Lipid Droplets in Cancer: Guardians of Fat in a Stressful World


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
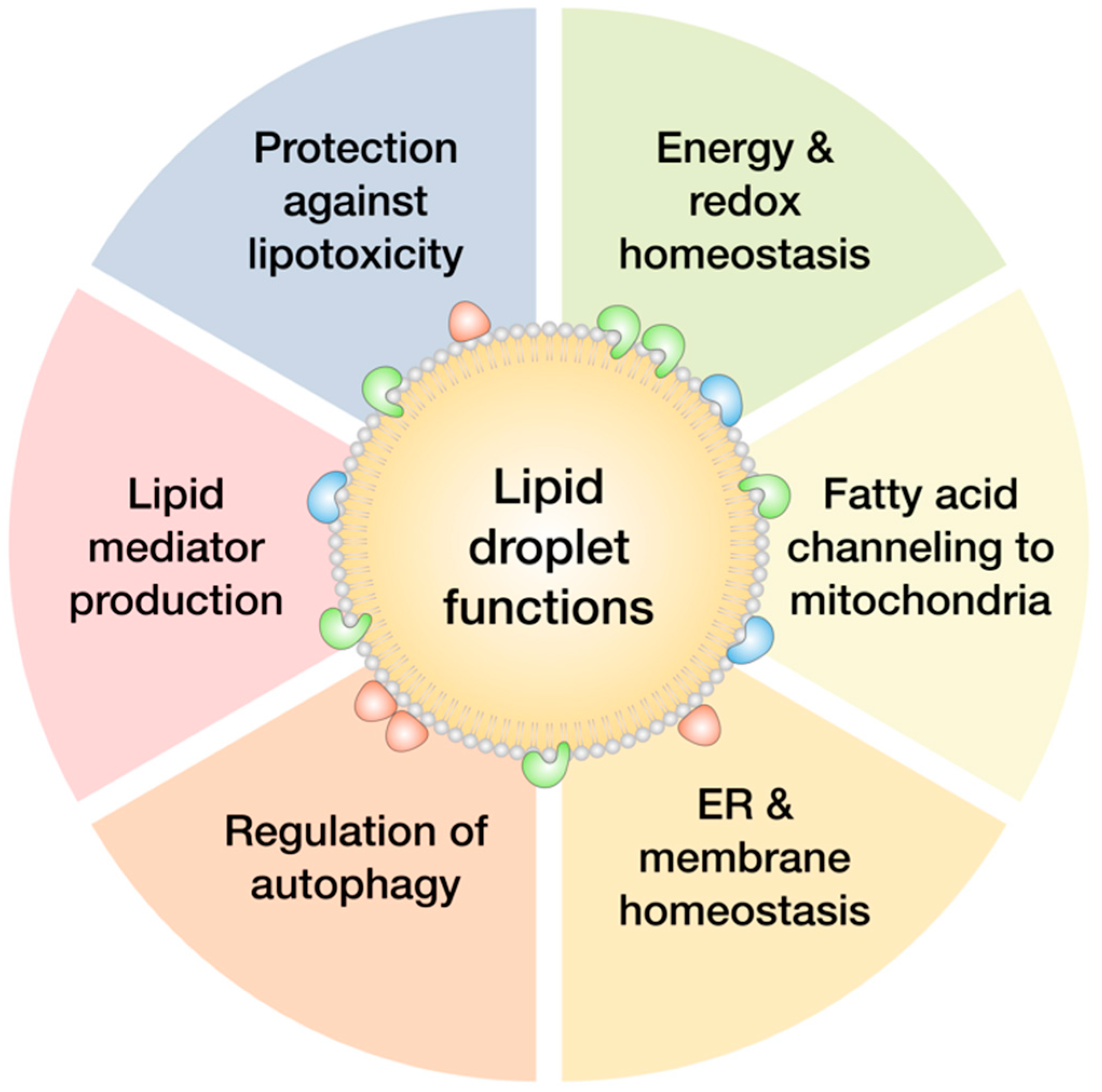
2. Lipid Droplets Basics
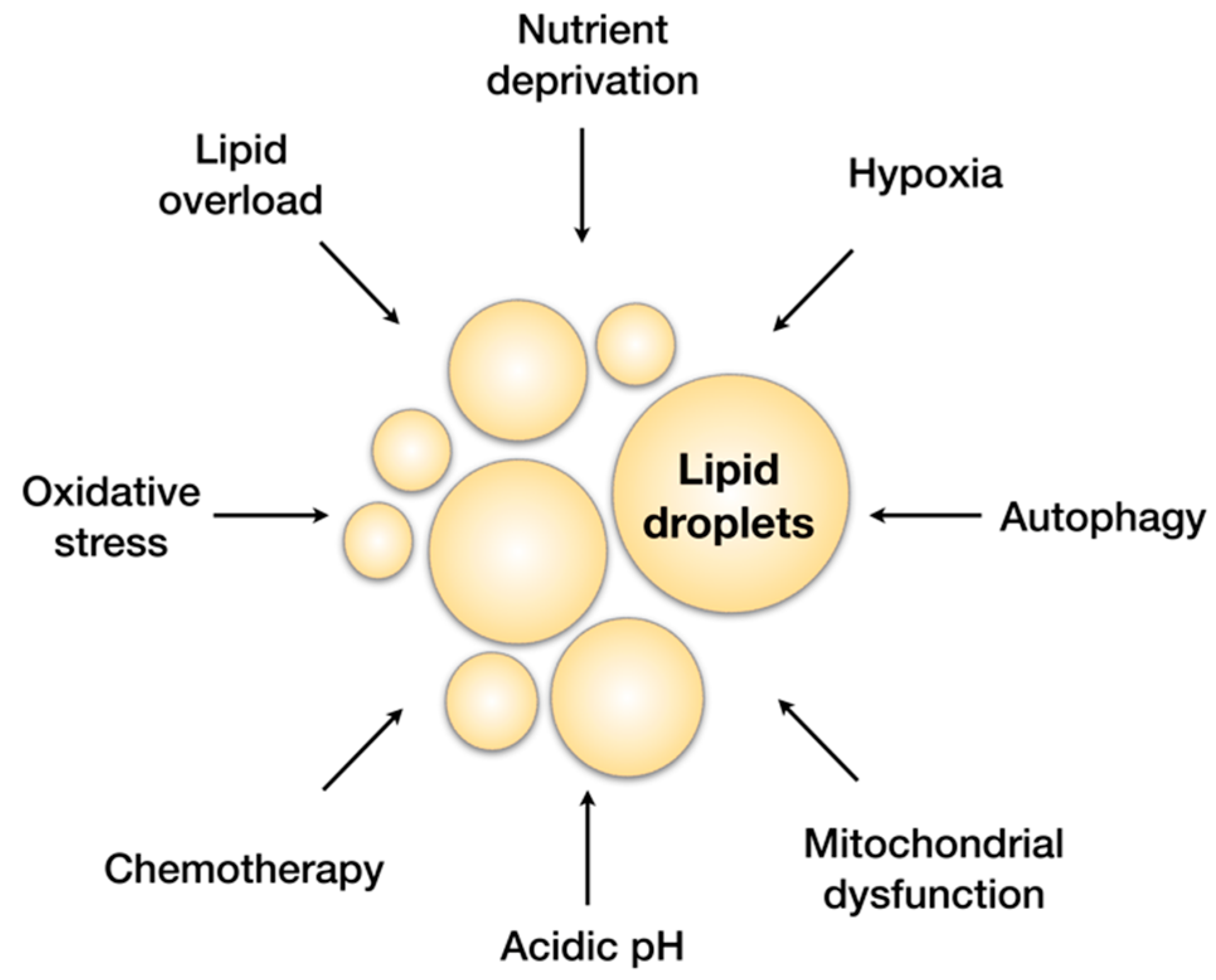
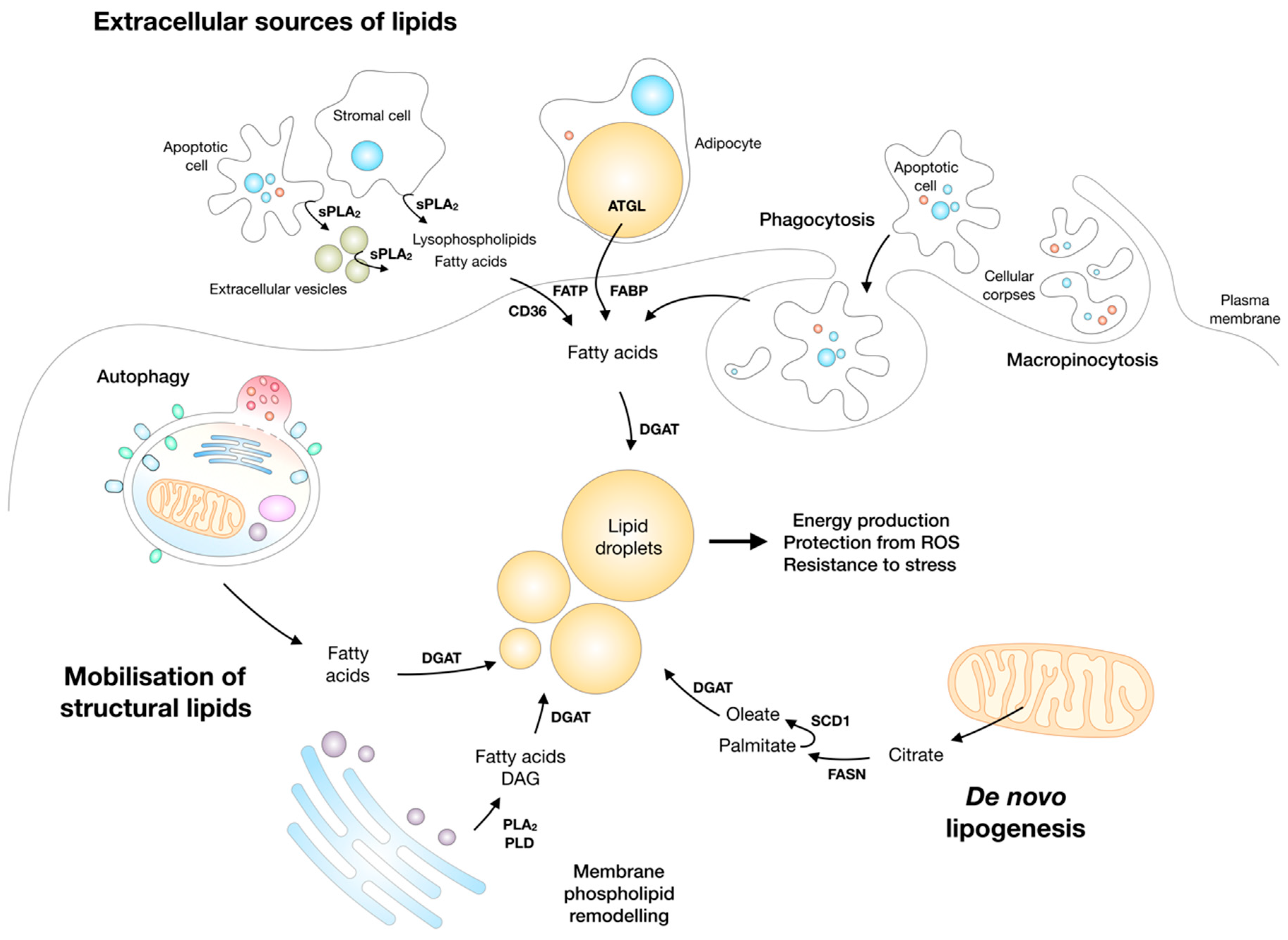
3. Cancer Cells Depend on Lipids to Survive and Grow in a Challenging Environment
Lipid Droplets Accumulate in Various Cancers
4. Lipid Droplet Biogenesis and Lipotoxicity
4.1. Lipid Droplet Biogenesis Protects from Lipotoxicity
4.2. Lipid Droplets Mediate the Protective Effects of Unsaturated FAs Against Lipotoxicity
4.3. Lipid Droplets Also Store Acylceramides and Reduce Ceramide Accumulation-Induced Cell Damage
4.4. Lipid Droplets Accumulate Cholesterol Esters to Regulate Cholesterol Availability and Promote Tumour Growth
4.5. Lipid Droplets Protect Cancer Cells From Chemotherapeutic Drugs
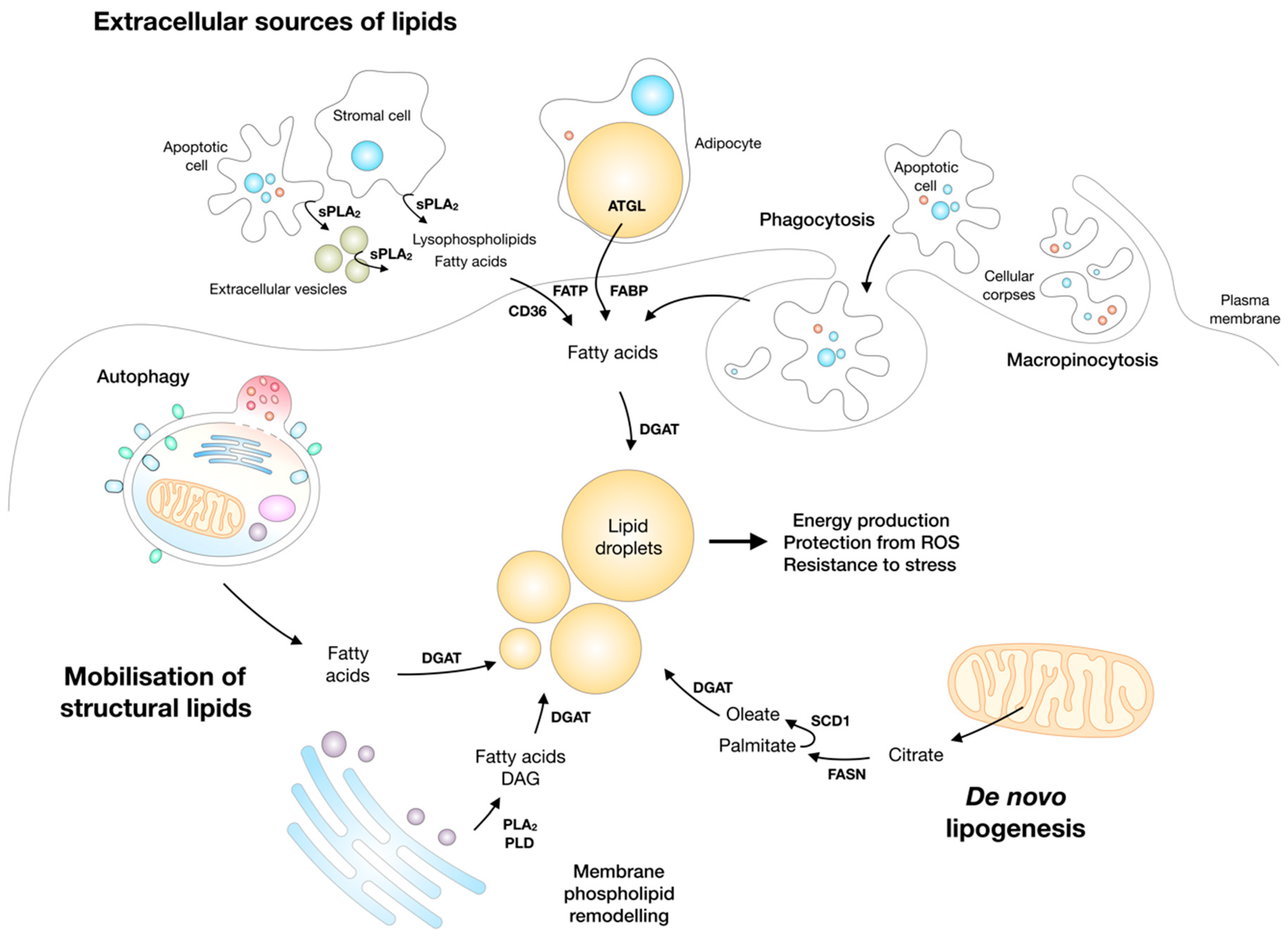
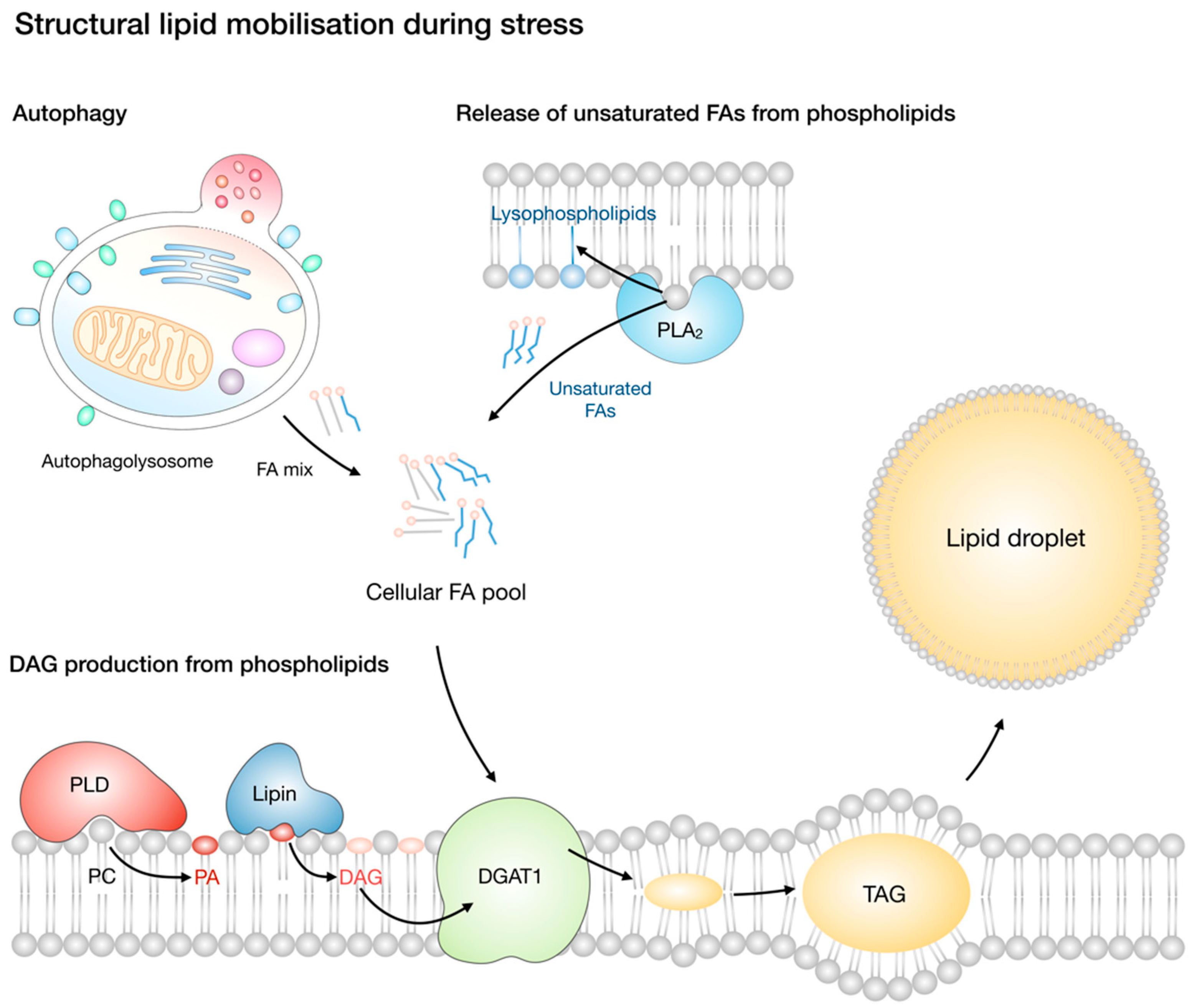
5. Lipolysis and Lipotoxic Stress
5.1. ATGL Regulates Fatty Acid-Mediated Energy Production, Signalling, and Lipotoxic Stress
5.2. ATGL and Cancer
6. Lipid Droplets are Synthesized and Broken Down during Nutrient Stress to Enable Cell Survival
6.1. Lipid Droplet Biogenesis is Enhanced during Nutrient Deprivation
6.2. Lipid Droplet Breakdown is Necessary for Cell Survival during Starvation
7. Lipid Droplets and Oxidative Stress
7.1. Lipid Droplets Accumulate in Hypoxia and Protect Cancer Cells from Oxidative Stress
7.2. Excessive Lipolysis is Damaging to Hypoxic Cancer Cells
7.3. Sequestration of PUFAs in Lipid Droplets Reduces Membrane Lipid Peroxidation and Protects Cells from Oxidative Stress
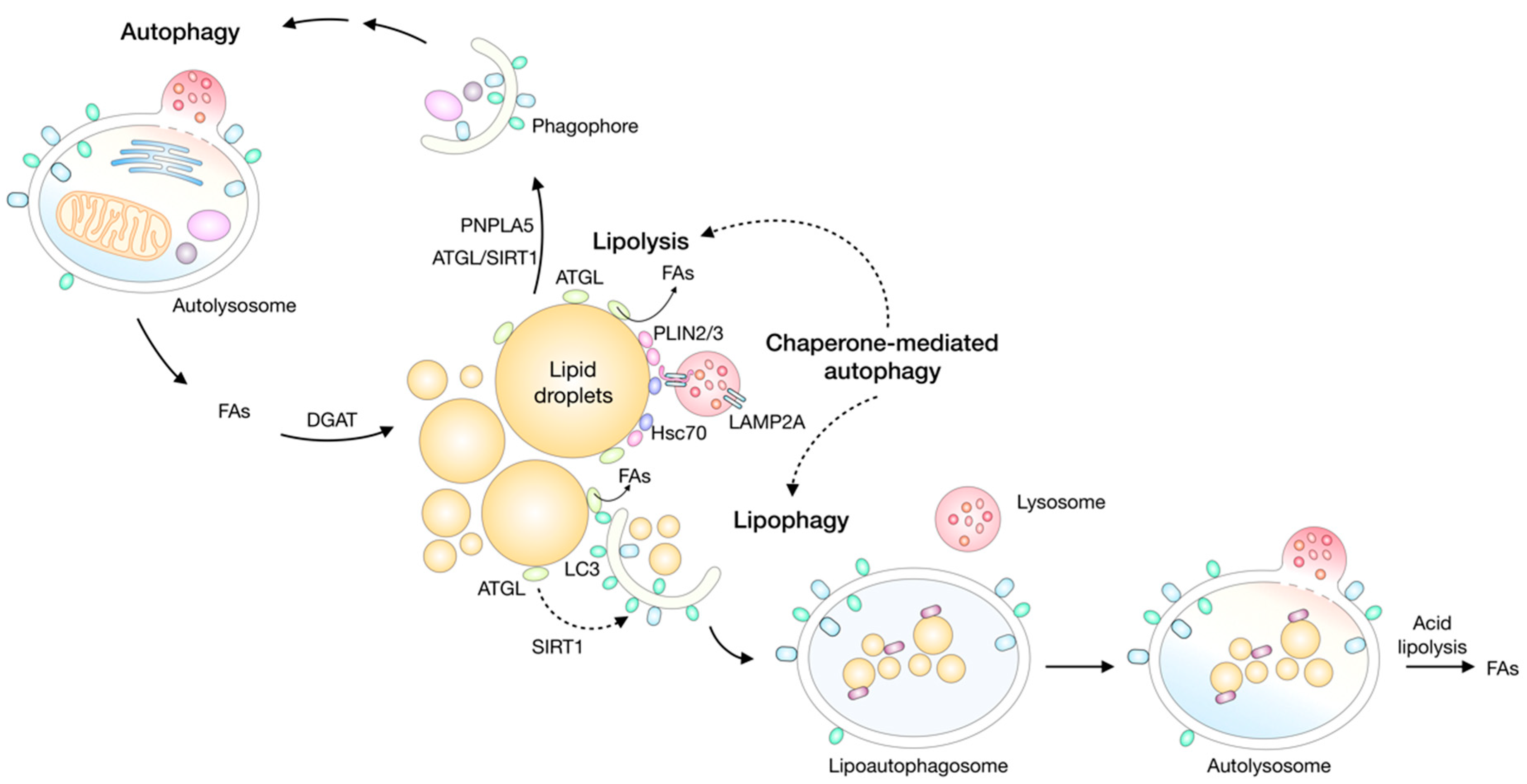
8. Lipid Droplets and Autophagy/Lipophagy
8.1. Lipophagy is an Important Pathway for Lipid Droplet Breakdown during Stress
8.2. Crosstalk between Lipolysis and Lipophagy/Autophagy
8.3. Autophagy Drives Lipid Droplet Biogenesis to Protect Cells from Stress
9. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wellen, K.E.; Thompson, C.B. Cellular metabolic stress: Considering how cells respond to nutrient excess. Mol. Cell 2010, 40, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulou, E.; Bulusu, V.; Kamphorst, J.J. Metabolic scavenging by cancer cells: When the going gets tough, the tough keep eating. Br. J. Cancer 2016, 115, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, D.; Simon, M.C. Hypoxia, lipids, and cancer: Surviving the harsh tumor microenvironment. Trends Cell Biol. 2014, 24, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, J.J.; Cross, J.R.; Fan, J.; de Stanchina, E.; Mathew, R.; White, E.P.; Thompson, C.B.; Rabinowitz, J.D. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. USA 2013, 110, 8882–8887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padanad, M.S.; Konstantinidou, G.; Venkateswaran, N.; Melegari, M.; Rindhe, S.; Mitsche, M.; Yang, C.; Batten, K.; Huffman, K.E.; Liu, J.; et al. Fatty Acid Oxidation Mediated by Acyl-CoA Synthetase Long Chain 3 Is Required for Mutant KRAS Lung Tumorigenesis. Cell Rep. 2016, 16, 1614–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pucer, A.; Brglez, V.; Payré, C.; Pungerčar, J.; Lambeau, G.; Petan, T. Group X secreted phospholipase A2 induces lipid droplet formation and prolongs breast cancer cell survival. Mol. Cancer 2013, 12, 111. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.-L.; et al. Fatty acid uptake and lipid storage induced by HIF-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [PubMed]
- Jarc, E.; Kump, A.; Malavašič, P.; Eichmann, T.O.; Zimmermann, R.; Petan, T. Lipid droplets induced by secreted phospholipase A2 and unsaturated fatty acids protect breast cancer cells from nutrient and lipotoxic stress. Biochim. Biophys. Acta 2018, 1863, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Nguyen, T.T.; Ravi, A.; Kubiniok, P.; Finicle, B.T.; Jayashankar, V.; Malacrida, L.; Hou, J.; Robertson, J.; Gao, D.; et al. PTEN deficiency and AMPK activation promote nutrient scavenging and anabolism in prostate cancer cells. Cancer Discov. 2018. [Google Scholar] [CrossRef] [PubMed]
- Delikatny, E.J.; Chawla, S.; Leung, D.-J.; Poptani, H. MR-visible lipids and the tumor microenvironment. NMR Biomed. 2011, 24, 592–611. [Google Scholar] [CrossRef] [PubMed]
- Koizume, S.; Miyagi, Y. Lipid Droplets: A Key Cellular Organelle Associated with Cancer Cell Survival under Normoxia and Hypoxia. Int. J. Mol. Sci. 2016, 17, 1430. [Google Scholar] [CrossRef] [PubMed]
- Bozza, P.T.; Viola, J.P.B. Lipid droplets in inflammation and cancer. Prostaglandins. Leukot. Essent. Fatty Acids 2010, 82, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Accioly, M.T.; Pacheco, P.; Maya-Monteiro, C.M.; Carrossini, N.; Robbs, B.K.; Oliveira, S.S.; Kaufmann, C.; Morgado-Diaz, J.A.; Bozza, P.T.; Viola, J.P.B. Lipid bodies are reservoirs of cyclooxygenase-2 and sites of prostaglandin-E2 synthesis in colon cancer cells. Cancer Res. 2008, 68, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Cabodevilla, A.G.; Sánchez-Caballero, L.; Nintou, E.; Boiadjieva, V.G.; Picatoste, F.; Gubern, A.; Claro, E. Cell survival during complete nutrient deprivation depends on lipid droplet-fueled β-oxidation of fatty acids. J. Biol. Chem. 2013, 288, 27777–27788. [Google Scholar] [CrossRef] [PubMed]
- Cotte, A.K.; Aires, V.; Fredon, M.; Limagne, E.; Derangère, V.; Thibaudin, M.; Humblin, E.; Scagliarini, A.; de Barros, J.-P.P.; Hillon, P.; et al. Lysophosphatidylcholine acyltransferase 2-mediated lipid droplet production supports colorectal cancer chemoresistance. Nat. Commun. 2018, 9, 322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Przybytkowski, E.; Joly, E.; Nolan, C.J.; Hardy, S.; Francoeur, A.-M.; Langelier, Y.; Prentki, M. Upregulation of cellular triacylglycerol—Free fatty acid cycling by oleate is associated with long-term serum-free survival of human breast cancer cells. Biochem. Cell Biol. 2007, 85, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Welte, M.A.; Gould, A.P. Lipid droplet functions beyond energy storage. Biochim. Biophys. Acta 2017, 1862, 1260–1272. [Google Scholar] [CrossRef] [PubMed]
- Henne, W.M.; Reese, M.L.; Goodman, J.M. The assembly of lipid droplets and their roles in challenged cells. EMBO J. 2018, 37, e98947. [Google Scholar] [CrossRef] [PubMed]
- Farese, R.V.; Walther, T.C. Lipid droplets finally get a little R-E-S-P-E-C-T. Cell 2009, 139, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.S.; Coleman, R.A.; Kraemer, F.B.; McManaman, J.L.; Obin, M.S.; Puri, V.; Yan, Q.; Miyoshi, H.; Mashek, D.G. The role of lipid droplets in metabolic disease in rodents and humans. J. Clin. Investig. 2011, 121, 2102–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walther, T.C.; Farese, R. V Lipid Droplets and Cellular Lipid Metabolism. Annu. Rev. Biochem. 2012, 81, 687–714. [Google Scholar] [CrossRef] [PubMed]
- Gubern, A.; Barceló-Torns, M.; Casas, J.; Barneda, D.; Masgrau, R.; Picatoste, F.; Balsinde, J.; Balboa, M.A.; Claro, E. Lipid droplet biogenesis induced by stress involves triacylglycerol synthesis that depends on group VIA phospholipase A2. J. Biol. Chem. 2009, 284, 5697–5708. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.P.; Koster, G.; Guillermier, C.; Hirst, E.M.A.; MacRae, J.I.; Lechene, C.P.; Postle, A.D.; Gould, A.P. Antioxidant Role for Lipid Droplets in a Stem Cell Niche of Drosophila. Cell 2015, 163, 340–353. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Cohen, S.; Lippincott-Schwartz, J. Fatty acid trafficking in starved cells: Regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 2015, 32, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Qiu, B.; Ackerman, D.; Sanchez, D.J.; Li, B.; Ochocki, J.D.; Grazioli, A.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Keith, B.; Simon, M.C. HIF2α-dependent lipid storage promotes endoplasmic reticulum homeostasis in clear-cell renal cell carcinoma. Cancer Discov. 2015, 5, 652–667. [Google Scholar] [CrossRef] [PubMed]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mylonis, I.; Sembongi, H.; Befani, C.; Liakos, P.; Siniossoglou, S.; Simos, G. Hypoxia causes triglyceride accumulation by HIF-1-mediated stimulation of lipin 1 expression. J. Cell Sci. 2012, 125, 3485–3493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlaepfer, I.R.; Nambiar, D.K.; Ramteke, A.; Kumar, R.; Dhar, D.; Agarwal, C.; Bergman, B.; Graner, M.; Maroni, P.; Singh, R.P.; et al. Hypoxia induces triglycerides accumulation in prostate cancer cells and extracellular vesicles supporting growth and invasiveness following reoxygenation. Oncotarget 2015, 6, 22836–22856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.; Tsai, T.H.; Li, L.; Saha, P.; Chan, L.; Chang, B.H.J. PLIN2 is a Key Regulator of the Unfolded Protein Response and Endoplasmic Reticulum Stress Resolution in Pancreatic β Cells. Sci. Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.B.; Louie, S.M.; Daniele, J.R.; Tran, Q.; Dillin, A.; Zoncu, R.; Nomura, D.K.; Olzmann, J.A. DGAT1-Dependent Lipid Droplet Biogenesis Protects Mitochondrial Function during Starvation-Induced Autophagy. Dev. Cell 2017, 42, 9–21.e5. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.B.; Olzmann, J.A. Lipid droplets and lipotoxicity during autophagy. Autophagy 2017, 13, 2002–2003. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Xiang, Y.; Wang, Y.; Baikati, K.; Cuervo, A.M.; Luu, Y.K.; Tang, Y.; Pessin, J.E.; Schwartz, G.J.; Czaja, M.J. Autophagy regulates adipose mass and differentiation in mice. J. Clin. Investig. 2009, 119, 3329–3339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roa-Mansergas, X.; Fadó, R.; Atari, M.; Mir, J.F.; Muley, H.; Serra, D.; Casals, N. CPT1C promotes human mesenchymal stem cells survival under glucose deprivation through the modulation of autophagy. Sci. Rep. 2018, 8, 6997. [Google Scholar] [CrossRef] [PubMed]
- Dichlberger, A.; Schlager, S.; Lappalainen, J.; Käkelä, R.; Hattula, K.; Butcher, S.J.; Schneider, W.J.; Kovanen, P.T. Lipid body formation during maturation of human mast cells. J. Lipid Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Boren, J.; Brindle, K.M. Apoptosis-induced mitochondrial dysfunction causes cytoplasmic lipid droplet formation. Cell Death Differ. 2012, 19, 1561–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Lizardo, D.Y.; Atilla-Gokcumen, G.E. Specific triacylglycerols accumulate via increased lipogenesis during 5-FU-induced apoptosis. ACS Chem. Biol. 2016, 11, 2583–2587. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; Zhang, J.; Choi, A.M.K.; Kim, H.P. Mitochondrial Dysfunction Induces Formation of Lipid Droplets as a Generalized Response to Stress. Oxid. Med. Cell. Longev. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Senkal, C.E.; Salama, M.F.; Snider, A.J.; Allopenna, J.J.; Rana, N.A.; Koller, A.; Hannun, Y.A.; Obeid, L.M. Ceramide Is Metabolized to Acylceramide and Stored in Lipid Droplets. Cell Metab. 2017, 25, 686–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosma, M.; Dapito, D.H.; Drosatos-Tampakaki, Z.; Huiping-Son, N.; Huang, L.-S.; Kersten, S.; Drosatos, K.; Goldberg, I.J. Sequestration of fatty acids in triglycerides prevents endoplasmic reticulum stress in an in vitro model of cardiomyocyte lipotoxicity. Biochim. Biophys. Acta 2014, 1841, 1648–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herms, A.; Bosch, M.; Reddy, B.J.N.; Schieber, N.L.; Fajardo, A.; Rupérez, C.; Fernández-Vidal, A.; Ferguson, C.; Rentero, C.; Tebar, F.; et al. AMPK activation promotes lipid droplet dispersion on detyrosinated microtubules to increase mitochondrial fatty acid oxidation. Nat. Commun. 2015, 6, 7176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbosa, A.D.; Sembongi, H.; Su, W.-M.; Abreu, S.; Reggiori, F.; Carman, G.M.; Siniossoglou, S. Lipid partitioning at the nuclear envelope controls membrane biogenesis. Mol. Biol. Cell 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurat, C.F.; Wolinski, H.; Petschnigg, J.; Kaluarachchi, S.; Andrews, B.; Natter, K.; Kohlwein, S.D. Cdk1/Cdc28-dependent activation of the major triacylglycerol lipase Tgl4 in yeast links lipolysis to cell-cycle progression. Mol. Cell 2009, 33, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Benador, I.Y.; Veliova, M.; Mahdaviani, K.; Petcherski, A.; Wikstrom, J.D.; Assali, E.A.; Acín-Pérez, R.; Shum, M.; Oliveira, M.F.; Cinti, S.; et al. Mitochondria Bound to Lipid Droplets Have Unique Bioenergetics, Composition, and Dynamics that Support Lipid Droplet Expansion. Cell Metab. 2018, 27, 869–885.e6. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kaufman, R.J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, E.J.; Vevea, J.D.; Pon, L.A. Lipid droplet autophagy during energy mobilization, lipid homeostasis and protein quality control. Front. Biosci. (Landmark Ed.) 2018, 23, 1552–1563. [Google Scholar] [CrossRef] [PubMed]
- Chitraju, C.; Mejhert, N.; Haas, J.T.; Diaz-Ramirez, L.G.; Grueter, C.A.; Imbriglio, J.E.; Pinto, S.; Koliwad, S.K.; Walther, T.C.; Farese, R.V.; et al. Triglyceride Synthesis by DGAT1 Protects Adipocytes from Lipid-Induced ER Stress during Lipolysis. Cell Metab. 2017, 26, 407–418.e3. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Cuervo, A.M. Lipophagy: Connecting autophagy and lipid metabolism. Int. J. Cell Biol. 2012, 2012, 282041. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Czaja, M.J. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 2013, 20, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Christian, P.; Sacco, J.; Adeli, K. Autophagy: Emerging roles in lipid homeostasis and metabolic control. Biochim. Biophys. Acta 2013, 1831, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-W. Lipid droplets, lipophagy, and beyond. Biochim. Biophys. Acta 2016, 1861, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lopez, N.; Singh, R. Autophagy and lipid droplets in the liver. Annu. Rev. Nutr. 2015, 35, 215–237. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, G.; Lass, A. Genetically modified mouse models to study hepatic neutral lipid mobilization. Biochim. Biophys. Acta 2018, 30205–30209. [Google Scholar] [CrossRef] [PubMed]
- Osumi, T.; Kuramoto, K. Heart lipid droplets and lipid droplet-binding proteins: Biochemistry, physiology, and pathology. Exp. Cell Res. 2016, 340, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Zechner, R.; Madeo, F.; Kratky, D. Cytosolic lipolysis and lipophagy: Two sides of the same coin. Nat. Rev. Mol. Cell Biol. 2017, 18, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Young, S.G.; Zechner, R. Biochemistry and pathophysiology of intravascular and intracellular lipolysis. Genes Dev. 2013, 27, 459–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupont, N.; Chauhan, S.; Arko-Mensah, J.; Castillo, E.F.; Masedunskas, A.; Weigert, R.; Robenek, H.; Proikas-Cezanne, T.; Deretic, V. Neutral lipid stores and lipase PNPLA5 contribute to autophagosome biogenesis. Curr. Biol. 2014, 24, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Shpilka, T.; Welter, E.; Borovsky, N.; Amar, N.; Mari, M.; Reggiori, F.; Elazar, Z. Lipid droplets and their component triglycerides and steryl esters regulate autophagosome biogenesis. EMBO J. 2015, 34, 2117–2131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velázquez, A.P.; Tatsuta, T.; Ghillebert, R.; Drescher, I.; Graef, M. Lipid droplet-mediated ER homeostasis regulates autophagy and cell survival during starvation. J. Cell Biol. 2016, 212, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Sathyanarayan, A.; Mashek, M.T.; Mashek, D.G. ATGL Promotes Autophagy/Lipophagy via SIRT1 to Control Hepatic Lipid Droplet Catabolism. Cell Rep. 2017, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dichlberger, A.; Schlager, S.; Maaninka, K.; Schneider, W.J.; Kovanen, P.T. Adipose triglyceride lipase regulates eicosanoid production in activated human mast cells. J. Lipid Res. 2014, 55, 2471–2478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlager, S.; Goeritzer, M.; Jandl, K.; Frei, R.; Vujic, N.; Kolb, D.; Strohmaier, H.; Dorow, J.; Eichmann, T.O.; Rosenberger, A.; et al. Adipose triglyceride lipase acts on neutrophil lipid droplets to regulate substrate availability for lipid mediator synthesis. J. Leukoc. Biol. 2015, 98, 837–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, W.-J.; Azhar, S.; Kraemer, F.B. Lipid droplets and steroidogenic cells. Exp. Cell Res. 2016, 340, 209–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haemmerle, G.; Moustafa, T.; Woelkart, G.; Büttner, S.; Schmidt, A.; van de Weijer, T.; Hesselink, M.; Jaeger, D.; Kienesberger, P.C.; Zierler, K.; et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat. Med. 2011, 17, 1076–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zechner, R.; Zimmermann, R.; Eichmann, T.O.; Kohlwein, S.D.; Haemmerle, G.; Lass, A.; Madeo, F. FAT SIGNALS—Lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 2012, 15, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Dichlberger, A.; Schlager, S.; Kovanen, P.T.; Schneider, W.J. Lipid droplets in activated mast cells—A significant source of triglyceride-derived arachidonic acid for eicosanoid production. Eur. J. Pharmacol. 2016, 785, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Wilfling, F.; Haas, J.T.; Walther, T.C.; Farese, R. V Lipid droplet biogenesis. Curr. Opin. Cell Biol. 2014, 29, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Peterson, C.W.H.; To, M.; Sahl, S.J.; Savikhin, V.; Grossman, E.A.; Nomura, D.K.; Olzmann, J.A. A Proximity Labeling Strategy Provides Insights into the Composition and Dynamics of Lipid Droplet Proteomes. Dev. Cell 2018, 44, 91–112. [Google Scholar] [CrossRef] [PubMed]
