New 2-Oxoindolin Phosphonates as Novel Agents to Treat Cancer: A Green Synthesis and Molecular Modeling


 ,
, 
Abstract
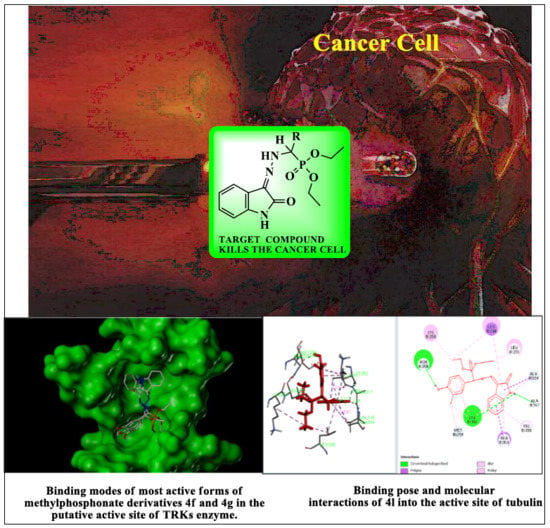
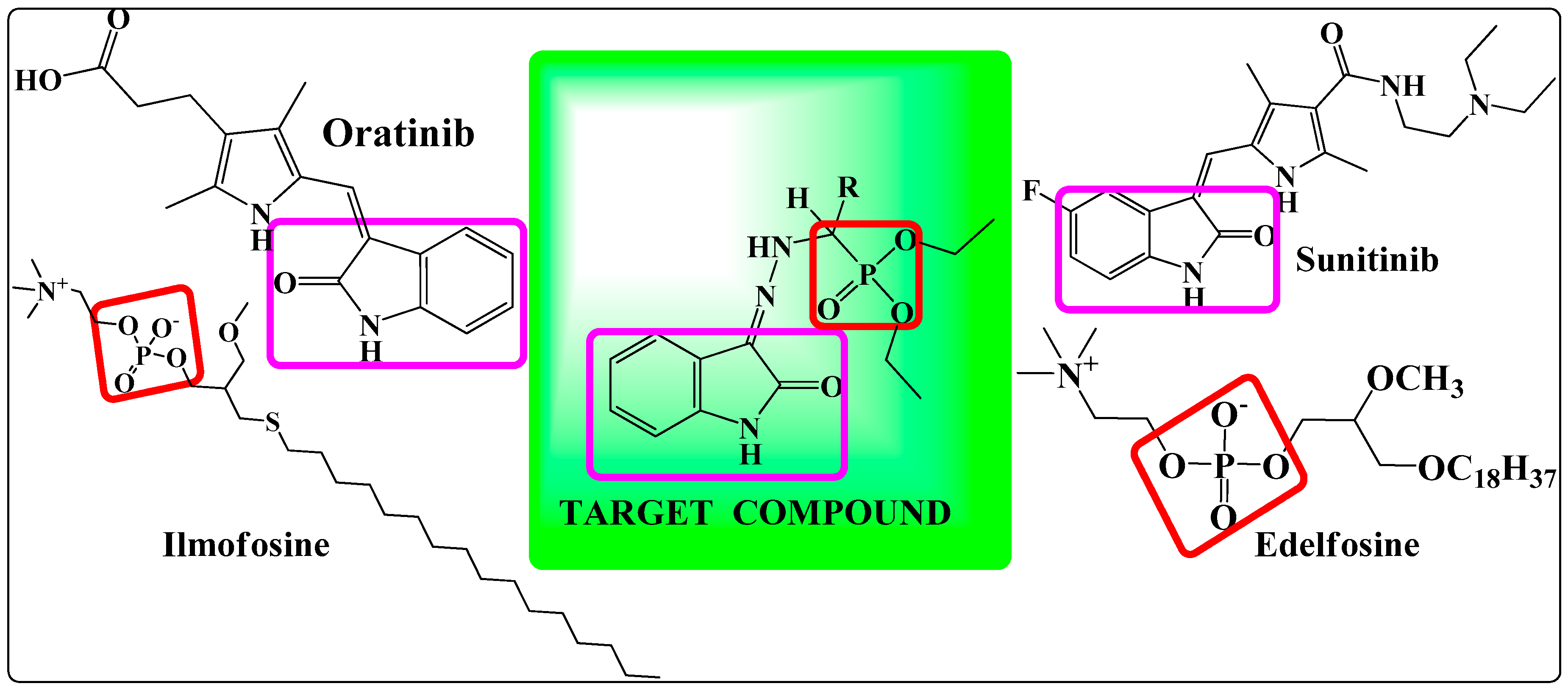
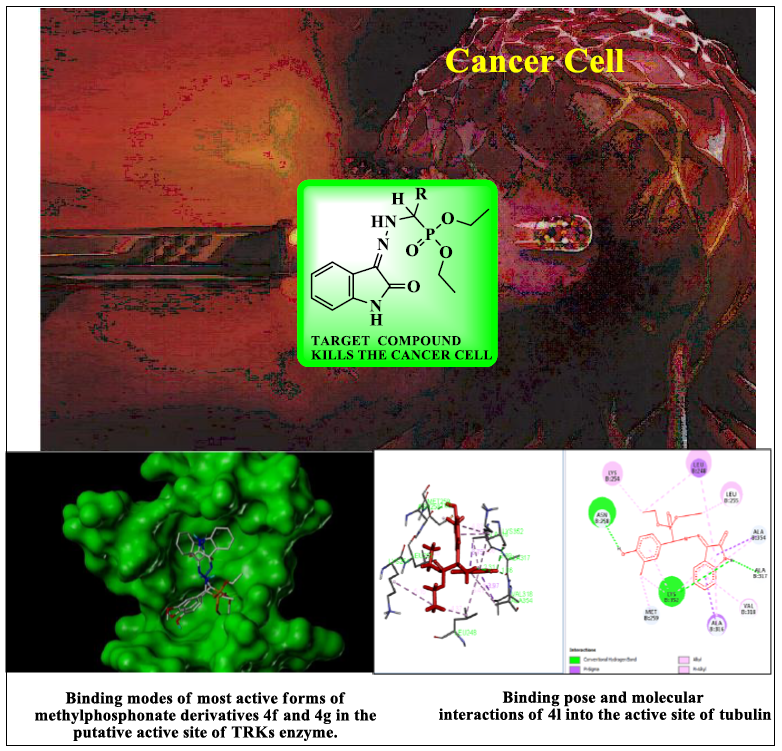
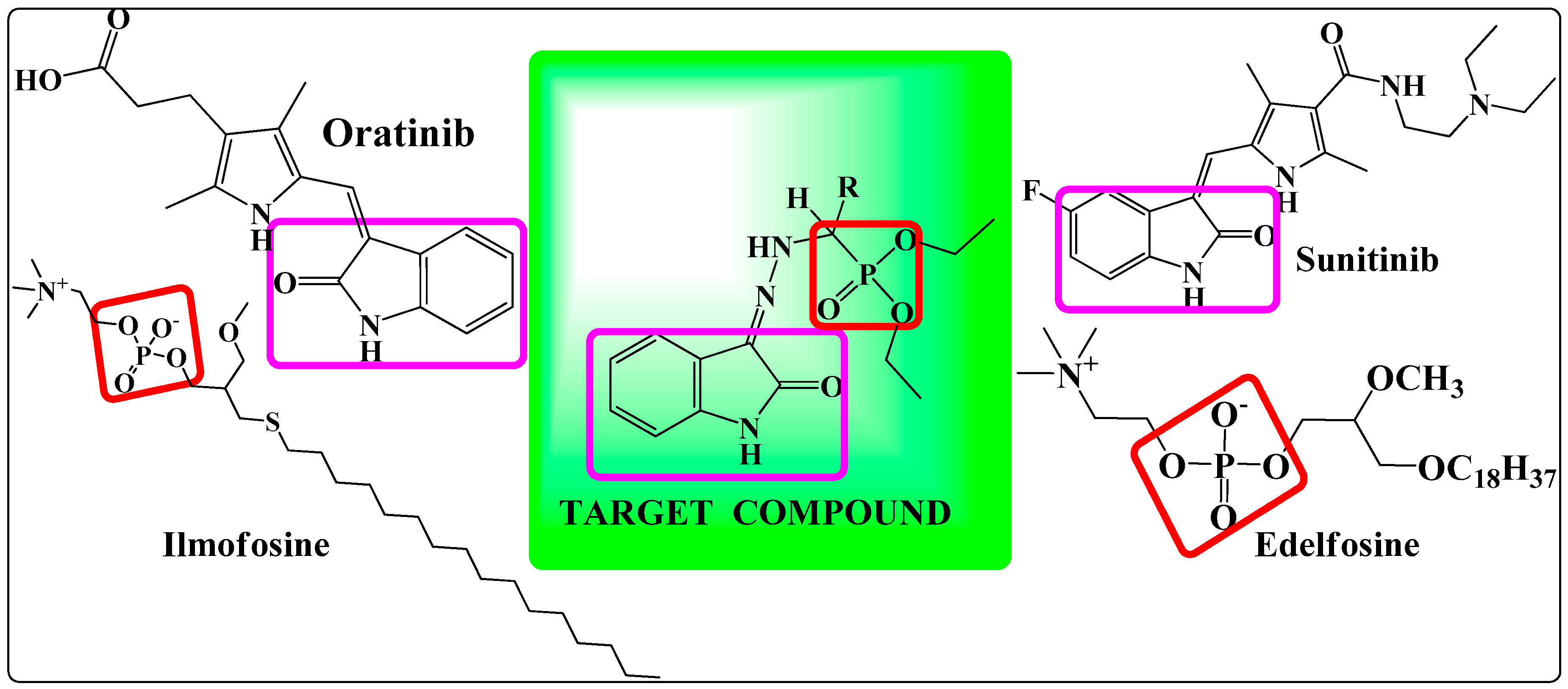
:
1. Introduction
2. Results
2.1. Chemistry
2.2. In Vitro Anticancer Screening
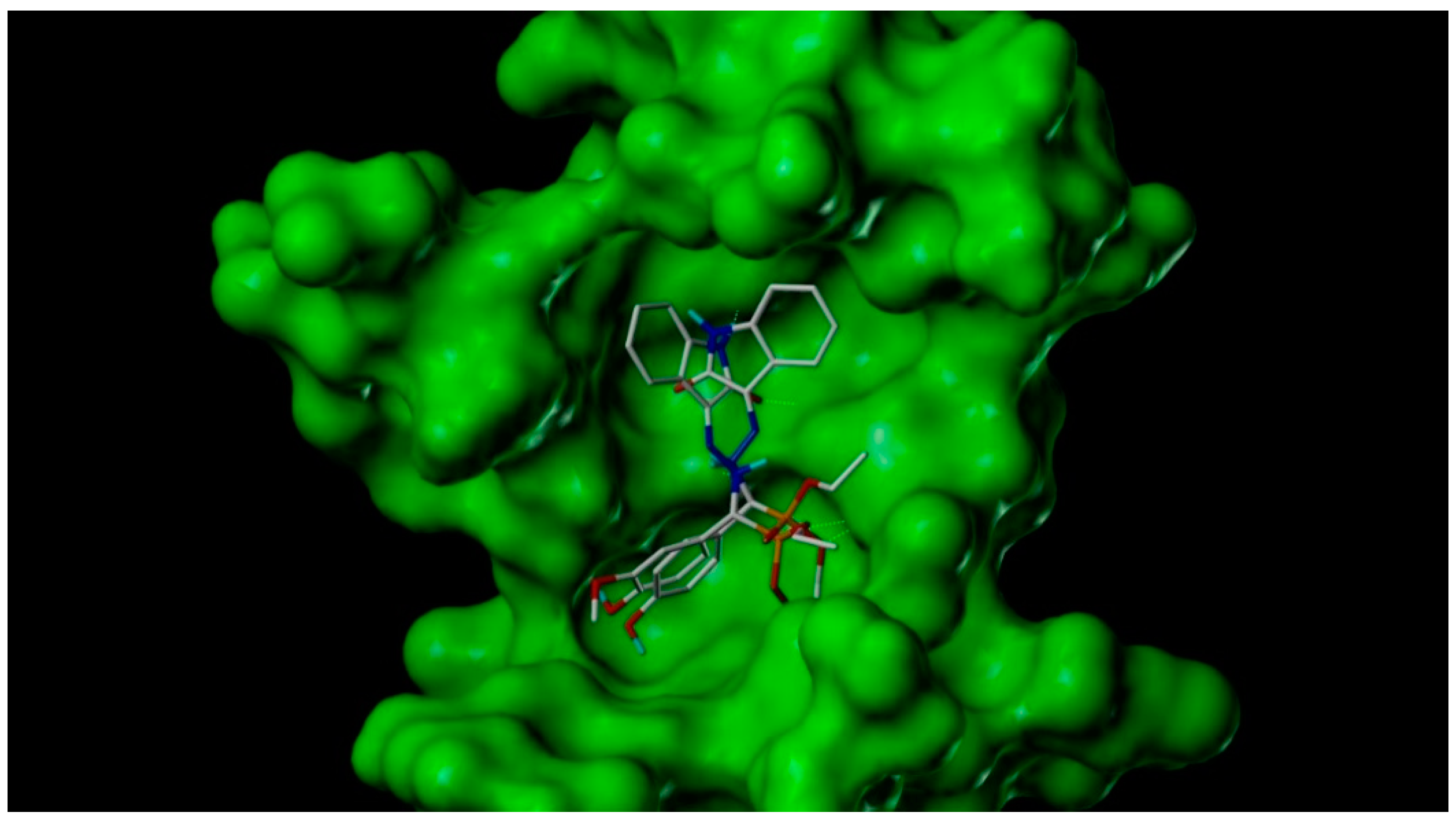
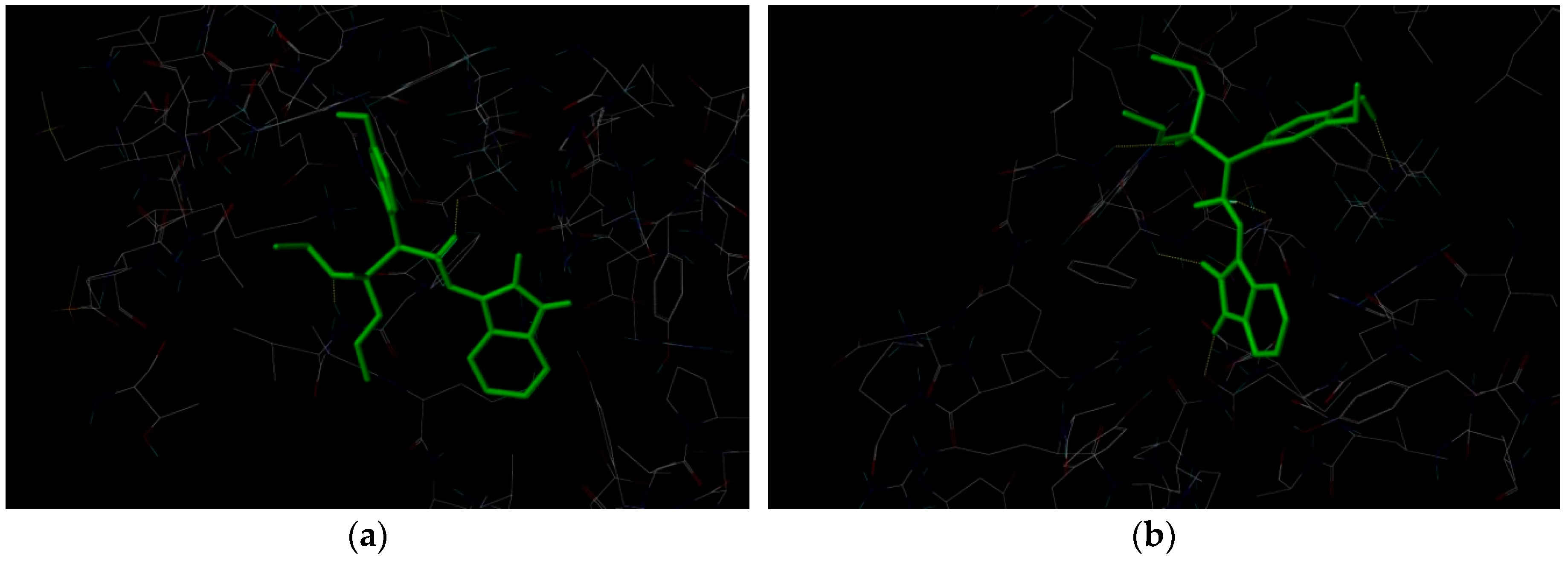
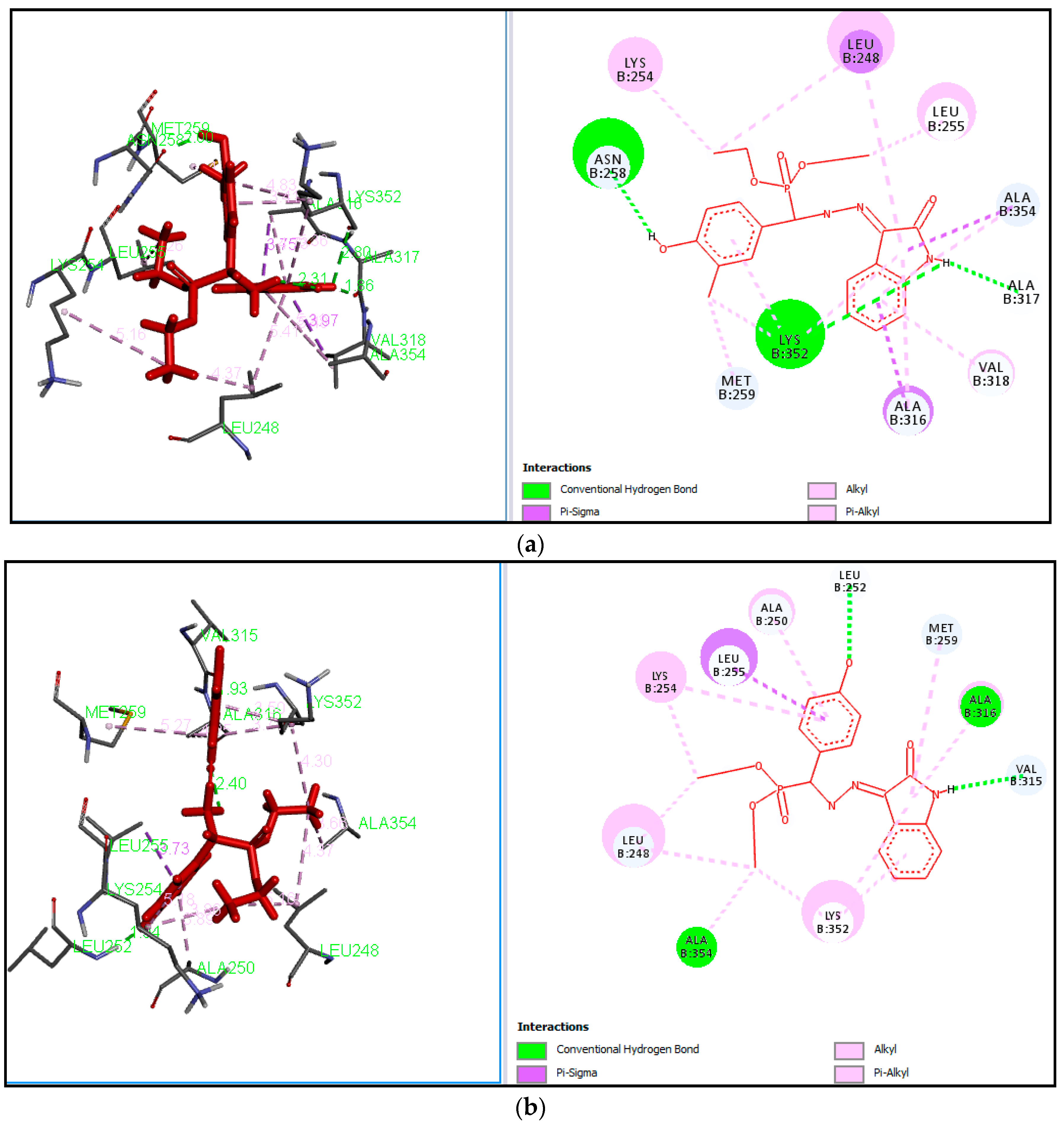
2.3. Docking Study
2.4. In Silico ADMET Predictions
2.5. In Vivo Acute Oral Toxicity Study and Gross Behavioral Studies
3. Materials and Methods
3.1. General
3.2. Instrumentation
