pH Stability and Antioxidant Power of CycloDOPA and Its Derivatives

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
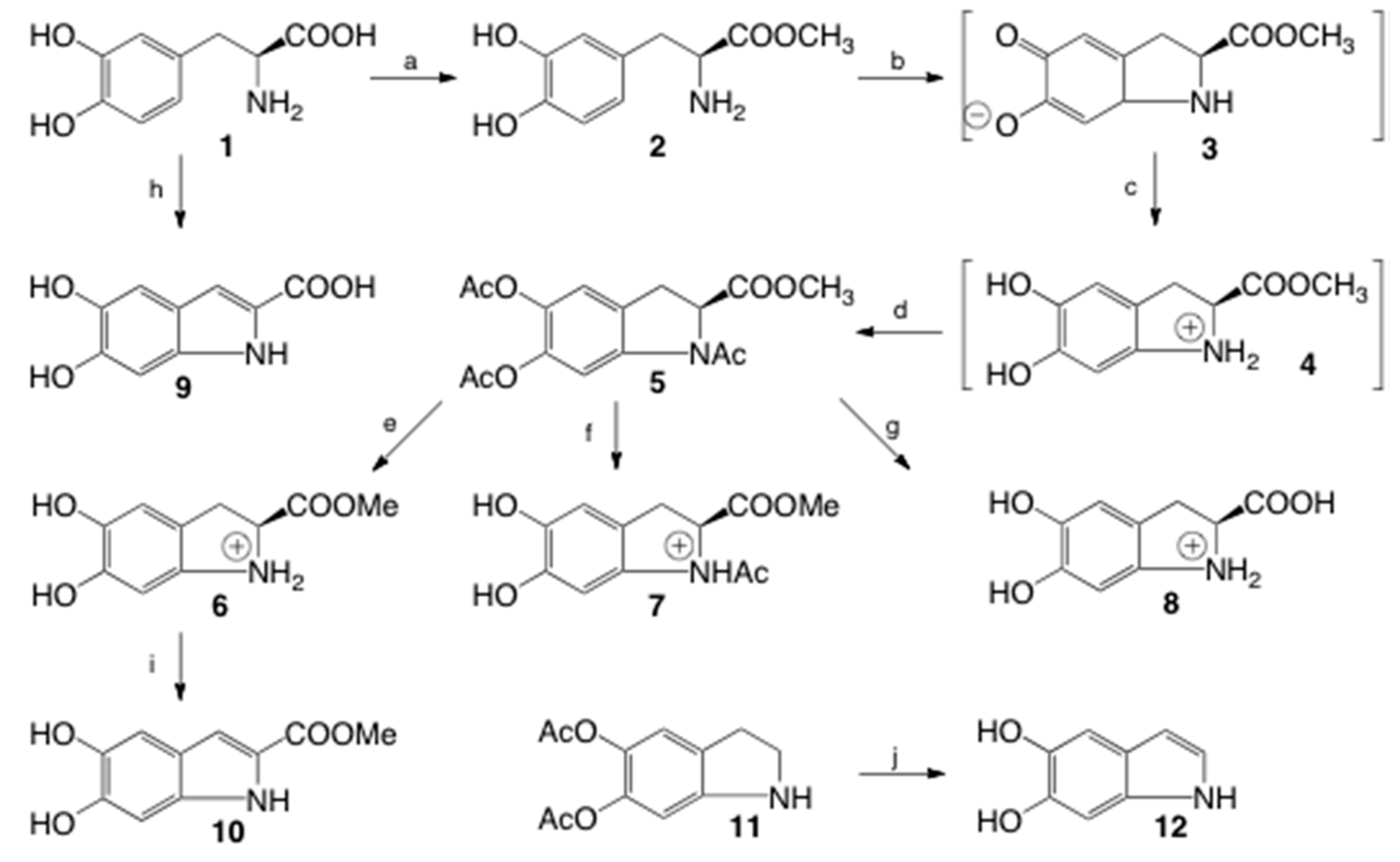
2.1. Synthesis
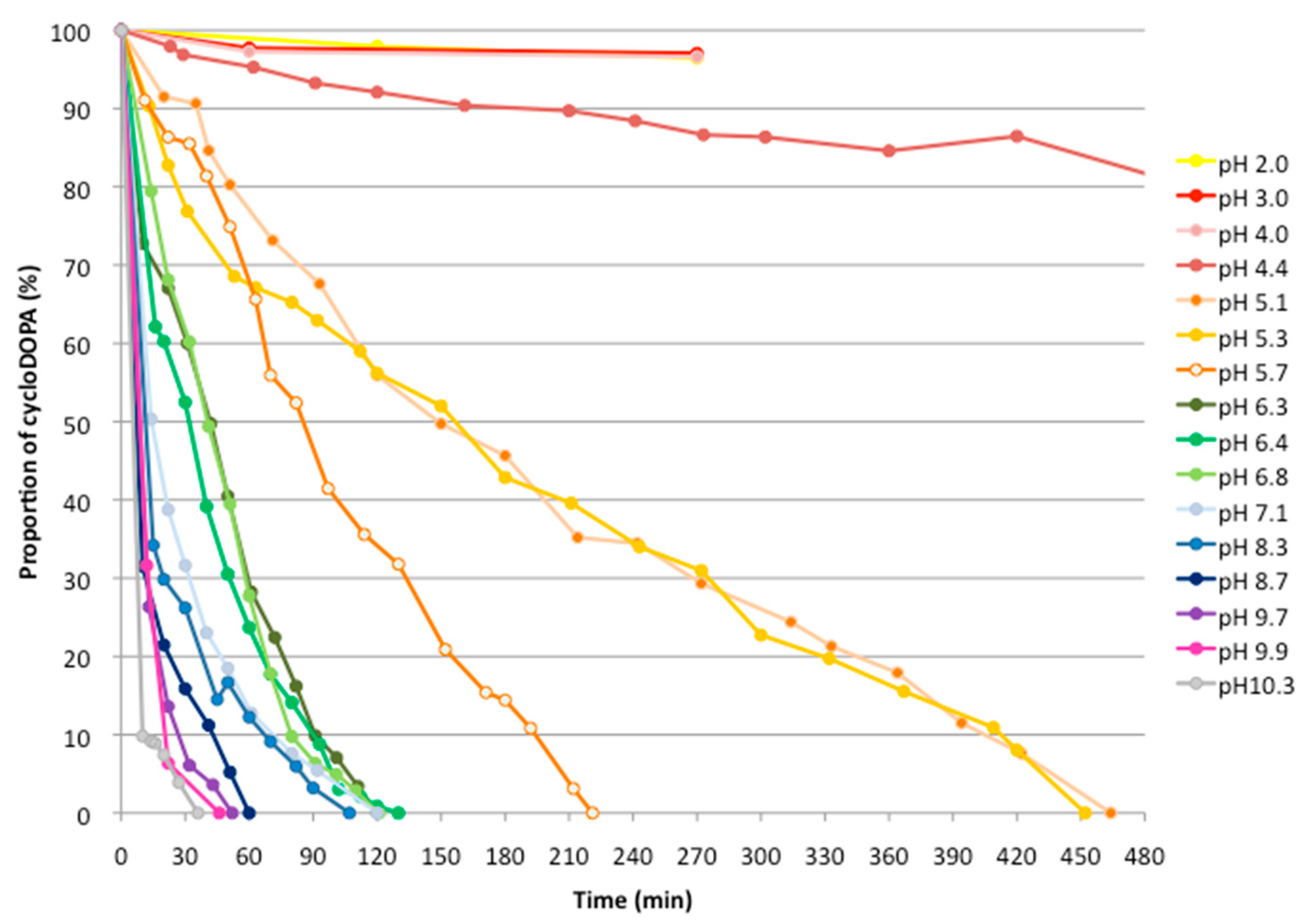
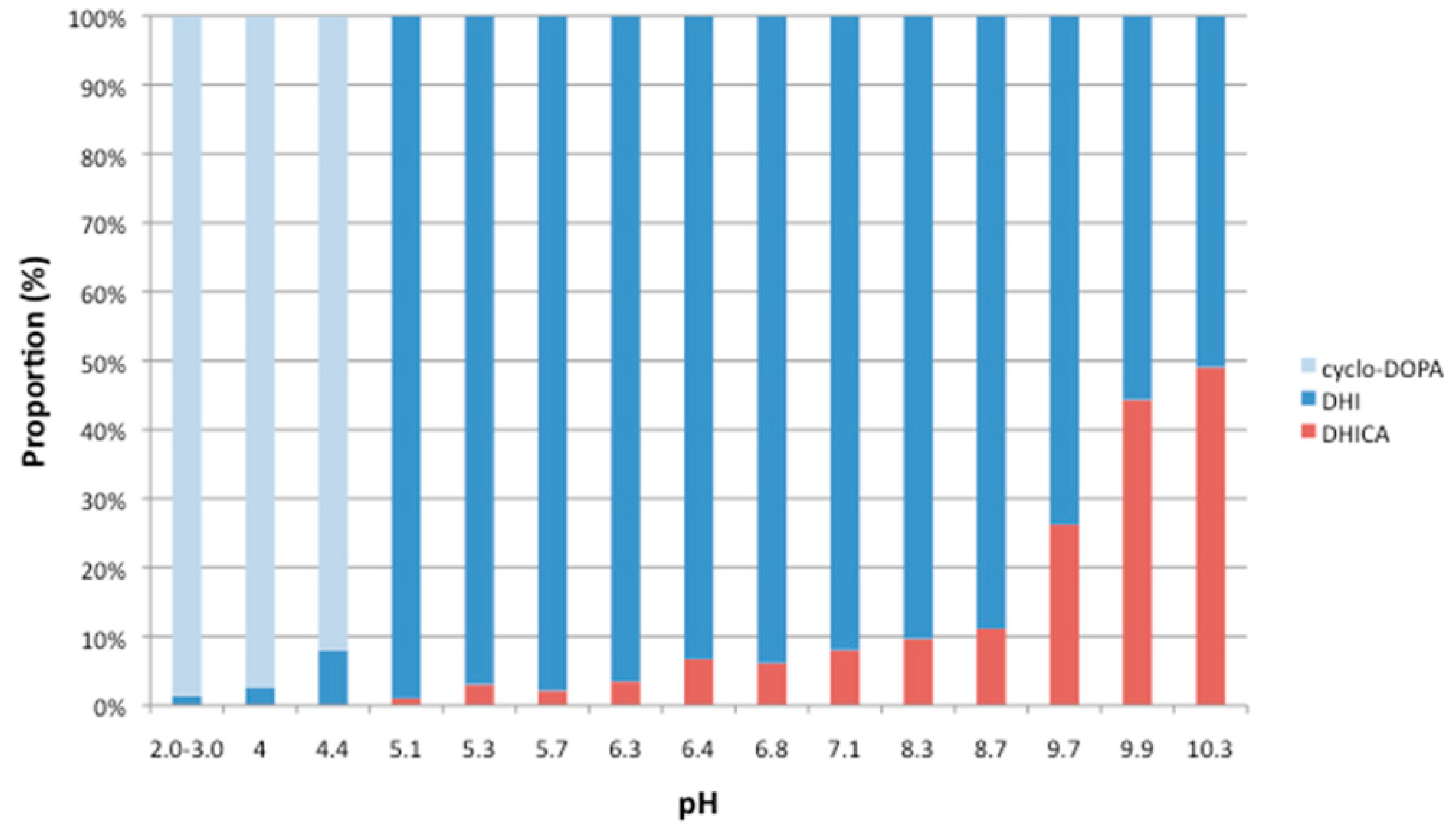
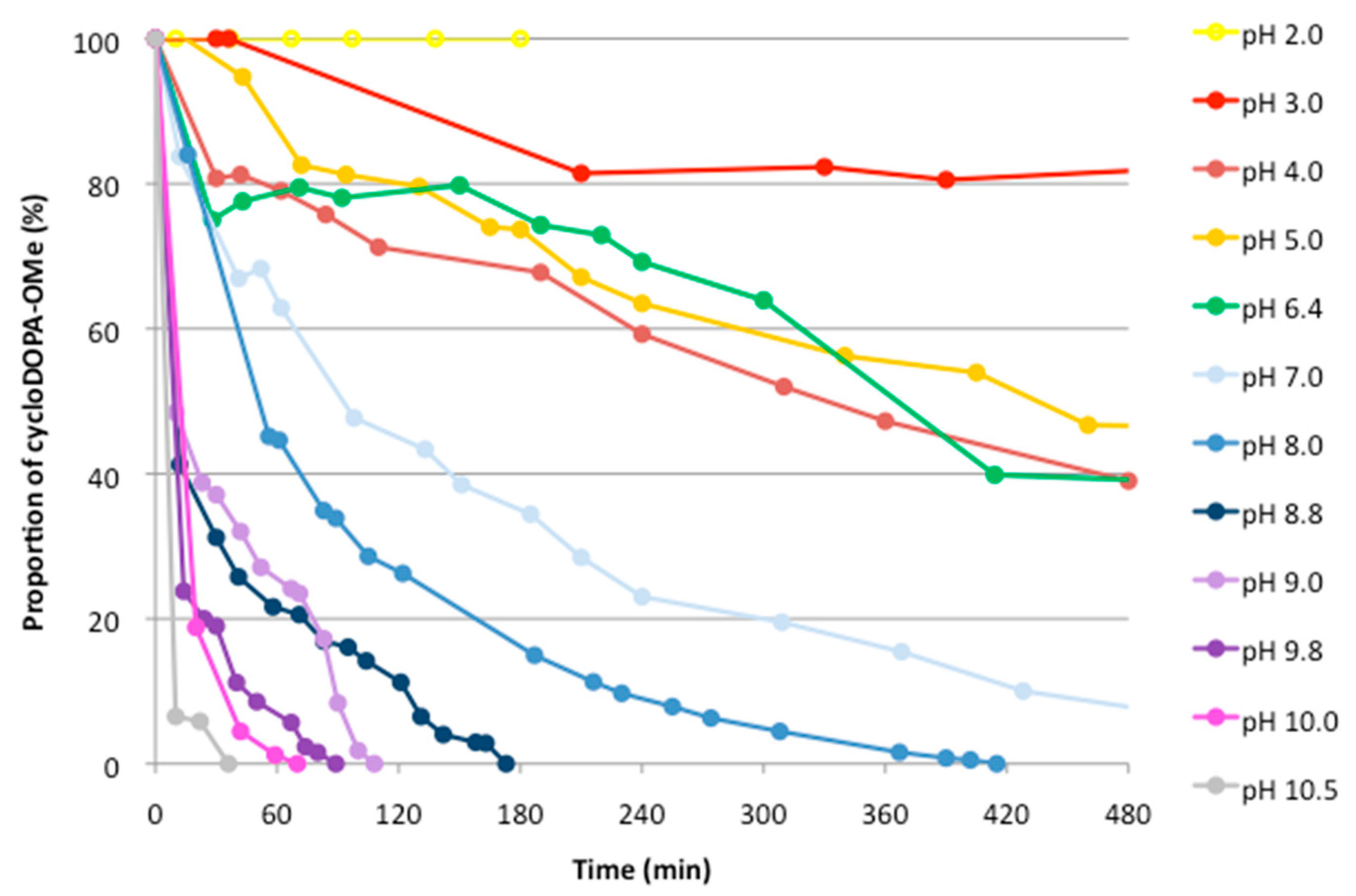
2.2. Analysis of The pH Stability of CycloDOPA (8) and CycloDOPA Methyl Ester (6)
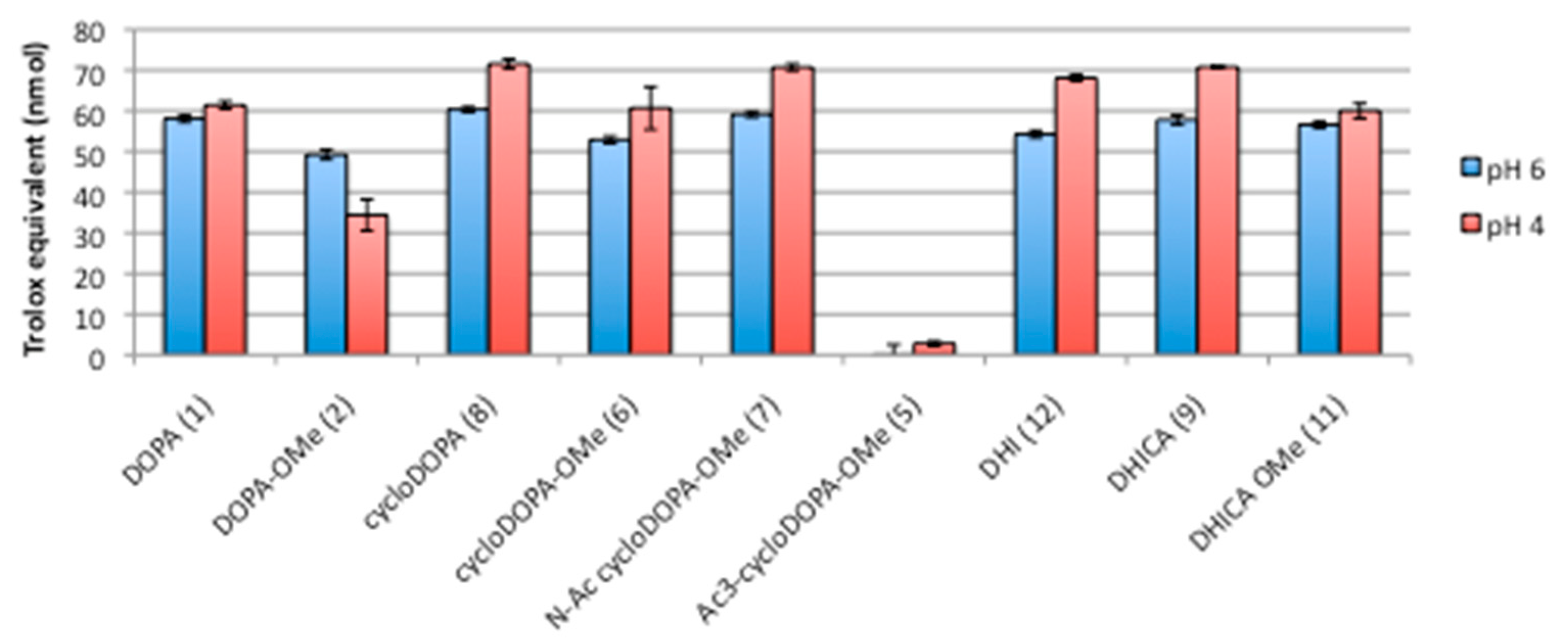
2.3. DPPH Assay for CycloDOPA and Its Related Products
3. Materials and Methods
3.1. General Procedures
3.2. Synthesis
3.3. pH Stability for CycloDOPA Derivatives
3.4. Evaluation of The DPPH Radical Scavenging Activity for CycloDOPA Derivatives