- Kory, N.; Farese, R.V.; Walther, T.C. Targeting Fat: Mechanisms of Protein Localization to Lipid Droplets. Trends Cell Biol. 2016, 26, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohsaki, Y.; Kawai, T.; Yoshikawa, Y.; Cheng, J.; Jokitalo, E.; Fujimoto, T. PML isoform II plays a critical role in nuclear lipid droplet formation. J. Cell Biol. 2016, 212, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanauska, A.; Köhler, A. The Inner Nuclear Membrane Is a Metabolically Active Territory that Generates Nuclear Lipid Droplets. Cell 2018, 174, 700–715. [Google Scholar] [CrossRef] [PubMed]
- Walther, T.C.; Chung, J.; Farese, R. V Lipid Droplet Biogenesis. Annu. Rev. Cell Dev. Biol. 2017, 33, 491–510. [Google Scholar] [CrossRef] [PubMed]
- Pol, A.; Gross, S.P.; Parton, R.G. Biogenesis of the multifunctional lipid droplet: Lipids, proteins, and sites. J. Cell Biol. 2014, 204, 635–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiam, A.R.; Farese, R.V.; Walther, T.C. The biophysics and cell biology of lipid droplets. Nat. Rev. Mol. Cell Biol. 2013, 14, 775. [Google Scholar] [CrossRef] [PubMed]
- Salo, V.T.; Belevich, I.; Li, S.; Karhinen, L.; Vihinen, H.; Vigouroux, C.; Magré, J.; Thiele, C.; Hölttä-Vuori, M.; Jokitalo, E.; Ikonen, E. Seipin regulates ER-lipid droplet contacts and cargo delivery. EMBO J. 2016, e201695170. [Google Scholar] [CrossRef] [PubMed]
- Thiam, A.R.; Beller, M. The why, when and how of lipid droplet diversity. J. Cell Sci. 2017, 130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, Y.; Cui, L.; Deng, Y.; Xu, S.; Yu, J.; Cichello, S.; Serrero, G.; Ying, Y.; Liu, P. Morphologically and Functionally Distinct Lipid Droplet Subpopulations. Sci. Rep. 2016, 6, 29539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molenaar, M.R.; Vaandrager, A.B.; Helms, J.B. Some lipid droplets are more equal than others: Different metabolic lipid droplet pools in hepatic stellate cells. Lipid Insights 2017, 10, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Herms, A.; Bosch, M.; Ariotti, N.; Reddy, B.J.; Fajardo, A.; Fernandez-Vidal, A.; Alvarez-Guaita, A.; Fernandez-Rojo, M.A.; Rentero, C.; Tebar, F.; et al. Cell-to-cell heterogeneity in lipid droplets suggests a mechanism to reduce lipotoxicity. Curr. Biol. 2013, 23, 1489–1496. [Google Scholar] [CrossRef] [PubMed]
- Schulze, R.J.; Sathyanarayan, A.; Mashek, D.G. Breaking fat: The regulation and mechanisms of lipophagy. Biochim. Biophys. Acta 2017, 1862, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Lass, A.; Zimmermann, R.; Oberer, M.; Zechner, R. Lipolysis—A highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog. Lipid Res. 2011, 50, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, R.; Strauss, J.G.; Haemmerle, G.; Schoiswohl, G.; Birner-Gruenberger, R.; Riederer, M.; Lass, A.; Neuberger, G.; Eisenhaber, F.; Hermetter, A.; et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 2004, 306, 1383–1386. [Google Scholar] [CrossRef] [PubMed]
- Schoiswohl, G.; Schweiger, M.; Schreiber, R.; Gorkiewicz, G.; Preiss-Landl, K.; Taschler, U.; Zierler, K.A.; Radner, F.P.W.; Eichmann, T.O.; Kienesberger, P.C.; et al. Adipose triglyceride lipase plays a key role in the supply of the working muscle with fatty acids. J. Lipid Res. 2010, 51, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Grabner, G.F.; Zimmermann, R.; Schicho, R.; Taschler, U. Monoglyceride lipase as a drug target: At the crossroads of arachidonic acid metabolism and endocannabinoid signaling. Pharmacol. Ther. 2017, 175, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.T.; Mashek, M.T.; Bu, S.Y.; Greenberg, A.S.; Mashek, D.G. Adipose triglyceride lipase is a major hepatic lipase that regulates triacylglycerol turnover and fatty acid signaling and partitioning. Hepatology 2011, 53, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Kuramoto, K.; Sakai, F.; Yoshinori, N.; Nakamura, T.Y.; Wakabayashi, S.; Kojidani, T.; Haraguchi, T.; Hirose, F.; Osumi, T. Deficiency of a lipid droplet protein, perilipin 5, suppresses myocardial lipid accumulation, thereby preventing type 1 diabetes-induced heart malfunction. Mol. Cell. Biol. 2014, 34, 2721–2731. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.D.; Claudel, T.; Kumari, P.; Haemmerle, G.; Pollheimer, M.J.; Stojakovic, T.; Scharnagl, H.; Halilbasic, E.; Gumhold, J.; Silbert, D.; et al. Absence of adipose triglyceride lipase protects from hepatic endoplasmic reticulum stress in mice. Hepatology 2012, 56, 270–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cingolani, F.; Czaja, M.J. Regulation and Functions of Autophagic Lipolysis. Trends Endocrinol. Metab. 2016, 27, 696–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, N.; Lupien, L.; Kuemmerle, N.B.; Kinlaw, W.B.; Swinnen, J.V.; Smans, K. Lipogenesis and lipolysis: The pathways exploited by the cancer cells to acquire fatty acids. Prog. Lipid Res. 2013, 52, 585–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R. V Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.A.; Brault, C.; Peck, B.; Bensaad, K.; Griffiths, B.; Mitter, R.; Chakravarty, P.; East, P.; Dankworth, B.; Alibhai, D.; et al. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene 2015, 34, 5128–5140. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, B.; Lewis, C.A.; Bensaad, K.; Ros, S.; Zhang, Q.; Ferber, E.C.; Konisti, S.; Peck, B.; Miess, H.; East, P.; et al. Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab. 2013, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661. [Google Scholar] [CrossRef] [PubMed]
- Balaban, S.; Shearer, R.F.; Lee, L.S.; van Geldermalsen, M.; Schreuder, M.; Shtein, H.C.; Cairns, R.; Thomas, K.C.; Fazakerley, D.J.; Grewal, T.; et al. Adipocyte lipolysis links obesity to breast cancer growth: Adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab. 2017, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.Y.; Attané, C.; Milhas, D.; Dirat, B.; Dauvillier, S.; Guerard, A.; Gilhodes, J.; Lazar, I.; Alet, N.; Laurent, V.; et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2017, 2, e87489. [Google Scholar] [CrossRef] [PubMed]
- Sulciner, M.L.; Serhan, C.N.; Gilligan, M.M.; Mudge, D.K.; Chang, J.; Gartung, A.; Lehner, K.A.; Bielenberg, D.R.; Schmidt, B.; Dalli, J.; et al. Resolvins suppress tumor growth and enhance cancer therapy. J. Exp. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Rysman, E.; Brusselmans, K.; Scheys, K.; Timmermans, L.; Derua, R.; Munck, S.; Van Veldhoven, P.P.; Waltregny, D.; Daniëls, V.W.; Machiels, J.; et al. V De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 2010, 70, 8117–8126. [Google Scholar] [CrossRef] [PubMed]
- Young, R.M.; Ackerman, D.; Quinn, Z.L.; Mancuso, A.; Gruber, M.; Liu, L.; Giannoukos, D.N.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Keith, B.; et al. Dysregulated mTORC1 renders cells critically dependent on desaturated lipids for survival under tumor-like stress. Genes Dev. 2013, 27, 1115–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brglez, V.; Lambeau, G.; Petan, T. Secreted phospholipases A2 in cancer: Diverse mechanisms of action. Biochimie 2014, 107 Pt A, 114–123. [Google Scholar] [CrossRef]
- Murakami, M.; Taketomi, Y.; Miki, Y.; Sato, H.; Yamamoto, K.; Lambeau, G. Emerging roles of secreted phospholipase A2 enzymes: The 3rd edition. Biochimie 2014, 107, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Lambeau, G.; Gelb, M.H. Biochemistry and physiology of mammalian secreted phospholipases A2. Annu. Rev. Biochem. 2008, 77, 495–520. [Google Scholar] [CrossRef] [PubMed]
- Boilard, E. Extracellular vesicles and their content in bioactive lipid mediators: More than a sack of microRNA. J. Lipid Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Guijas, C.; Rodríguez, J.P.; Rubio, J.M.; Balboa, M.A.; Balsinde, J. Phospholipase A2 regulation of lipid droplet formation. Biochim. Biophys. Acta 2014, 1841, 1661–1671. [Google Scholar] [CrossRef] [PubMed]
- Novikoff, A.B. A transplantable rat liver tumor induced by 4-dimethylaminoazobenzene. Cancer Res. 1957, 17, 1010–1027. [Google Scholar] [PubMed]