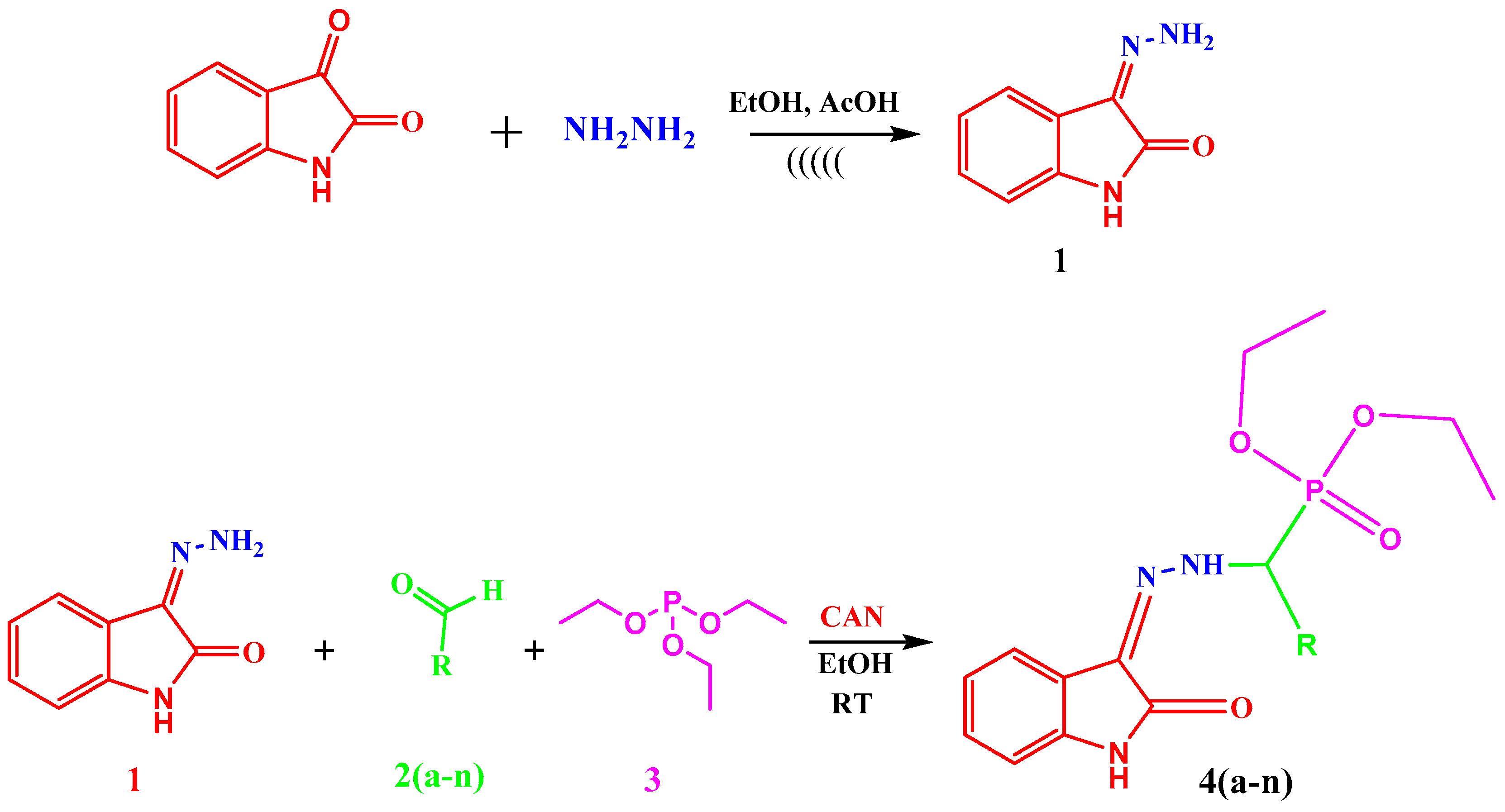
3.3. Synthesis
3.3.1. Synthesis of 3-Hydrazonoindolin-2-one (1)
3.3.2. General Procedure for the Synthesis of Diethyl(substituted phenyl/heteroaryl)(2-(2-oxoindolin-3-ylidene)hydrazinyl)methylphosphonates
3.4. In Vitro Anticancer Activity
3.5. Docking Study
3.6. In-Silico Bioavailability Predictions
3.7. In Vivo Acute Oral Toxicity Study and Gross Behavioral Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Notes
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef]
- Zhang, X.; Raghavan, S.; Ihnat, M.; Hamel, E.; Zammiello, C.; Bastian, A.; Mooberry, S.L.; Gangjee, A. The design, synthesis and biological