G.; Egorov, M.V.; Karger, E.M.; Milanovsky, G.E.; Galkin, I.I.; Skulachev, V.P.; et al. Age-associated murine cardiac lesions are attenuated by the mitochondria-targeted antioxidant SkQ1. Histol. Histopathol. 2015, 30, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Brzheskiy, V.V.; Efimova, E.L.; Vorontsova, T.N.; Alekseev, V.N.; Gusarevich, O.G.; Shaidurova, K.N.; Ryabtseva, A.A.; Andryukhina, O.M.; Kamenskikh, T.G.; Sumarokova, E.S.; et al. Results of a Multicenter, Randomized, Double-Masked, Placebo-Controlled Clinical Study of the Efficacy and Safety of Visomitin Eye Drops in Patients with Dry Eye Syndrome. Adv. Ther. 2015, 32, 1263–1279. [Google Scholar] [CrossRef] [PubMed]
- Jansen, E.M.; Low, W.C. Long-term effects of neonatal ischemic-hypoxic brain injury on sensorimotor and locomotor tasks in rats. Behav. Brain Res. 1996, 78, 189–194. [Google Scholar] [CrossRef]
- Schallert, T. Behavioral tests for preclinical intervention assessment. NeuroRx 2006, 3, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Azzopardi, D.V.; Strohm, B.; Edwards, A.D.; Dyet, L.; Halliday, H.L.; Juszczak, E.; Kapellou, O.; Levene, M.; Marlow, N.; Porter, E.; et al. Moderate hypothermia to treat perinatal asphyxial encephalopathy. N. Engl. J. Med. 2009, 361, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Bayir, H.; Adelson, P.D.; Wisniewski, S.R.; Shore, P.; Lai, Y.; Brown, D.; Janesko-Feldman, K.L.; Kagan, V.E.; Kochanek, P.M. Therapeutic hypothermia preserves antioxidant defenses after severe traumatic brain injury in infants and children. Crit. Care Med. 2009, 37, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Arteaga, O.; Álvarez, A.; Revuelta, M.; Santaolalla, F.; Urtasun, A.; Hilario, E. Role of Antioxidants in Neonatal Hypoxic-Ischemic Brain Injury: New Therapeutic Approaches. Int. J. Mol. Sci. 2017, 18, 265. [Google Scholar] [CrossRef] [PubMed]
- Tataranno, M.L.; Perrone, S.; Longini, M.; Buonocore, G. New antioxidant drugs for neonatal brain injury. Oxid. Med. Cell. Longev. 2015, 2015, 108251. [Google Scholar] [CrossRef] [PubMed]
- Bavarsad Shahripour, R.; Harrigan, M.R.; Alexandrov, A.V. N-acetylcysteine (NAC) in neurological disorders: Mechanisms of action and therapeutic opportunities. Brain Behav. 2014, 4, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Jatana, M.; Singh, I.; Singh, A.K.; Jenkins, D. Combination of systemic hypothermia and N-acetylcysteine attenuates hypoxic-ischemic brain injury in neonatal rats. Pediatr. Res. 2006, 59, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Jantzie, L.L.; Oppong, A.Y.; Conteh, F.S.; Yellowhair, T.R.; Kim, J.; Fink, G.; Wolin, A.R.; Northington, F.J.; Robinson, S. Repetitive Neonatal Erythropoietin and Melatonin Combinatorial Treatment Provides Sustained Repair of Functional Deficits in a Rat Model of Cerebral Palsy. Front. Neurol. 2018, 9, 233. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.-X.; Lv, Y.; Li, Y.-H.; Ding, X.; Wang, Y.; Han, X.; Liu, M.-H.; Sun, B.; Feng, X. Melatonin alleviates brain and peripheral tissue edema in a neonatal rat model of hypoxic-ischemic brain damage: The involvement of edema related proteins. BMC Pediatr. 2017, 17, 90. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Jiang, Z.-H.; Zhang, X.-F.; Yang, Q.-Z. Effects of erythropoietin on neonatal hypoxia-ischemia brain injury in rat model. Physiol. Behav. 2017, 169, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Juul, S.E.; Pet, G.C. Erythropoietin and Neonatal Neuroprotection. Clin. Perinatol. 2015, 42, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.-X.; Manchester, L.C.; Esteban-Zubero, E.; Zhou, Z.; Reiter, R.J. Melatonin as a Potent and Inducible Endogenous Antioxidant: Synthesis and Metabolism. Molecules 2015, 20, 18886–18906. [Google Scholar] [CrossRef] [PubMed]