- Aboumrad, M.H.; Horn, R.C.; Fine, G. Lipid-secreting mammary carcinoma. Report of a case associated with Paget’s disease of the nipple. Cancer 1963, 16, 521–525. [Google Scholar] [CrossRef]
- Ramos, C.V.; Taylor, H.B. Lipid-rich carcinoma of the breast. A clinicopathologic analysis of 13 examples. Cancer 1974, 33, 812–819. [Google Scholar] [CrossRef]
- Abbas, M.K.; Yoo, T.J.; Viles, J. Ultrastructure and fatty acid composition of fatty acid-modified Morris 7777 hepatoma cells. Cancer Res. 1982, 42, 4639–4649. [Google Scholar] [PubMed]
- Lantos, P.L. Macrophages in brain tumours induced transplacentally by N-ethyl-N-nitrosourea in rats: An electron-microscope study. J. Pathol. 1975, 116, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Spence, A.M.; Rubinstein, L.J. Cerebellar capillary hemangioblastoma: Its histogenesis studied by organ culture and electron microscopy. Cancer 1975, 35, 326–341. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Melkoumian, Z.K.; Lucktong, A.; Moniwa, M.; Davie, J.R.; Strobl, J.S. Rapid induction of histone hyperacetylation and cellular differentiation in human breast tumor cell lines following degradation of histone deacetylase-1. J. Biol. Chem. 2000, 275, 35256–35263. [Google Scholar] [CrossRef] [PubMed]
- Dessi, S.; Batetta, B.; Pulisci, D.; Spano, O.; Cherchi, R.; Lanfranco, G.; Tessitore, L.; Costelli, P.; Baccino, F.M.; Anchisi, C. Altered pattern of lipid metabolism in patients with lung cancer. Oncology 1992, 49, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Than, N.G.; Sumegi, B.; Bellyei, S.; Berki, T.; Szekeres, G.; Janaky, T.; Szigeti, A.; Bohn, H.; Than, G.N. Lipid droplet and milk lipid globule membrane associated placental protein 17b (PP17b) is involved in apoptotic and differentiation processes of human epithelial cervical carcinoma cells. Eur. J. Biochem. 2003, 270, 1176–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hager, M.H.; Solomon, K.R.; Freeman, M.R. The role of cholesterol in prostate cancer. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Straub, B.K.; Herpel, E.; Singer, S.; Zimbelmann, R.; Breuhahn, K.; Macher-Goeppinger, S.; Warth, A.; Lehmann-Koch, J.; Longerich, T.; Heid, H.; et al. Lipid droplet-associated PAT-proteins show frequent and differential expression in neoplastic steatogenesis. Mod. Pathol. 2010, 23, 480–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundelin, J.P.; Ståhlman, M.; Lundqvist, A.; Levin, M.; Parini, P.; Johansson, M.E.; Borén, J. Increased expression of the very low-density lipoprotein receptor mediates lipid accumulation in clear-cell renal cell carcinoma. PLoS ONE 2012, 7, e48694. [Google Scholar] [CrossRef] [PubMed]
- Guillaumond, F.; Bidaut, G.; Ouaissi, M.; Servais, S.; Gouirand, V.; Olivares, O.; Lac, S.; Borge, L.; Roques, J.; Gayet, O.; et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, 2473–2478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Gonzalo-Calvo, D.; López-Vilaró, L.; Nasarre, L.; Perez-Olabarria, M.; Vázquez, T.; Escuin, D.; Badimon, L.; Barnadas, A.; Lerma, E.; Llorente-Cortés, V. Intratumor cholesteryl ester accumulation is associated with human breast cancer proliferation and aggressive potential: A molecular and clinicopathological study. BMC Cancer 2015, 15, 460. [Google Scholar] [CrossRef] [PubMed]
- Freitas, I.; Bono, B.; Bertone, V.; Griffini, P.; Baronzio, G.F.; Bonandrini, L.; Gerzeli, G. Characterization of the metabolism of perinecrotic cells in solid tumors by enzyme histochemistry. Anticancer Res. 1996, 16, 1491–1502. [Google Scholar] [PubMed]
- Tirinato, L.; Pagliari, F.; Limongi, T.; Marini, M.; Falqui, A.; Seco, J.; Candeloro, P.; Liberale, C.; Di Fabrizio, E. An Overview of Lipid Droplets in Cancer and Cancer Stem Cells. Stem Cells Int. 2017, 2017, 1656053. [Google Scholar] [CrossRef] [PubMed]
- Jaishy, B.; Abel, E.D. Lipids, lysosomes, and autophagy. J. Lipid Res. 2016, 57, 1619–1635. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, J.E. Lipotoxicity: When tissues overeat. Curr. Opin. Lipidol. 2003, 14, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Ertunc, M.E.; Hotamisligil, G.S. Lipid signaling and lipotoxicity in metaflammation: Indications for metabolic disease pathogenesis and treatment. J. Lipid Res. 2016, 57, 2099–2114. [Google Scholar] [CrossRef] [PubMed]
- Aon, M.A.; Bhatt, N.; Cortassa, S.C. Mitochondrial and cellular mechanisms for managing lipid excess. Front. Physiol. 2014, 5, 282. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Font-Burgada, J.; Sun, B.; Karin, M. Obesity and Cancer: The Oil that Feeds the Flame. Cell Metab. 2016, 23, 48–62. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosma, M.; Kersten, S.; Hesselink, M.K.C.; Schrauwen, P. Re-evaluating lipotoxic triggers in skeletal muscle: Relating intramyocellular lipid metabolism to insulin sensitivity. Prog. Lipid Res. 2012, 51, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Summers, S.A. Ceramides in insulin resistance and lipotoxicity. Prog. Lipid Res. 2006, 45, 42–72. [Google Scholar] [CrossRef] [PubMed]
- Erion, D.M.; Shulman, G.I. Diacylglycerol-mediated insulin resistance. Nat. Med. 2010, 16, 400–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuramoto, K.; Okamura, T.; Yamaguchi, T.; Nakamura, T.Y.; Wakabayashi, S.; Morinaga, H.; Nomura, M.; Yanase, T.; Otsu, K.; Usuda, N.; et al. Perilipin 5, a lipid droplet-binding protein, protects heart from oxidative burden by sequestering fatty acid from excessive oxidation. J. Biol. Chem. 2012, 287, 23852–23863. [Google Scholar] [CrossRef] [PubMed]
- Drosatos, K.; Bharadwaj, K.G.; Lymperopoulos, A.; Ikeda, S.; Khan, R.; Hu, Y.; Agarwal, R.; Yu, S.; Jiang, H.; Steinberg, S.F.; et al. Cardiomyocyte lipids impair β-adrenergic receptor function via PKC activation. Am. J. Physiol. Endocrinol. Metab. 2011, 300, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Bosma, M.; Hesselink, M.K.C.; Sparks, L.M.; Timmers, S.; Ferraz, M.J.; Mattijssen, F.; van Beurden, D.; Schaart, G.; de Baets, M.H.; Verheyen, F.K.; et al. Perilipin 2 improves insulin sensitivity in skeletal muscle despite elevated intramuscular lipid levels. Diabetes 2012, 61, 2679–2690. [Google Scholar] [CrossRef] [PubMed]
- Schenk, S.; Horowitz, J.F. Acute exercise increases triglyceride synthesis in skeletal muscle and prevents fatty acid-induced insulin resistance. J. Clin. Investig. 2007, 117, 1690–1698. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Betters, J.L.; Lord, C.; Ma, Y.; Han, X.; Yang, K.; Alger, H.M.; Melchior, J.; Sawyer, J.; Shah, R.; et al. CGI-58 knockdown in mice causes hepatic steatosis but prevents diet-induced obesity and glucose intolerance. J. Lipid Res. 2010, 51, 3306–3315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koliwad, S.K.; Streeper, R.S.; Monetti, M.; Cornelissen, I.; Chan, L.; Terayama, K.; Naylor, S.; Rao, M.; Hubbard, B.; Farese, R.V. DGAT1-dependent triacylglycerol storage by macrophages protects mice from diet-induced insulin resistance and inflammation. J. Clin. Investig. 2010, 120, 756–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.C.; Stone, S.J.; Zhou, P.; Buhman, K.K.; Farese, R. V Dissociation of obesity and impaired glucose disposal in mice overexpressing acyl coenzyme a:diacylglycerol acyltransferase 1 in white adipose tissue. Diabetes 2002, 51, 3189–3195. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Shi, X.; Bharadwaj, K.G.; Ikeda, S.; Yamashita, H.; Yagyu, H.; Schaffer, J.E.; Yu, Y.-H.; Goldberg, I.J. DGAT1 expression increases heart triglyceride content but ameliorates lipotoxicity. J. Biol. Chem. 2009, 284, 36312–36323. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Trent, C.M.; Fang, X.; Son, N.-H.; Jiang, H.; Blaner, W.S.; Hu, Y.; Yin, Y.-X.; Farese, R.V.; Homma, S.; et al. Cardiomyocyte-specific loss of diacylglycerol acyltransferase 1 (DGAT1) reproduces the abnormalities in lipids found in severe heart failure. J. Biol. Chem. 2014, 289, 29881–29891. [Google Scholar] [CrossRef] [PubMed]
- Monetti, M.; Levin, M.C.; Watt, M.J.; Sajan, M.P.; Marmor, S.; Hubbard, B.K.; Stevens, R.D.; Bain, J.R.; Newgard, C.B.; Farese, R.V.; et al. Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab. 2007, 6, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, Y.; Chen, N.; Shi, X.; Tsang, B.; Yu, Y.-H. Upregulation of myocellular DGAT1 augments triglyceride synthesis in skeletal muscle and protects against fat-induced insulin resistance. J. Clin. Investig. 2007, 117, 1679–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chokshi, A.; Drosatos, K.; Cheema, F.H.; Ji, R.; Khawaja, T.; Yu, S.; Kato, T.; Khan, R.; Takayama, H.; Knöll, R.; et al. Ventricular assist device implantation corrects myocardial lipotoxicity, reverses insulin resistance, and normalizes cardiac metabolism in patients with advanced heart failure. Circulation 2012, 125, 2844–2853. [Google Scholar] [CrossRef] [PubMed]