evaluation of conformationally restricted 4-substituted-2,6-dimethylfuro[2,3-d]pyrimidines as multi-targeted receptor tyrosine kinase and microtubule inhibitors as potential antitumor agents. Bioorg. Med. Chem. 2015, 23, 2408–2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerbel, R.; Folkman, J. Clinical translation of angiogenesis inhibitors. Nat. Rev. Cancer 2002, 2, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, R.K.; Carmeliet, P. SnapShot: Tumor angiogenesis. Cell 2012, 149, 1408–1411. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Waxman, D.J. Combination of antiangiogenesis with chemotherapy for more effective cancer treatment. Mol. Cancer Ther. 2008, 7, 3670–3684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.; Gourley, C.; McNeish, I.; Ledermann, J.; Gore, M.; Jayson, G.; Perren, T.; Rustin, G.; Kaye, S. Targeted anti-vascular therapies for ovarian cancer: Current evidence. Br. J. Cancer 2013, 108, 250–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Glaxo, S.K.; Combination of Lapatinib with Carboplatin, Paclitaxel and Trastuzumab in Metastatic Breast Cancer. In ClinicalTrials.gov [Internet]. In Bethesda (MD): National Library of Medicine (US). Available online: http://clinicaltrials.gov/show/NCT00367471 NLM Identifier: NCT00367471 (accessed on 6 March 2015).