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Ito, S. A chemist’s view of melanogenesis. Pigment. Cell Res. 2003, 16, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Herrero, F.G.; Carmona, F.G. Biosynthesis of betalains: Yellow and violet plant pigments. Trends Plant. Sci. 2013, 18, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Strack, D.; Vogt, T.; Schliemann, W. Recent advances in betalain research. Phytochemistry 2003, 62, 247–269. [Google Scholar] [CrossRef]
- Wyler, H.; Chiovini, J. Die Synthese von Cyclodopa (Leukodopachrom). Helv. Chim. Acta 1968, 51, 1476–1494. [Google Scholar] [CrossRef] [PubMed]
- Schliemann, W.; Steiner, U.; Strack, D. Betanidin formation from dihydroxyphenylalanine in a model assay system. Phytochemistry 1998, 49, 1593–1598. [Google Scholar] [CrossRef]
- Sasaki, N.; Wada, K.; Koda, T.; Kasahara, K.; Adachi, T.; Ozeki, Y. Isolation and characterization of cDNAs encoding an enzyme with glucosyltransferase activity for cyclo-DOPA from four o’clocks and feather cockscombs. Plant. Cell Physiol. 2005, 46, 666–670. [Google Scholar] [CrossRef] [PubMed]
- Suh, D.H.; Lee, S.; Heo, D.Y.; Kim, Y.-S.; Cho, S.K.; Lee, S.; Lee, C.H. Metabolite profiling of red and white pitayas (Hylocereus polyrhizus and Hylocereus undatus) for comparing betalain biosynthesis and antioxidant activity. J. Agric. Food Chem. 2014, 62, 8764–8771. [Google Scholar] [CrossRef] [PubMed]