- Kuo, A.; Lee, M.Y.; Sessa, W.C. Lipid Droplet Biogenesis and Function in the Endothelium. Circ. Res. 2017, 120, 1289–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henique, C.; Mansouri, A.; Fumey, G.; Lenoir, V.; Girard, J.; Bouillaud, F.; Prip-Buus, C.; Cohen, I. Increased mitochondrial fatty acid oxidation is sufficient to protect skeletal muscle cells from palmitate-induced apoptosis. J. Biol. Chem. 2010, 285, 36818–36827. [Google Scholar] [CrossRef] [PubMed]
- Coll, T.; Eyre, E.; Rodríguez-Calvo, R.; Palomer, X.; Sánchez, R.M.; Merlos, M.; Laguna, J.C.; Vázquez-Carrera, M. Oleate reverses palmitate-induced insulin resistance and inflammation in skeletal muscle cells. J. Biol. Chem. 2008, 283, 11107–11116. [Google Scholar] [CrossRef] [PubMed]
- Leamy, A.K.; Hasenour, C.M.; Egnatchik, R.A.; Trenary, I.A.; Yao, C.-H.; Patti, G.J.; Shiota, M.; Young, J.D. Knockdown of triglyceride synthesis does not enhance palmitate lipotoxicity or prevent oleate-mediated rescue in rat hepatocytes. Biochim. Biophys. Acta 2016, 1861, 1005–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robblee, M.M.; Kim, C.C.; Porter Abate, J.; Valdearcos, M.; Sandlund, K.L.M.; Shenoy, M.K.; Volmer, R.; Iwawaki, T.; Koliwad, S.K. Saturated Fatty Acids Engage an IRE1α-Dependent Pathway to Activate the NLRP3 Inflammasome in Myeloid Cells. Cell Rep. 2016, 14, 2611–2623. [Google Scholar] [CrossRef] [PubMed]
- Hardy, S.; Langelier, Y.; Prentki, M. Oleate activates phosphatidylinositol 3-kinase and promotes proliferation and reduces apoptosis of MDA-MB-231 breast cancer cells, whereas palmitate has opposite effects. Cancer Res. 2000, 60, 6353–6358. [Google Scholar] [PubMed]
- Hardy, S.; El-Assaad, W.; Przybytkowski, E.; Joly, E.; Prentki, M.; Langelier, Y. Saturated fatty acid-induced apoptosis in MDA-MB-231 breast cancer cells. A role for cardiolipin. J. Biol. Chem. 2003, 278, 31861–31870. [Google Scholar] [CrossRef] [PubMed]
- Guštin, E.; Jarc, E.; Kump, A.; Petan, T. Lipid Droplet Formation in HeLa Cervical Cancer Cells Depends on Cell Density and the Concentration of Exogenous Unsaturated Fatty Acids. Acta Chim. Slov. 2017, 64, 549–554. [Google Scholar] [CrossRef] [PubMed]
- D’Eliseo, D.; Velotti, F. Omega-3 Fatty Acids and Cancer Cell Cytotoxicity: Implications for Multi-Targeted Cancer Therapy. J. Clin. Med. 2016, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Chénais, B.; Blanckaert, V. The janus face of lipids in human breast cancer: How polyunsaturated Fatty acids affect tumor cell hallmarks. Int. J. Breast Cancer 2012, 2012, 712536. [Google Scholar] [CrossRef] [PubMed]
- Schley, P.D.; Jijon, H.B.; Robinson, L.E.; Field, C.J. Mechanisms of omega-3 fatty acid-induced growth inhibition in MDA-MB-231 human breast cancer cells. Breast Cancer Res. Treat. 2005, 92, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, Y.; Jia, C.; Wang, Y.; Lai, P.; Zhou, X.; Wang, Y.; Song, Q.; Lin, J.; Ren, Z.; et al. mTORC1/2 targeted by n-3 polyunsaturated fatty acids in the prevention of mammary tumorigenesis and tumor progression. Oncogene 2014, 33, 4548–4557. [Google Scholar] [CrossRef] [PubMed]
- Mouradian, M.; Kikawa, K.D.; Dranka, B.P.; Komas, S.M.; Kalyanaraman, B.; Pardini, R.S. Docosahexaenoic acid attenuates breast cancer cell metabolism and the Warburg phenotype by targeting bioenergetic function. Mol. Carcinog. 2015, 54, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Serini, S.; Fasano, E.; Piccioni, E.; Cittadini, A.R.M.; Calviello, G. Dietary n-3 polyunsaturated fatty acids and the paradox of their health benefits and potential harmful effects. Chem. Res. Toxicol. 2011, 24, 2093–2105. [Google Scholar] [CrossRef] [PubMed]
- Quehenberger, O.; Armando, A.M.; Brown, A.H.; Milne, S.B.; Myers, D.S.; Merrill, A.H.; Bandyopadhyay, S.; Jones, K.N.; Kelly, S.; Shaner, R.L.; et al. Lipidomics reveals a remarkable diversity of lipids in human plasma. J. Lipid Res. 2010, 51, 3299–3305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, S.; Li, J.; Lee, S.-Y.; Lee, H.J.; Shao, T.; Song, B.; Cheng, L.; Masterson, T.A.; Liu, X.; Ratliff, T.L.; et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 2014, 19, 393–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Gu, D.; Lee, S.S.Y.; Song, B.; Bandyopadhyay, S.; Chen, S.; Konieczny, S.F.; Ratliff, T.L.; Liu, X.; Xie, J.; et al. Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer. Oncogene 2016, 35, 6378–6388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, F.; Cheng, X.; Wu, X.; Yoo, J.Y.; Cheng, C.; Guo, J.Y.; Mo, X.; Ru, P.; Hurwitz, B.; Kim, S.H.; et al. Inhibition of SOAT1 suppresses glioblastoma growth via blocking SREBP-1-mediated lipogenesis. Clin. Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Antalis, C.J.; Uchida, A.; Buhman, K.K.; Siddiqui, R.A. Migration of MDA-MB-231 breast cancer cells depends on the availability of exogenous lipids and cholesterol esterification. Clin. Exp. Metastasis 2011, 28, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Antalis, C.J.; Arnold, T.; Rasool, T.; Lee, B.; Buhman, K.K.; Siddiqui, R.A. High ACAT1 expression in estrogen receptor negative basal-like breast cancer cells is associated with LDL-induced proliferation. Breast Cancer Res. Treat. 2010, 122, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, T.; Parton, R.G. Not just fat: The structure and function of the lipid droplet. Cold Spring Harb. Perspect. Biol. 2011, 3, a004838. [Google Scholar] [CrossRef] [PubMed]
- Mottillo, E.P.; Bloch, A.E.; Leff, T.; Granneman, J.G. Lipolytic products activate peroxisome proliferator-activated receptor (PPAR) α and δ in brown adipocytes to match fatty acid oxidation with supply. J. Biol. Chem. 2012, 287, 25038–25048. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, M.; Lass, A.; Zimmermann, R.; Eichmann, T.O.; Zechner, R. Neutral lipid storage disease: Genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI-58/ABHD5. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E289–E296. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, G.; Lass, A.; Zimmermann, R.; Gorkiewicz, G.; Meyer, C.; Rozman, J.; Heldmaier, G.; Maier, R.; Theussl, C.; Eder, S.; et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 2006, 312, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Kienesberger, P.C.; Lee, D.; Pulinilkunnil, T.; Brenner, D.S.; Cai, L.; Magnes, C.; Koefeler, H.C.; Streith, I.E.; Rechberger, G.N.; Haemmerle, G.; et al. Adipose triglyceride lipase deficiency causes tissue-specific changes in insulin signaling. J. Biol. Chem. 2009, 284, 30218–30229. [Google Scholar] [CrossRef] [PubMed]
- Hoy, A.J.; Bruce, C.R.; Turpin, S.M.; Morris, A.J.; Febbraio, M.A.; Watt, M.J. Adipose triglyceride lipase-null mice are resistant to high-fat diet-induced insulin resistance despite reduced energy expenditure and ectopic lipid accumulation. Endocrinology 2011, 152, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.; Hofer, P.; Taschler, U.; Voshol, P.J.; Rechberger, G.N.; Kotzbeck, P.; Jaeger, D.; Preiss-Landl, K.; Lord, C.C.; Brown, J.M.; et al. Hypophagia and metabolic adaptations in mice with defective ATGL-mediated lipolysis cause resistance to HFD-induced obesity. Proc. Natl. Acad. Sci. USA 2015, 112, 13850–13855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweiger, M.; Romauch, M.; Schreiber, R.; Grabner, G.F.; Hütter, S.; Kotzbeck, P.; Benedikt, P.; Eichmann, T.O.; Yamada, S.; Knittelfelder, O.; et al. Pharmacological inhibition of adipose triglyceride lipase corrects high-fat diet-induced insulin resistance and hepatosteatosis in mice. Nat. Commun. 2017, 8, 14859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Zhao, Y.; Gao, X.; Li, L.; Yuan, Y.; Liu, F.; Zhang, L.; Wu, J.; Hu, P.; Zhang, X.; et al. Perilipin 5 improves hepatic lipotoxicity by inhibiting lipolysis. Hepatology 2015, 61, 870–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, P.; Xie, Z.; Yuan, Y.; Sui, W.; Wang, C.; Gao, X.; Zhao, Y.; Zhang, F.; Gu, Y.; Hu, P.; et al. Plin5 alleviates myocardial ischaemia/reperfusion injury by reducing oxidative stress through inhibiting the lipolysis of lipid droplets. Sci. Rep. 2017, 7, 42574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, R.R.; Mokhtar, R.; Matzaris, M.; Selathurai, A.; Kowalski, G.M.; Mokbel, N.; Meikle, P.J.; Bruce, C.R.; Watt, M.J. PLIN5 deletion remodels intracellular lipid composition and causes insulin resistance in muscle. Mol. Metab. 2014, 3, 652–663. [Google Scholar] [CrossRef] [PubMed]