- Twenty Eight Clinical Trials Were Found on Clinicaltrials.gov (accessed in March 2015) Site in Which RTK Inhibitors Were Being Used in Combination with Antitubulins and Other Chemotherapeutic Agents. Of These, 16 Trials Are Currently in Progress and the Identification Numbers in Clinical Trials.Gov Are Provided Below: NCT01855750; NCT01804530; NCT01606878; NCT01746277; NCT02326285; NCT01620190; NCT01974440; NCT01683994; NCT02378389; NCT01939054; NCT01719302; NCT02191059; NCT02191059; NCT01876082; NCT00567554; NCT00367471NCT02191059; NCT02191059; NCT01876082; NCT00567554; NCT00367471. Available online: https://clinicaltrials.gov/ct2/results?term=receptor+tyrosine+kinase+AND+tubulin&Search=Search (accessed on 3 June 2015).
- Vermont, U.O. Docetaxel, Gemcitabine and Pazopanib as Treatment for Soft Tissue Sarcoma. In ClinicalTrials.gov [Internet]; National Library of Medicine (US): Bethesda, MD, USA, 2015. Available online: http://clinicaltrials.gov/show/NCT01719302 NLM Identifier: NCT01719302 (accessed on 6 March 2015).
- Gangjee, A.; Zaware, N.; Raghavan, S.; Ihnat, M.; Shenoy, S.; Kisliuk, R.L. Single agents with designed combination chemotherapy potential: Synthesis and evaluation of substituted pyrimido[4,5-b]indoles as receptor tyrosine kinase and thymidylate synthase inhibitors and as antitumor agents. J. Med. Chem. 2010, 53, 1563–1578. [Google Scholar] [CrossRef] [PubMed]
- Nikalje, A.G.; Tiwari, S.V.; Sangshetti, J.N.; Damale, M.D. Ultrasound Mediated Synthesis, Biological Evaluation, Docking Study and in vivo acute oral toxicity study of Novel Indolin-2-one Coupled Pyrimidine Derivatives. Res. Chem. Intermed. 2018, 44, 3031–3059. [Google Scholar] [CrossRef]
- Rathi, A.K.; Syed, R.V.; Singh, H.; Shin, H.S.; Patel, R.V. Kinase Inhibitor Indole Derivatives as Anticancer Agents. A Patent Review. Recent Pat. Anticancer Drug Discov. 2016, 12, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Tyrosine-Kinase Inhib. Available online: https://en.wikipedia.org/wiki/Tyrosine-kinase_inhib (accessed on 7 February 2018).
- Li, C.; Song, B.; Yan, K.; Xu, G.; Hu, D.; Yang, S.; Jin, L.; Xue, W.; Lu, P. One Pot Synthesis of α-Aminophosphonates Containing Bromo and 3,4,5-Trimethoxybenzyl Groups under Solvent-free Conditions. Molecules 2007, 12, 163–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, G.S.; Krishna, J.R.; Manjunath, M.; Reddy, O.V.S.; Krishnaiah, M.; Reddy, C.S.; Puranikd, V.G. Synthesis, NMR, X-ray crystallography and bioactivity of some α-aminophosphonates. Arkivoc 2007, 13, 133–141. [Google Scholar]
- Rao, X.; Song, Z.; He, L. Synthesis and antitumor activity of novel α-aminophosphonates from di-terpenicdehydroabietylamine. Heteroat. Chem. 2008, 19, 512–516. [Google Scholar] [CrossRef]
- Naydenova, E.D.; Todorov, P.T.; Mateeva, P.I.; Zamfirova, R.N.; Pavlov, N.D.; Todorov, S.B. Synthesis and biological activity of novel small peptides with aminophosphonates moiety as NOP receptor ligands. Amino Acids 2010, 39, 1537–1543. [Google Scholar] [CrossRef] [PubMed]
- Tusek-bozic, L.; Juribasic, M.; Traldi, P.; Scarcia, V.; Furlani, A. Synthesis, characterization and antitumor activity of palladium(II) complexes of monoethyl 8-quinolylmethylphosphonate. Polyhedron 2008, 27, 1317–1328. [Google Scholar] [CrossRef]
- Wang, B.; Miao, Z.W.; Wang, J.; Chen, R.Y.; Zhang, X.D. Synthesis and biological evaluation of novel naphthoquinone fused cyclic aminoalkylphosphonates and aminoalkylphosphonic monoester. Amino Acids 2008, 35, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, Z.; Firouzabadi, H.; Iranpoor, N.; Ghaderi, A.; Jafari, M.R.; Jafari, A.A.; Zare, H.R. Design and one-pot synthesis of alpha-aminophosphonates and bis(alpha-aminophosphonates) by iron(III) chloride and cytotoxic activity. See comment in PubMed Commons below. Eur. J. Med. Chem. 2009, 44, 4266–4275. [Google Scholar] [CrossRef] [PubMed]
- Onita, N.; Sisu, I.; Penescu, M.; Purcarea, V.L.; Kurunczi, L. Synthesis, characterization and biological activity of some α-aminophosphonates. Farmacia 2010, 58, 531–545. [Google Scholar]
- Zhang, X.; Qu, Y.; Fan, X.; Bores, C.; Feng, D.; Andrei, G.; Snoeck, R.; De Clercq, E.; Loiseau, P.M. Solvent-free synthesis of pyrimidine nucleoside-aminophosphonate hybrids and their biological activity evaluation. Nucleosides Nucleotides Nucleic Acids 2010, 29, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, S.; Li, X.; Fan, H.; Bhadury, P.; Xu, W.; Wu, J.; Wang, Z. Synthesis and antiviral bioactivity of chiral thioureas containing leucine and phosphonate moieties. Molecules 2010, 15, 5112–5123. [Google Scholar] [CrossRef] [PubMed]
- Biswal, S.; Sahoo, U.; Sethy, S.; Kumar, H.K.S.; Banerjee, M. Indole: The Molecule of Diverse Biological Activities. Asian J. Pharm. Clin. Res. 2012, 5, 1–6. [Google Scholar]
- Vine, K.L.; Matesic, L.; Locke, J.M.; Skropeta, D. Recent Highlights in the Development of Isatin-Based Anticancer Agents. In Advances in Anticancer Agents in Medicinal; Bentham Science Publishers: Sharjah, UAE, 2013; Volume 59, pp. 254–312. [Google Scholar]
- Prakash, R.C.; Raja, S. Indolinones as Promising Scaffold as Kinase Inhibitors: A Review. Mini Rev. Med. Chem. 2012, 12, 98–119. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, H.R.; Pireddu, R.; Chen, L.; Luo, Y.; Sung, S.S.; Szymanski, A.M.; Yip, M.L.R.; Guida, W.C.; Sebti, S.M.; Wu, J.; et al. Inhibitors of Src homology-2 domain containing protein tyrosine phosphatase-2 (Shp2) based on oxindole scaffolds. J. Med. Chem. 2008, 51, 4948–4956. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.G.; Bostjan, S.; Knox, J.J. Sunitinib in solid tumors. Expert Opin. Investig. Drugs 2009, 18, 821–834. [Google Scholar]