- Guimond, N.; Mayer, P.; Trauner, D. Development of an iron(II)-catalyzed aerobic catechol cleavage and biomimetic synthesis of betanidin. Chem. Eur. J. 2014, 20, 9519–9523. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, L.Q.; Simon, J.D. Ultra-low temperature oxidation of 5,6-dihydroxyindole: A Novel approach to study synthetic melanogenesis. Photochem. Photobiol. 2008, 84, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Charkoudian, L.K.; Franz, K.J. Fe (III)-coordination properties of neuromelanin components: 5,6-Dihydroxyindole and 5,6-dihydroxyindole-2-carboxylic acid. Inorg. Chem. 2006, 45, 3657–3664. [Google Scholar] [CrossRef] [PubMed]
- Pyrzynska, K.; Pękal, A. Application of free radical diphenylpicrylhydrazyl (DPPH) to estimate the antioxidant capacity of food samples. Anal. Methods 2013, 5, 4288–4295. [Google Scholar] [CrossRef]
- Takebayashi, J.; Tai, A.; Gohda, E.; Yamamoto, I. Characterization of the radical-scavenging reaction of 2-O-substituted ascorbic acid derivatives, AA-2G, AA-2P, and AA-2S: A Kinetic and stoichiometric study. Biol. Pharm. Bull. 2006, 29, 766–771. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakagawa, S.; Tachrim, Z.P.; Kurokawa, N.; Ohashi, F.; Sakihama, Y.; Suzuki, T.; Hashidoko, Y.; Hashimoto, M. pH Stability and Antioxidant Power of CycloDOPA and Its Derivatives. Molecules 2018, 23, 1943. https://doi.org/10.3390/molecules23081943
Nakagawa S, Tachrim ZP, Kurokawa N, Ohashi F, Sakihama Y, Suzuki T, Hashidoko Y, Hashimoto M. pH Stability and Antioxidant Power of CycloDOPA and Its Derivatives. Molecules. 2018; 23(8):1943. https://doi.org/10.3390/molecules23081943
Chicago/Turabian StyleNakagawa, Shiori, Zetryana Puteri Tachrim, Natsumi Kurokawa, Fumina Ohashi, Yasuko Sakihama, Takeyuki Suzuki, Yasuyuki Hashidoko, and Makoto Hashimoto. 2018. "pH Stability and Antioxidant Power of CycloDOPA and Its Derivatives" Molecules 23, no. 8: 1943. https://doi.org/10.3390/molecules23081943