- Pollak, N.M.; Schweiger, M.; Jaeger, D.; Kolb, D.; Kumari, M.; Schreiber, R.; Kolleritsch, S.; Markolin, P.; Grabner, G.F.; Heier, C.; et al. Cardiac-specific overexpression of perilipin 5 provokes severe cardiac steatosis via the formation of a lipolytic barrier. J. Lipid Res. 2013, 54, 1092–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Bell, M.; Sreenivasan, U.; Sreenevasan, U.; Hu, H.; Liu, J.; Dalen, K.; Londos, C.; Yamaguchi, T.; Rizzo, M.A.; et al. Unique regulation of adipose triglyceride lipase (ATGL) by perilipin 5, a lipid droplet-associated protein. J. Biol. Chem. 2011, 286, 15707–15715. [Google Scholar] [CrossRef] [PubMed]
- Laurens, C.; Bourlier, V.; Mairal, A.; Louche, K.; Badin, P.-M.; Mouisel, E.; Montagner, A.; Marette, A.; Tremblay, A.; Weisnagel, J.S.; et al. Perilipin 5 fine-tunes lipid oxidation to metabolic demand and protects against lipotoxicity in skeletal muscle. Sci. Rep. 2016, 6, 38310. [Google Scholar] [CrossRef] [PubMed]
- Gallardo-Montejano, V.I.; Saxena, G.; Kusminski, C.M.; Yang, C.; McAfee, J.L.; Hahner, L.; Hoch, K.; Dubinsky, W.; Narkar, V.A.; Bickel, P.E. Nuclear Perilipin 5 integrates lipid droplet lipolysis with PGC-1α/SIRT1-dependent transcriptional regulation of mitochondrial function. Nat. Commun. 2016, 7, 12723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aflaki, E.; Radovic, B.; Chandak, P.G.; Kolb, D.; Eisenberg, T.; Ring, J.; Fertschai, I.; Uellen, A.; Wolinski, H.; Kohlwein, S.-D.; et al. Triacylglycerol accumulation activates the mitochondrial apoptosis pathway in macrophages. J. Biol. Chem. 2011, 286, 7418–7428. [Google Scholar] [CrossRef] [PubMed]
- Nomura, D.K.; Long, J.Z.; Niessen, S.; Hoover, H.S.; Ng, S.-W.; Cravatt, B.F. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 2010, 140, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Nomura, D.K.; Lombardi, D.P.; Chang, J.W.; Niessen, S.; Ward, A.M.; Long, J.Z.; Hoover, H.H.; Cravatt, B.F. Monoacylglycerol lipase exerts dual control over endocannabinoid and fatty acid pathways to support prostate cancer. Chem. Biol. 2011, 18, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Vegliante, R.; Di Leo, L.; Ciccarone, F.; Ciriolo, M.R. Hints on ATGL implications in cancer: Beyond bioenergetic clues. Cell Death Dis. 2018, 9, 316. [Google Scholar] [CrossRef] [PubMed]
- Zagani, R.; El-Assaad, W.; Gamache, I.; Teodoro, J.G. Inhibition of adipose triglyceride lipase (ATGL) by the putative tumor suppressor G0S2 or a small molecule inhibitor attenuates the growth of cancer cells. Oncotarget 2015, 6, 28282–28295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, J.; Miao, H.; Ma, Y.; Guo, F.; Deng, J.; Wei, X.; Zhou, J.; Xie, G.; Shi, H.; Xue, B.; et al. Loss of abhd5 promotes colorectal tumor development and progression by inducing aerobic glycolysis and epithelial-mesenchymal transition. Cell Rep. 2014, 9, 1798–1811. [Google Scholar] [CrossRef] [PubMed]
- Grace, S.A.; Meeks, M.W.; Chen, Y.; Cornwell, M.; Ding, X.; Hou, P.; Rutgers, J.K.; Crawford, S.E.; Lai, J.-P. Adipose Triglyceride Lipase (ATGL) Expression Is Associated with Adiposity and Tumor Stromal Proliferation in Patients with Pancreatic Ductal Adenocarcinoma. Anticancer Res. 2017, 37, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Al-Zoughbi, W.; Pichler, M.; Gorkiewicz, G.; Guertl-Lackner, B.; Haybaeck, J.; Jahn, S.W.; Lackner, C.; Liegl-Atzwanger, B.; Popper, H.; Schauer, S.; et al. Loss of adipose triglyceride lipase is associated with human cancer and induces mouse pulmonary neoplasia. Oncotarget 2016, 7, 33832–33840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Fu, A.Q.; McNerney, M.E.; White, K.P. Widespread genetic epistasis among cancer genes. Nat. Commun. 2014, 5, 4828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, L.; Hickey, R.W.; Bayir, H.; Watkins, S.C.; Tyurin, V.A.; Guo, F.; Kochanek, P.M.; Jenkins, L.W.; Ren, J.; Gibson, G.; et al. Starving neurons show sex difference in autophagy. J. Biol. Chem. 2009, 284, 2383–2396. [Google Scholar] [CrossRef] [PubMed]
- Ramanadham, S.; Ali, T.; Ashley, J.W.; Bone, R.N.; Hancock, W.D.; Lei, X. Calcium-independent phospholipases A2 and their roles in biological processes and diseases. J. Lipid Res. 2015, 56, 1643–1668. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Taketomi, Y.; Miki, Y.; Sato, H.; Hirabayashi, T.; Yamamoto, K. Recent progress in phospholipase A2 research: From cells to animals to humans. Prog. Lipid Res. 2011, 50, 152–192. [Google Scholar] [CrossRef] [PubMed]
- Gubern, A.; Casas, J.; Barceló-Torns, M.; Barneda, D.; de la Rosa, X.; Masgrau, R.; Picatoste, F.; Balsinde, J.; Balboa, M.A.; Claro, E. Group IVA phospholipase A2 is necessary for the biogenesis of lipid droplets. J. Biol. Chem. 2008, 283, 27369–27382. [Google Scholar] [CrossRef] [PubMed]
- Akiba, S.; Yoneda, Y.; Ohno, S.; Nemoto, M.; Sato, T. Oxidized LDL activates phospholipase A2 to supply fatty acids required for cholesterol esterification. J. Lipid Res. 2003, 44, 1676–1685. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.B.; Chandel, N.S. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014, 2, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, J.J.; Chung, M.K.; Fan, J.; Rabinowitz, J.D. Quantitative analysis of acetyl-CoA production in hypoxic cancer cells reveals substantial contribution from acetate. Cancer Metab. 2014, 2, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurenti, G.; Benedetti, E.; D’Angelo, B.; Cristiano, L.; Cinque, B.; Raysi, S.; Alecci, M.; Cerù, M.P.; Cifone, M.G.; Galzio, R.; et al. Hypoxia induces peroxisome proliferator-activated receptor α (PPARα) and lipid metabolism peroxisomal enzymes in human glioblastoma cells. J. Cell. Biochem. 2011, 112, 3891–3901. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Saarinen, A.M.; Hitosugi, T.; Wang, Z.; Wang, L.; Ho, T.H.; Liu, J. Inhibition of intracellular lipolysis promotes human cancer cell adaptation to hypoxia. eLife 2017, 6, e31132. [Google Scholar] [CrossRef] [PubMed]
- Pouysségur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Höckel, M.; Mayer, A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid. Redox Signal. 2007, 9, 1221–1235. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Rofstad, E.K.; Gaustad, J.-V.; Egeland, T.A.M.; Mathiesen, B.; Galappathi, K. Tumors exposed to acute cyclic hypoxic stress show enhanced angiogenesis, perfusion and metastatic dissemination. Int. J. Cancer 2010, 127, 1535–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazure, N.M.; Pouysségur, J. Hypoxia-induced autophagy: Cell death or cell survival? Curr. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Zoula, S.; Rijken, P.F.J.W.; Peters, J.P.W.; Farion, R.; Van der Sanden, B.P.J.; Van der Kogel, A.J.; Décorps, M.; Rémy, C. Pimonidazole binding in C6 rat brain glioma: Relation with lipid droplet detection. Br. J. Cancer 2003, 88, 1439–1444. [Google Scholar] [CrossRef] [PubMed]
- Rémy, C.; Fouilhé, N.; Barba, I.; Sam-Laï, E.; Lahrech, H.; Cucurella, M.G.; Izquierdo, M.; Moreno, A.; Ziegler, A.; Massarelli, R.; et al. Evidence that mobile lipids detected in rat brain glioma by 1H nuclear magnetic resonance correspond to lipid droplets. Cancer Res. 1997, 57, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Gimm, T.; Wiese, M.; Teschemacher, B.; Deggerich, A.; Schödel, J.; Knaup, K.X.; Hackenbeck, T.; Hellerbrand, C.; Amann, K.; Wiesener, M.S.; et al. Hypoxia-inducible protein 2 is a novel lipid droplet protein and a specific target gene of hypoxia-inducible factor-1. FASEB J. 2010, 24, 4443–4458. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Wang, C.; Liu, D.; Zhang, X.; Wang, L.; Yan, M.; Zhang, W.; Zhu, J.; Li, Z.-C.; Mi, C.; et al. Hypoxia upregulates HIG2 expression and contributes to bevacizumab resistance in glioblastoma. Oncotarget 2016, 7, 47808–47820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padmanabha Das, K.M.; Wechselberger, L.; Liziczai, M.; De la Rosa Rodriguez, M.; Grabner, G.F.; Heier, C.; Viertlmayr, R.; Radler, C.; Lichtenegger, J.; Zimmermann, R.; et al. Hypoxia-inducible lipid droplet-associated protein inhibits adipose triglyceride lipase. J. Lipid Res. 2018, 59, 531–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Zhou, G.; Aras, S.; He, Z.; Lucas, S.; Podgorski, I.; Skar, W.; Granneman, J.G.; Wang, J. Loss of ABHD5 promotes the aggressiveness of prostate cancer cells. Sci. Rep. 2017, 7, 13021. [Google Scholar] [CrossRef] [PubMed]