- Yancey, M.F.; Merritt, D.A.; White, J.A.; Marsh, S.A.; Locuson, C.W. Distribution, metabolism, and excretion of toceranib phosphate (Palladia™, SU11654), a novel tyrosine kinase inhibitor, in dogs. J. Vet. Pharmacol. Ther. 2010, 33, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.V.; Seijas, J.A.; Vazquez-Tato, M.P.; Sarkate, A.P.; Karnik, K.S.; Nikalje, A.P.G. Facile synthesis of novel coumarin derivatives, antimicrobial analysis, enzyme assay, docking study, ADMET prediction and toxicity study. Molecules 2017, 22, 1172. [Google Scholar] [CrossRef] [PubMed]
- Nikalje, A.P.G.; Tiwar, S.V.; Tupe, J.G.; Vyas, K.V.; Qureshi, G. Ultrasound Assisted-synthesis and Biological Evaluation of Piperazinylprop- 1-en-2-yloxy-2H-chromen-2-ones as Cytotoxic Agents. Lett. Drug Des. Discov. 2017, 14, 1195–1205. [Google Scholar] [CrossRef]
- Shailee, V.T.; Siddiqui, S.; Seijas, J.A.; Vazquez-Tato, M.P.; Sarkate, A.P.; Lokwani, D.K.; Nikalje, A.P.G. Microwave-Assisted Facile Synthesis, Anticancer Evaluation and Docking Study of N-((5-(Substituted methylene amino)-1,3,4-thiadiazol-2-yl)methyl) Benzamide Derivatives. Molecules 2017, 22, 995. [Google Scholar] [CrossRef]
- Nimbalkar, U.D.; Seijas, J.A.; Vazquez-Tato, M.P.; Damale, M.G.; Sangshetti, J.N.; Nikalje, A.P.G. Ionic Liquid-Catalyzed Green Protocol for Multi-Component Synthesis of Dihydropyrano[2,3-c]pyrazoles as Potential Anticancer Scaffolds. Molecules 2017, 22, 1628. [Google Scholar] [CrossRef] [PubMed]
- Matveeva, E.D.; Zefirov, N.S. Catalytic Kabachnik-Fields reaction: New horizons for old reaction. Arkivoc 2008, 1, 1–17. [Google Scholar]
- Sayed, I.E.; Kosy, S.M.E.; Magied, M.F.A.; Hamed, M.A.; Gokha, A.A.A.; Sattar, M.A. One-pot Synthesis of Novel α-Aminophosphonate Derivatives Containing a Pyrazole Moiety. J. Am. Sci. 2011, 7, 604–608. [Google Scholar]
- Mandhane, P.G.; Joshi, R.S.; Nagargoje, D.R.; Gill, C.H. Thiamine hydrochloride (VB1): An efficient catalyst for one-pot synthesis of α-aminophosphonates under ultrasonic irradiation. Chin. Chem. Lett. 2011, 22, 563–566. [Google Scholar] [CrossRef]
- Huang, X.C.; Wang, M.; Pan, Y.M.; Tian, X.Y.; Wang, H.S.; Zhang, Y. Synthesis and antitumor activities of novel α-aminophosphonatesdehydroabietic acid derivatives. Bioorg. Med. Chem. Lett. 2013, 23, 5283–5289. [Google Scholar] [CrossRef] [PubMed]
- Bhagat, S.; Chakraborti, A.K. Zirconium(IV) Compounds As Efficient Catalysts for Synthesis of α-Aminophosphonates. J. Org. Chem. 2008, 73, 6029–6032. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Luo, Y.; Wu, J.; Shen, Q.; Chen, H. Facile one-pot synthesis of α-aminophosphonates using lanthanide chloride as catalys. Heteroat. Chem. 2006, 17, 389–392. [Google Scholar] [CrossRef]
- Qian, C.; Huang, T.J. One-Pot Synthesis of α-Amino Phosphonates from Aldehydes Using Lanthanide Triflate as a Catalyst. J. Org. Chem. 1998, 63, 4125–4128. [Google Scholar] [CrossRef]
- Wu, J.; Sun, W.; Xia, H.G.; Sun, X. A facile and highly efficient route to α-amino phosphonates via three-component reactions catalyzed by Mg(ClO4)2 or molecular iodine. Org. Biomol. Chem. 2006, 4, 1663–1666. [Google Scholar] [CrossRef] [PubMed]
- Azizi, N.; Rajabi, F.; Saidi, M.R. A mild and highly efficient protocol for the one-pot synthesis of primary α-amino phosphonates under solvent-free conditions. Tetrahedron Lett. 2004, 45, 9233–9235. [Google Scholar] [CrossRef]
- Sangshetti, J.N.; Kokare, N.D.; Kotharkar, S.A.; Shinde, D.B. Ceric ammonium nitrate catalysed three component one-pot efficient synthesis of 2,4,5-triaryl-1Himidazoles. J. Chem. Sci. 2008, 120, 463–467. [Google Scholar] [CrossRef]
- Lin, C.M.; Ho, H.H.; Pettit, G.R.; Hamel, E. Antimitotic natural products combretastatin A-4 and combretastatin A-2: Studies on the mechanism of their inhibition of the binding of colchicine to tubulin. Biochemistry 1989, 28, 6984–6991. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.A.; Gilmartin, M.E.; Hall, J.L.; Cowan, N.J. Three expressed sequences within the human beta tubulin multigene family each define a distinct isotype. J. Mol. Biol. 1985, 182, 11–20. [Google Scholar] [CrossRef]
- Nicoletti, M.I.; Valoti, G.; Giannakakou, P.; Zhan, Z.; Kim, J.H.; Lucchini, V.; Landoni, F.; Mayo, J.G.; Giavazzi, R.; Fojo, T. Expression of beta-tubulin isotypes in human ovarian carcinoma xenografts and in a sub-panel of human cancer cell lines from the NCI-Anticancer Drug Screen: Correlation with sensitivity to microtubule active agents. Clin. Cancer Res. 2001, 7, 2912–2922. [Google Scholar] [PubMed]
- McKean, P.G.; Vaughan, S.; Gull, K. The extended tubulin super family. J. Cell Sci. 2001, 114, 2723–2733. [Google Scholar] [PubMed]
- Perez, E.A. Microtubule inhibitors: Differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol. Cancer Ther. 2009, 8, 2086–2095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarroll, J.A.; Gan, P.P.; Liu, M.; Kavallaris, M. βIII-Tubulin Is a Multifunctional Protein Involved in Drug Sensitivity and Tumorigenesis in Non–Small Cell Lung Cancer. Cancer Rev. 2010, 70, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lagorce, D.; Sperandio, O.; Baell, J.B.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs3: A web server for compound property calculation and chemical library design. Nucleic Acids Res. 2015, 43, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, L.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 225–226. [Google Scholar] [CrossRef]
- Zhao, Y.; Abraham, M.H.; Lee, J.; Hersey, A.; Luscombe, N.C.; Beck, G.; Sherborne, B.; Cooper, I. Rate-limited steps of human oral absorption and QSAR studies. Pharm. Res. 2002, 19, 1446–1457. [Google Scholar] [CrossRef] [PubMed]
- Ajitha, M.; Rajnarayana, K.; Sarangapani, M. Synthesis of new 2-substituted-[1,3,4]-oxadiazino-[5,6-b]-indoles with H1-antihistaminic, antimuscarinic and antimicrobial activity. Pharmazie 2002, 57, 796–799. [Google Scholar] [CrossRef] [PubMed]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- SYBYL. Tripos Molecular Modeling Program Package. Available online: http://www.tripos.com/data/SYBYL (accessed on 2 January 2018).