- Elmahdy, H.; El-Mashad, A.-R.; El-Bahrawy, H.; El-Gohary, T.; El-Barbary, A.; Aly, H. Human recombinant erythropoietin in asphyxia neonatorum: Pilot trial. Pediatrics 2010, 125, e1135–e1142. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.W.; Bauer, L.A.; Ballard, R.A.; Ferriero, D.M.; Glidden, D.V.; Mayock, D.E.; Chang, T.; Durand, D.J.; Song, D.; Bonifacio, S.L.; et al. Erythropoietin for neuroprotection in neonatal encephalopathy: Safety and pharmacokinetics. Pediatrics 2012, 130, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.W.; Mathur, A.M.; Chang, T.; McKinstry, R.C.; Mulkey, S.B.; Mayock, D.E.; Van Meurs, K.P.; Rogers, E.E.; Gonzalez, F.F.; Comstock, B.A.; et al. High-Dose Erythropoietin and Hypothermia for Hypoxic-Ischemic Encephalopathy: A Phase II Trial. Pediatrics 2016, 137, e20160191. [Google Scholar] [CrossRef] [PubMed]
- Malla, R.R.; Asimi, R.; Teli, M.A.; Shaheen, F.; Bhat, M.A. Erythropoietin monotherapy in perinatal asphyxia with moderate to severe encephalopathy: A randomized placebo-controlled trial. J. Perinatol. 2017, 37, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Rokitskaya, T.I.; Klishin, S.S.; Severina, I.I.; Skulachev, V.P.; Antonenko, Y.N. Kinetic analysis of permeation of mitochondria-targeted antioxidants across bilayer lipid membranes. J. Membr. Biol. 2008, 224, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Stefanova, N.A.; Muraleva, N.A.; Maksimova, K.Y.; Rudnitskaya, E.A.; Kiseleva, E.; Telegina, D.V.; Kolosova, N.G. An antioxidant specifically targeting mitochondria delays progression of Alzheimer’s disease-like pathology. Aging 2016, 8, 2713–2733. [Google Scholar] [CrossRef] [PubMed]
- Stefano, G.B.; Snyder, C.; Kream, R.M. Mitochondria, Chloroplasts in Animal and Plant Cells: Significance of Conformational Matching. Med. Sci. Monit. 2015, 21, 2073–2078. [Google Scholar] [CrossRef] [PubMed]
- Agati, G.; Brunetti, C.; Di Ferdinando, M.; Ferrini, F.; Pollastri, S.; Tattini, M. Functional roles of flavonoids in photoprotection: New evidence, lessons from the past. Plant Physiol. Biochem. 2013, 72, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Mubarakshina, M.M.; Ivanov, B.N. The production and scavenging of reactive oxygen species in the plastoquinone pool of chloroplast thylakoid membranes. Physiol. Plant. 2010, 140, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Severin, F.F.; Severina, I.I.; Antonenko, Y.N.; Rokitskaya, T.I.; Cherepanov, D.A.; Mokhova, E.N.; Vyssokikh, M.Y.; Pustovidko, A.V.; Markova, O.V.; Yaguzhinsky, L.S.; et al. Penetrating cation/fatty acid anion pair as a mitochondria-targeted protonophore. Proc. Natl. Acad. Sci. USA 2010, 107, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Northington, F.J.; Ferriero, D.M.; Graham, E.M.; Traystman, R.J.; Martin, L.J. Early Neurodegeneration after Hypoxia-Ischemia in Neonatal Rat Is Necrosis while Delayed Neuronal Death Is Apoptosis. Neurobiol. Dis. 2001, 8, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Rathelot, J.-A.; Strick, P.L. Subdivisions of primary motor cortex based on cortico-motoneuronal cells. Proc. Natl. Acad. Sci. USA 2009, 106, 918–923. [Google Scholar] [CrossRef] [PubMed]
- Azim, E.; Fink, A.J.P.; Jessell, T.M. Internal and External Feedback Circuits for Skilled Forelimb Movement. Cold Spring Harb. Symp. Quant. Biol. 2014, 79, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, J.; Wang, L.; Lu, M.; Chopp, M. Treatment of stroke in rat with intracarotid administration of marrow stromal cells. Neurology 2001, 56, 1666–1672. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, C.E.; Murphy, M.P.; Smith, R.A.J.; Oorschot, D.E. Neonatal rat hypoxia-ischemia: Effect of the anti-oxidant mitoquinol, and S-PBN. Pediatr. Int. 2008, 50, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Mittal, R.; Khanna, H.D.; Basu, S. Free radical injury and blood-brain barrier permeability in hypoxic-ischemic encephalopathy. Pediatrics 2008, 122, e722–e727. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.; Fernandes, E.; Lima, J.L.F.C. Fluorescence probes used for detection of reactive oxygen species. J. Biochem. Biophys. Methods 2005, 65, 45–80. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Scorziello, A.; Duchen, M.R. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J. Neurosci. 2007, 27, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Odorcyk, F.K.; Kolling, J.; Sanches, E.F.; Wyse, A.T.S.; Netto, C.A. Experimental neonatal hypoxia ischemia causes long lasting changes of oxidative stress parameters in the hippocampus and the spleen. J. Perinat. Med. 2018, 46, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.O.; Nabinger, P.M.; Strapasson, A.C.P.; Nardin, P.; Gonçalves, C.A.S.; Siqueira, I.R.; Netto, C.A. Long-term effects of environmental stimulation following hypoxia-ischemia on the oxidative state and BDNF levels in rat hippocampus and frontal cortex. Brain Res. 2009, 1247, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Hanumanthappa, P.; Densi, A.; Krishnamurthy, R.G. Glycogen synthase kinase-β3 in ischemic neuronal death. Curr. Neurovasc. Res. 2014, 11, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.E.; Vannucci, R.C.; Brierley, J.B. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann. Neurol. 1981, 9, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Silachev, D.N.; Uchevatkin, A.A.; Pirogov, Y.A.; Zorov, D.B.; Isaev, N.K. Comparative evaluation of two methods for studies of experimental focal ischemia: Magnetic resonance tomography and triphenyltetrazoleum detection of brain injuries. Bull. Exp. Biol. Med. 2009, 147, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Montoya, C.P.; Campbell-Hope, L.J.; Pemberton, K.D.; Dunnett, S.B. The “staircase test”: A measure of independent forelimb reaching and grasping abilities in rats. J. Neurosci. Methods 1991, 36, 219–228. [Google Scholar] [CrossRef]
- Silachev, D.N.; Shubina, M.I.; Yankauskas, S.S.; Mkrtchyan, V.P.; Manskikh, V.N.; Gulyaev, M.V.; Zorov, D.B. Assessment of Long-Term Sensorimotor Deficit after Cerebral Ischemia/Hypoxia in Neonatal Rats. Neurosci. Behav. Physiol. 2014, 44, 879–887. [Google Scholar] [CrossRef]
Sample Availability: Not available. |






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silachev, D.N.; Plotnikov, E.Y.; Pevzner, I.B.; Zorova, L.D.; Balakireva, A.V.; Gulyaev, M.V.; Pirogov, Y.A.; Skulachev, V.P.; Zorov, D.B. Neuroprotective Effects of Mitochondria-Targeted Plastoquinone in a Rat Model of Neonatal Hypoxic–Ischemic Brain Injury. Molecules 2018, 23, 1871. https://doi.org/10.3390/molecules23081871
Silachev DN, Plotnikov EY, Pevzner IB, Zorova LD, Balakireva AV, Gulyaev MV, Pirogov YA, Skulachev VP, Zorov DB. Neuroprotective Effects of Mitochondria-Targeted Plastoquinone in a Rat Model of Neonatal Hypoxic–Ischemic Brain Injury. Molecules. 2018; 23(8):1871. https://doi.org/10.3390/molecules23081871
Chicago/Turabian StyleSilachev, Denis N., Egor Y. Plotnikov, Irina B. Pevzner, Ljubava D. Zorova, Anastasia V. Balakireva, Mikhail V. Gulyaev, Yury A. Pirogov, Vladimir P. Skulachev, and Dmitry B. Zorov. 2018. "Neuroprotective Effects of Mitochondria-Targeted Plastoquinone in a Rat Model of Neonatal Hypoxic–Ischemic Brain Injury" Molecules 23, no. 8: 1871. https://doi.org/10.3390/molecules23081871
APA StyleSilachev, D. N., Plotnikov, E. Y., Pevzner, I. B., Zorova, L. D., Balakireva, A. V., Gulyaev, M. V., Pirogov, Y. A., Skulachev, V. P., & Zorov, D. B. (2018). Neuroprotective Effects of Mitochondria-Targeted Plastoquinone in a Rat Model of Neonatal Hypoxic–Ischemic Brain Injury. Molecules, 23(8), 1871. https://doi.org/10.3390/molecules23081871