- Hashani, M.; Witzel, H.R.; Pawella, L.M.; Lehmann-Koch, J.; Schumacher, J.; Mechtersheimer, G.; Schnölzer, M.; Schirmacher, P.; Roth, W.; Straub, B.K. Widespread expression of perilipin 5 in normal human tissues and in diseases is restricted to distinct lipid droplet subpopulations. Cell Tissue Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Barrera, G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol. 2012, 2012, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, K.; Sandoval, H.; Yamamoto, S.; Jaiswal, M.; Sanz, E.; Li, Z.; Hui, J.; Graham, B.H.; Quintana, A.; et al. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 2015, 160, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; MacKenzie, K.R.; Putluri, N.; Maletić-Savatić, M.; Bellen, H.J. The Glia-Neuron Lactate Shuttle and Elevated ROS Promote Lipid Synthesis in Neurons and Lipid Droplet Accumulation in Glia via APOE/D. Cell Metab. 2017, 26. [Google Scholar] [CrossRef] [PubMed]
- Tuohetahuntila, M.; Spee, B.; Kruitwagen, H.S.; Wubbolts, R.; Brouwers, J.F.; van de Lest, C.H.; Molenaar, M.R.; Houweling, M.; Helms, J.B.; Vaandrager, A.B. Role of long-chain acyl-CoA synthetase 4 in formation of polyunsaturated lipid species in hepatic stellate cells. Biochim. Biophys. Acta 2015, 1851, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Tuohetahuntila, M.; Molenaar, M.R.; Spee, B.; Brouwers, J.F.; Houweling, M.; Vaandrager, A.B.; Helms, J.B. ATGL and DGAT1 are involved in the turnover of newly synthesized triacylglycerols in hepatic stellate cells. J. Lipid Res. 2016, 57, 1162–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Chitraju, C.; Trötzmüller, M.; Hartler, J.; Wolinski, H.; Thallinger, G.G.; Lass, A.; Zechner, R.; Zimmermann, R.; Köfeler, H.C.; Spener, F. Lipidomic analysis of lipid droplets from murine hepatocytes reveals distinct signatures for nutritional stress. J. Lipid Res. 2012. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Wilson, M.; McConville, C.; Arvanitis, T.N.; Griffin, J.L.; Kauppinen, R.A.; Peet, A.C. Increased unsaturation of lipids in cytoplasmic lipid droplets in DAOY cancer cells in response to cisplatin treatment. Metabolomics 2013, 9, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Liesenfeld, D.B.; Grapov, D.; Fahrmann, J.F.; Salou, M.; Scherer, D.; Toth, R.; Habermann, N.; Böhm, J.; Schrotz-King, P.; Gigic, B.; et al. Metabolomics and transcriptomics identify pathway differences between visceral and subcutaneous adipose tissue in colorectal cancer patients: The ColoCare study. Am. J. Clin. Nutr. 2015, 102, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Al-Saffar, N.M.S.; Titley, J.C.; Robertson, D.; Clarke, P.A.; Jackson, L.E.; Leach, M.O.; Ronen, S.M. Apoptosis is associated with triacylglycerol accumulation in Jurkat T-cells. Br. J. Cancer 2002, 86, 963–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Levine, B.; Kroemer, G. Metabolic control of autophagy. Cell 2014, 159, 1263–1276. [Google Scholar] [CrossRef] [PubMed]
- Sica, V.; Galluzzi, L.; Bravo-San Pedro, J.M.; Izzo, V.; Maiuri, M.C.; Kroemer, G. Organelle-specific initiation of autophagy. Mol. Cell 2015, 59, 522–539. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef] [PubMed]
- Niso-Santano, M.; Malik, S.A.; Pietrocola, F.; Bravo-San Pedro, J.M.; Mariño, G.; Cianfanelli, V.; Ben-Younès, A.; Troncoso, R.; Markaki, M.; Sica, V.; et al. Unsaturated fatty acids induce non-canonical autophagy. EMBO J. 2015, 34, 1025–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Vevea, J.D.; Garcia, E.J.; Chan, R.B.; Zhou, B.; Schultz, M.; Di Paolo, G.; McCaffery, J.M.; Pon, L.A. Role for Lipid Droplet Biogenesis and Microlipophagy in Adaptation to Lipid Imbalance in Yeast. Dev. Cell 2015, 35. [Google Scholar] [CrossRef] [PubMed]
- Chu, K.Y.; O’Reilly, L.; Ramm, G.; Biden, T.J. High-fat diet increases autophagic flux in pancreatic beta cells in vivo and ex vivo in mice. Diabetologia 2015, 58, 2074–2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, C.; Martinez-Lopez, N.; Otten, E.G.; Carroll, B.; Maetzel, D.; Singh, R.; Sarkar, S.; Korolchuk, V.I. Autophagy, lipophagy and lysosomal lipid storage disorders. Biochim. Biophys. Acta 2016, 1861, 269–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efeyan, A.; Comb, W.C.; Sabatini, D.M. Nutrient-sensing mechanisms and pathways. Nature 2015, 517, 302–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, M.; Yoshimura, K.; Furuya, N.; Koike, M.; Ueno, T.; Komatsu, M.; Arai, H.; Tanaka, K.; Kominami, E.; Uchiyama, Y. The MAP1-LC3 conjugation system is involved in lipid droplet formation. Biochem. Biophys. Res. Commun. 2009, 382, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chao, X.; Yang, L.; Lu, Q.; Li, T.; Ding, W.-X.; Ni, H.-M. Impaired fasting-induced adaptive lipid droplet biogenesis in liver-specific Atg5-deficient mouse liver is mediated by persistent nuclear factor-like 2 activation. Am. J. Pathol. 2018, 188, 1833–1846. [Google Scholar] [CrossRef] [PubMed]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Czaja, M.J. Regulation of lipid droplets by autophagy. Trends Endocrinol. Metab. 2011, 22, 234–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Singh, R.; Xiang, Y.; Czaja, M.J. Macroautophagy and chaperone-mediated autophagy are required for hepatocyte resistance to oxidant stress. Hepatology 2010, 52, 266–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexaki, A.; Gupta, S.D.; Majumder, S.; Kono, M.; Tuymetova, G.; Harmon, J.M.; Dunn, T.M.; Proia, R.L. Autophagy regulates sphingolipid levels in the liver. J. Lipid Res. 2014, 55, 2521–2531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Y.; Cheng, J.; Yu, F.; Li, H.; Guo, C.; Fan, X. Ghrelin attenuated lipotoxicity via autophagy induction and nuclear factor-κB inhibition. Cell. Physiol. Biochem. 2015, 37, 563–576. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Li, M.; Chen, X.; Ni, H.; Lin, C.; Gao, W.; Lu, B.; Stolz, D.B.; Clemens, D.L.; Yin, X. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology 2010, 139, 1740–1752. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-N.; Kwon, H.-J.; Akindehin, S.; Jeong, H.W.; Lee, Y.-H. Effects of epigallocatechin-3-gallate on autophagic lipolysis in adipocytes. Nutrients 2017, 9, 680. [Google Scholar] [CrossRef] [PubMed]
- Ouimet, M.; Franklin, V.; Mak, E.; Liao, X.; Tabas, I.; Marcel, Y.L. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011, 13, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Ouimet, M.; Marcel, Y.L. Regulation of lipid droplet cholesterol efflux from macrophage foam cells. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Khaldoun, S.A.; Emond-Boisjoly, M.-A.; Chateau, D.; Carrière, V.; Lacasa, M.; Rousset, M.; Demignot, S.; Morel, E. Autophagosomes contribute to intracellular lipid distribution in enterocytes. Mol. Biol. Cell 2014, 25. [Google Scholar] [CrossRef] [PubMed]
- Minami, S.; Yamamoto, T.; Takabatake, Y.; Takahashi, A.; Namba, T.; Matsuda, J.; Kimura, T.; Kaimori, J.-Y.; Matsui, I.; Hamano, T.; et al. Lipophagy maintains energy homeostasis in the kidney proximal tubule during prolonged starvation. Autophagy 2017, 13, 1629–1647. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Rodriguez-Navarro, J.A.; Arias, E.; Kiffin, R.; Sahu, S.; Schwartz, G.J.; Cuervo, A.M.; Singh, R. Autophagy in hypothalamic AgRP neurons regulates food intake and energy balance. Cell Metab. 2011, 14, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lopez, N.; Garcia-Macia, M.; Sahu, S.; Athonvarangkul, D.; Liebling, E.; Merlo, P.; Cecconi, F.; Schwartz, G.J.; Singh, R. Autophagy in the CNS and periphery coordinate lipophagy and lipolysis in the brown adipose tissue and liver. Cell Metab. 2016, 23, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, T.; Takayama, K.; Ishii, S.; Yamamoto, A.; Hara, T.; Minami, N.; Miyasaka, N.; Kubota, T.; Matsuura, A.; Itakura, E.; et al. Forced lipophagy reveals that lipid droplets are required for early embryonic development in mouse. Development 2018, 145, dev161893. [Google Scholar] [CrossRef] [PubMed]
- Seo, A.Y.; Lau, P.W.; Feliciano, D.; Sengupta, P.; Le Gros, M.A.; Cinquin, B.; Larabell, C.A.; Lippincott-Schwartz, J. AMPK and vacuole-associated Atg14p orchestrate μ-lipophagy for energy production and long-term survival under glucose starvation. eLife 2017, 6, e21690. [Google Scholar] [CrossRef] [PubMed]