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Nucleic Acids Res. 2002, 58, 899–907. [Google Scholar] [CrossRef] [Green Version]
- OECD. Guideline for Testing of Chemicals. In No. 425: Acute Oral Toxicity Up-and Down-Procedure (UDP); Organisation for Economic Co-Operation and Development: Paris, France, 2008. [Google Scholar]
Sample Availability: Samples of the compounds 4(a–n) are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | R | Molecular Formula | Molecular Weight (gm) | Time Required (min) | % Yield | Melting Point (°C) |
|---|---|---|---|---|---|---|
| 4a | Phenyl | C19H22N3O4P | 387.37 | 75 | 90 | 195–196 |
| 4b | 4-Chlorophenyl | C19H21ClN3O4P | 421.81 | 70 | 92 | 150–152 |
| 4c | 4-Fluorophenyl | C19H21FN3O4P | 405.36 | 75 | 95 | 176–180 |
| 4d | 4-Methoxyphenyl | C20H24N3O5P | 417.40 | 85 | 89 | 178–179 |
| 4e | 3,4-Dimethoxyphenyl | C21H26N3O6P | 447.42 | 90 | 90 | 189–190 |
| 4f | 4-Hydroxyphenyl | C19H22N3O5P | 403.37 | 80 | 88 | 140–142 |
| 4g | 4-Hydroxy-3-methoxyphenyl | C20H24N3O6P | 433.39 | 75 | 94 | 112–114 |
| 4h | 4-hyroxy-3-ethoxyphenyl | C21H26N3O6P | 447.44 | 80 | 92 | 160–162 |
| 4i | Thiophen-2-yl | C17H20N3O4PS | 393.40 | 80 | 87 | 179–182 |
| 4j | Furan-2-yl | C17H20N3O5P | 377.33 | 80 | 84 | 176–178 |
| 4k | 2-Hydroxyphenyl | C19H22N3O5P | 403.37 | 75 | 88 | 152–154 |
| 4l | 4-Hydroxy-3-methylphenyl | C20H24N3O6P | 417.40 | 82 | 75 | 190–192 |
| 4m | 4-Nitrophenyl | C19H21N4O6P | 432.37 | 89 | 90 | 172–174 |
| 4n | 4-Methylthiazole-5-yl | C17H 21N4O4PS | 408.10 | 75 | 70 | 188–190 |
| Compounds | GI50 (µM) | |||||||
|---|---|---|---|---|---|---|---|---|
| R | MCF-7 | IMR-32 | SKMEL-2 | MG-63 | HT-29 | Hep-G2 | NIH/3T3 | |
| 4a | Phenyl | >100 | >100 | 56.9 | <0.1 | 56.9 | 55.2 | >250 |
| 4b | 4-Chlorophenyl | 67.2 | >100 | 41.4 | <0.1 | 46.9 | 18.6 | >250 |
| 4c | 4-Fluorophenyl | >100 | >100 | 57.5 | <0.1 | 20.8 | 32.0 | >250 |
| 4d | 4-Methoxyphenyl | >100 | >100 | 65.7 | <0.1 | <0.1 | 45.3 | >250 |
| 4e | 3,4-Dimethoxyphenyl | >100 | >100 | 55.2 | <0.1 | <0.1 | 12.2 | >250 |
| 4f | 4-Hydroxyphenyl | >100 | <0.1 | 51.6 | <0.1 | <0.1 | 12.2 | >250 |
| 4g | 4-Hydroxy-3-methoxyphenyl | >100 | >100 | 24.0 | <0.1 | 12.2 | 32.0 | >250 |
| 4h | 4-hyroxy-3-ethoxyphenyl | 70.7 | >100 | 51.0 | <0.1 | 4.1 | 43.8 | >250 |
| 4i | Thiophen-2-yl | >100 | >100 | 55.5 | <0.1 | <0.1 | <0.1 | >250 |
| 4j | Furan -2-yl | >100 | >100 | 93.9 | <0.1 | <0.1 | 44.6 | >250 |
| 4k | 2-Hydroxyphenyl | >100 | 44.1 | 66.2 | <0.1 | 26.1 | 11.7 | >250 |
| 4l | 4-Hydroxy-3-methylphenyl | 78.4 | 89.8 | 76.4 | <0.1 | <0.1 | <0.1 | >250 |
| 4m | 4-Nitrophenyl | 77.2 | 56.6 | 44.2 | <0.1 | <0.1 | 13.3 | >250 |
| 4n | 4-Methylthiazole-5-yl | 58.2 | 67.8 | 33.1 | <0.1 | <0.1 | 24.9 | >250 |
| aADR | - | <0.1 | <0.1 | <0.1 | <0.1 | <0.1 | <0.1 | 82.2 |
| Compound ID | Total Score −Log(ki) | Crash | Polar |
|---|---|---|---|
| 4a | 5.1189 | −1.2354 | 1.997 |
| 4b | 5.0299 | −0.889 | 3.0602 |
| 4c | 4.7873 | −0.8462 | 1.6198 |
| 4d | 5.4734 | −1.1481 | 1.6983 |
| 4e | 5.3336 | −1.9652 | 3.1869 |
| 4f | 6.0396 | −0.7543 | 2.4298 |
| 4g | 5.5786 | −1.5587 | 3.8036 |
| 4h | 4.8916 | −2.8753 | 3.8417 |
| 4i | 3.5991 | −1.0332 | 3.0929 |
| 4j | 4.5136 | −0.6275 | 3.1716 |
| 4k | 5.7714 | −1.0431 | 1.5553 |
| 4l | 4.9855 | −0.6461 | 1.4008 |
| 4m | 5.6219 | −0.919 | 3.4772 |
| 4n | 5.5030 | −0.9370 | 3.7706 |
| Sr. No | Total Score (−Log Ki) | Crash Score | Ploar Score |
|---|---|---|---|
| 4a | 4.09 | −1.14 | 1.01 |
| 4b | 4.19 | −2.77 | 1.65 |