- Sahu, R.; Kaushik, S.; Clement, C.C.; Cannizzo, E.S.; Scharf, B.; Follenzi, A.; Potolicchio, I.; Nieves, E.; Cuervo, A.M.; Santambrogio, L. Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 2011, 20, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Kaini, R.R.; Sillerud, L.O.; Zhaorigetu, S.; Hu, C.-A.A. Autophagy regulates lipolysis and cell survival through lipid droplet degradation in androgen-sensitive prostate cancer cells. Prostate 2012, 72, 1412–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assumpção, J.A.F.; Magalhães, K.G.; Corrêa, J.R. The role of pparγ and autophagy in ros production, lipid droplets biogenesis and its involvement with colorectal cancer cells modulation. Cancer Cell Int. 2017, 17, 82. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Jiang, Y.; Xiao, Y.; Liu, X.D.; Yue, F.; Li, W.; Li, X.; He, Y.; Jiang, X.; Huang, H.; et al. Fast clearance of lipid droplets through MAP1S-activated autophagy suppresses clear cell renal cell carcinomas and promotes patient survival. Oncotarget 2016, 7, 6255–6265. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Schlaepfer, I.R.; Bergman, B.C.; Panda, P.K.; Praharaj, P.P.; Naik, P.P.; Agarwal, R.; Bhutia, S.K. ATG14 facilitated lipophagy in cancer cells induce ER stress mediated mitoptosis through a ROS dependent pathway. Free Radic. Biol. Med. 2017, 104, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Zhao, T.; Ding, X.; Yan, C. Hepatocyte-specific expression of human lysosome acid lipase corrects liver inflammation and tumor metastasis in lal−/− mice. Am. J. Pathol. 2015, 185, 2379–2389. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Ding, X.; Du, H.; Yan, C. Lung epithelial cell–specific expression of human lysosomal acid lipase ameliorates lung inflammation and tumor metastasis in lipa−/− mice. Am. J. Pathol. 2016, 186, 2183–2192. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.-A.; Xing, X.; Harris, J.W.; Zaytseva, Y.Y.; Mitov, M.I.; Napier, D.L.; Weiss, H.L.; Mark Evers, B.; Gao, T. Adipocytes activate mitochondrial fatty acid oxidation and autophagy to promote tumor growth in colon cancer. Cell Death Dis. 2017, 8, e2593. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, I.; Rotter, V. Regulation of lipid metabolism by p53-fighting two villains with one sword. Trends Endocrinol. Metab. 2012, 23, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Kenzelmann Broz, D.; Spano Mello, S.; Bieging, K.T.; Jiang, D.; Dusek, R.L.; Brady, C.A.; Sidow, A.; Attardi, L.D. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013, 27, 1016–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; Ballabio, A. Lysosome: Regulator of lipid degradation pathways. Trends Cell Biol. 2014, 24, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat. Cell Biol. 2015, 17, 759–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, J.L.; Suh, Y.; Cuervo, A.M. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab. 2014, 20, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. AMPK-dependent phosphorylation of lipid droplet protein PLIN2 triggers its degradation by CMA. Autophagy 2016, 12, 432–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Q.; Deng, Y.; Chen, S.; Chen, R.; Yang, M.; Zhang, Z.; Sun, X.; Wang, W.; He, Y.; Wang, F.; et al. Downregulation of ATG5-dependent macroautophagy by chaperone-mediated autophagy promotes breast cancer cell metastasis. Sci. Rep. 2017, 7, 4759. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V. Autophagosomes and lipid droplets: No longer just chewing the fat. EMBO J. 2015, 34, 2111–2113. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Miao, H.; Wu, S.; Yang, W.; Zhang, Y.; Xie, G.; Xie, X.; Li, J.; Shi, C.; Ye, L.; et al. ABHD5 interacts with BECN1 to regulate autophagy and tumorigenesis of colon cancer independent of PNPLA2. Autophagy 2016, 12, 2167–2182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.Y.; Jang, H.J.; Yang, Y.R.; Park, K.I.; Seo, J.K.; Shin, I.W.; Jeon, T.I.; Ahn, S.C.; Suh, P.G.; Osborne, T.F.; et al. SREBP-2/PNPLA8 axis improves non-alcoholic fatty liver disease through activation of autophagy. Sci. Rep. 2016, 6, 35732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancuso, D.J.; Jenkins, C.M.; Gross, R.W. The genomic organization, complete mRNA sequence, cloning, and expression of a novel human intracellular membrane-associated calcium-independent phospholipase A2. J. Biol. Chem. 2000, 275, 9937–9945. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, D.J.; Han, X.; Jenkins, C.M.; Lehman, J.J.; Sambandam, N.; Sims, H.F.; Yang, J.; Yan, W.; Yang, K.; Green, K.; et al. Dramatic accumulation of triglycerides and precipitation of cardiac hemodynamic dysfunction during brief caloric restriction in transgenic myocardium expressing human calcium-independent phospholipase A2γ. J. Biol. Chem. 2007, 282, 9216–9227. [Google Scholar] [CrossRef] [PubMed]
- Baerga, R.; Zhang, Y.; Chen, P.-H.; Goldman, S.; Jin, S. Targeted deletion of autophagy-related 5 (atg5) impairs adipogenesis in a cellular model and in mice. Autophagy 2009, 5, 1118–1130. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Yoshimura, K.; Tamura, H.; Ueno, T.; Nishimura, T.; Inoue, T.; Sasaki, M.; Koike, M.; Arai, H.; Kominami, E.; et al. LC3, a microtubule-associated protein1A/B light chain3, is involved in cytoplasmic lipid droplet formation. Biochem. Biophys. Res. Commun. 2010, 393, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yamamoto, M. Stress-sensing mechanisms and the physiological roles of the Keap1-Nrf2 system during cellular stress. J. Biol. Chem. 2017, 292, 16817–16824. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-S.; Mendez, R.; Heng, H.H.; Yang, Z.-Q.; Zhang, K. Pharmacological ER stress promotes hepatic lipogenesis and lipid droplet formation. Am. J. Transl. Res. 2012, 4, 102–113. [Google Scholar] [PubMed]
- Xu, G.; Sztalryd, C.; Lu, X.; Tansey, J.T.; Gan, J.; Dorward, H.; Kimmel, A.R.; Londos, C. Post-translational regulation of adipose differentiation-related protein by the ubiquitin/proteasome pathway. J. Biol. Chem. 2005, 280, 42841–42847. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Sztalryd, C.; Londos, C. Degradation of perilipin is mediated through ubiquitination-proteasome pathway. Biochim. Biophys. Acta 2006, 1761, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Cosin-Roger, J.; Simmen, S.; Melhem, H.; Atrott, K.; Frey-Wagner, I.; Hausmann, M.; de Vallière, C.; Spalinger, M.R.; Spielmann, P.; Wenger, R.H.; et al. Hypoxia ameliorates intestinal inflammation through NLRP3/mTOR downregulation and autophagy activation. Nat. Commun. 2017, 8, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardi, R.; Guernah, I.; Jin, D.; Grisendi, S.; Alimonti, A.; Teruya-Feldstein, J.; Cordon-Cardo, C.; Simon, M.C.; Rafii, S.; Pandolfi, P.P. PML inhibits HIF-1alpha translation and neoangiogenesis through repression of mTOR. Nature 2006, 442, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Casals, N.; Zammit, V.; Herrero, L.; Fadó, R.; Rodríguez-Rodríguez, R.; Serra, D. Carnitine palmitoyltransferase 1C: From cognition to cancer. Prog. Lipid Res. 2016, 61, 134–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaugg, K.; Yao, Y.; Reilly, P.T.; Kannan, K.; Kiarash, R.; Mason, J.; Huang, P.; Sawyer, S.K.; Fuerth, B.; Faubert, B.; et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011, 25, 1041–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madeira, J.B.; Masuda, C.A.; Maya-Monteiro, C.M.; Matos, G.S.; Montero-Lomelí, M.; Bozaquel-Morais, B.L. TORC1 inhibition induces lipid droplet replenishment in yeast. Mol. Cell. Biol. 2015, 35, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Couso, I.; Pérez-Pérez, M.E.; Martínez-Force, E.; Kim, H.-S.; He, Y.; Umen, J.G.; Crespo, J.L. Autophagic flux is required for the synthesis of triacylglycerols and ribosomal protein turnover in Chlamydomonas. J. Exp. Bot. 2018, 69, 1355–1367. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petan, T.; Jarc, E.; Jusović, M. Lipid Droplets in Cancer: Guardians of Fat in a Stressful World. Molecules 2018, 23, 1941. https://doi.org/10.3390/molecules23081941
Petan T, Jarc E, Jusović M. Lipid Droplets in Cancer: Guardians of Fat in a Stressful World. Molecules. 2018; 23(8):1941. https://doi.org/10.3390/molecules23081941
Chicago/Turabian StylePetan, Toni, Eva Jarc, and Maida Jusović. 2018. "Lipid Droplets in Cancer: Guardians of Fat in a Stressful World" Molecules 23, no. 8: 1941. https://doi.org/10.3390/molecules23081941
APA StylePetan, T., Jarc, E., & Jusović, M. (2018). Lipid Droplets in Cancer: Guardians of Fat in a Stressful World. Molecules, 23(8), 1941. https://doi.org/10.3390/molecules23081941