| 4c | 4.40 | −1.56 | 0.78 |
| 4d | 4.60 | −1.22 | 1.15 |
| 4e | 3.62 | −2.25 | 1.46 |
| 4f | 5.28 | −1.08 | 2.45 |
| 4g | 5.11 | −1.01 | 3.01 |
| 4h | 4.27 | −1.38 | 1.82 |
| 4i | 3.52 | −1.53 | 2.10 |
| 4j | 4.32 | −1.34 | 1.99 |
| 4k | 3.52 | −2.35 | 1.29 |
| 4l | 6.12 | −1.15 | 2.11 |
| 4m | 4.98 | −0.93 | 1.67 |
| 4n | 4.72 | −2.08 | 1.48 |
| ADR | 3.77 | −1.58 | 3.47 |
| Entry | % ABS | TPSA(A2) | n-ROTB | MV | MW | miLogP | n-ON | n-OHNH | Lipinski Violation | Toxicity |
|---|---|---|---|---|---|---|---|---|---|---|
| Rule | - | - | - | - | <500 | <5 | <10 | <5 | <1 | - |
| 4a | 76.98 | 92.79 | 8 | 341.53 | 387.38 | 2.92 | 7 | 2 | 0 | Non Toxic |
| 4b | 76.98 | 92.79 | 8 | 355.06 | 421.82 | 3.59 | 7 | 2 | 0 | Non Toxic |
| 4c | 76.98 | 92.79 | 8 | 346.46 | 405.37 | 3.08 | 7 | 2 | 1 | Non Toxic |
| 4d | 73.70 | 102.3 | 9 | 367.07 | 417.40 | 2.97 | 8 | 2 | 0 | Non Toxic |
| 4e | 70.61 | 111.26 | 10 | 392.62 | 447.43 | 2.56 | 9 | 2 | 0 | Non Toxic |
| 4f | 69.94 | 113.2 | 8 | 349.55 | 403.38 | 2.44 | 3 | 3 | 0 | Non Toxic |
| 4g | 66.82 | 122.25 | 9 | 375.09 | 433.40 | 2.25 | 9 | 3 | 0 | Non Toxic |
| 4h | 66.82 | 122.25 | 10 | 391.89 | 447.43 | 2.63 | 9 | 3 | 0 | Non Toxic |
| 4i | 76.98 | 92.79 | 8 | 332.34 | 393.40 | 2.81 | 7 | 2 | 0 | Non Toxic |
| 4j | 72.45 | 105.93 | 8 | 323.10 | 377.34 | 2.17 | 8 | 2 | 1 | Non Toxic |
| 4k | 67.92 | 113.2 | 9 | 375.09 | 403.39 | 2.25 | 9 | 3 | 0 | Non Toxic |
| 4l | 67.92 | 119.06 | 9 | 367.07 | 417.39 | 2.97 | 8 | 2 | 0 | Non Toxic |
| 4m | 59.48 | 92.79 | 9 | 364.86 | 432.37 | 2.86 | 10 | 3 | 0 | Non Toxic |
| 4n | 50.05 | 170.86 | 9 | 349.04 | 407.43 | 1.77 | 8 | 1 | 0 | Non Toxic |
| Additional Observation | Behavioral Observation | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Toxicity | No. of Death | Skin and Fur | Eyes Lacrimation | Salivation | Diarrhea | Respiration | Straub Tail | Pilo Erection | Convulsions | Motor Activity | Stereotypy | Tremors | Catalepsy | Sedation | Hypnosis | Writhing | Muscle Spasm | Analgesia | Arching & Rolling | Writhing |
| 4b | Nil | Nil | N | N | N | N | N | N | Y | N | N | N | N | N | N | N | N | N | N | N | N |
| 4h | Nil | Nil | N | N | N | N | N | N | Y | N | Y | N | N | N | N | N | N | N | N | N | N |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tiwari, S.V.; Sharif, N.S.; Gajare, R.I.; Vazquez, J.A.S.; Sangshetti, J.N.; Damale, M.D.; Nikalje, A.P.G. New 2-Oxoindolin Phosphonates as Novel Agents to Treat Cancer: A Green Synthesis and Molecular Modeling. Molecules 2018, 23, 1981. https://doi.org/10.3390/molecules23081981
Tiwari SV, Sharif NS, Gajare RI, Vazquez JAS, Sangshetti JN, Damale MD, Nikalje APG. New 2-Oxoindolin Phosphonates as Novel Agents to Treat Cancer: A Green Synthesis and Molecular Modeling. Molecules. 2018; 23(8):1981. https://doi.org/10.3390/molecules23081981
Chicago/Turabian StyleTiwari, Shailee V., Nawaz S. Sharif, Rekha I. Gajare, Julio A. Seijas Vazquez, Jaiprakash N. Sangshetti, Manoj D. Damale, and Anna Pratima G. Nikalje. 2018. "New 2-Oxoindolin Phosphonates as Novel Agents to Treat Cancer: A Green Synthesis and Molecular Modeling" Molecules 23, no. 8: 1981. https://doi.org/10.3390/molecules23081981
APA StyleTiwari, S. V., Sharif, N. S., Gajare, R. I., Vazquez, J. A. S., Sangshetti, J. N., Damale, M. D., & Nikalje, A. P. G. (2018). New 2-Oxoindolin Phosphonates as Novel Agents to Treat Cancer: A Green Synthesis and Molecular Modeling. Molecules, 23(8), 1981. https://doi.org/10.3390/molecules23081981