
Synthesis and Biological Evaluation of New Cholinesterase Inhibitors for Alzheimer’s Disease

 and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Enzymatic Inhibition
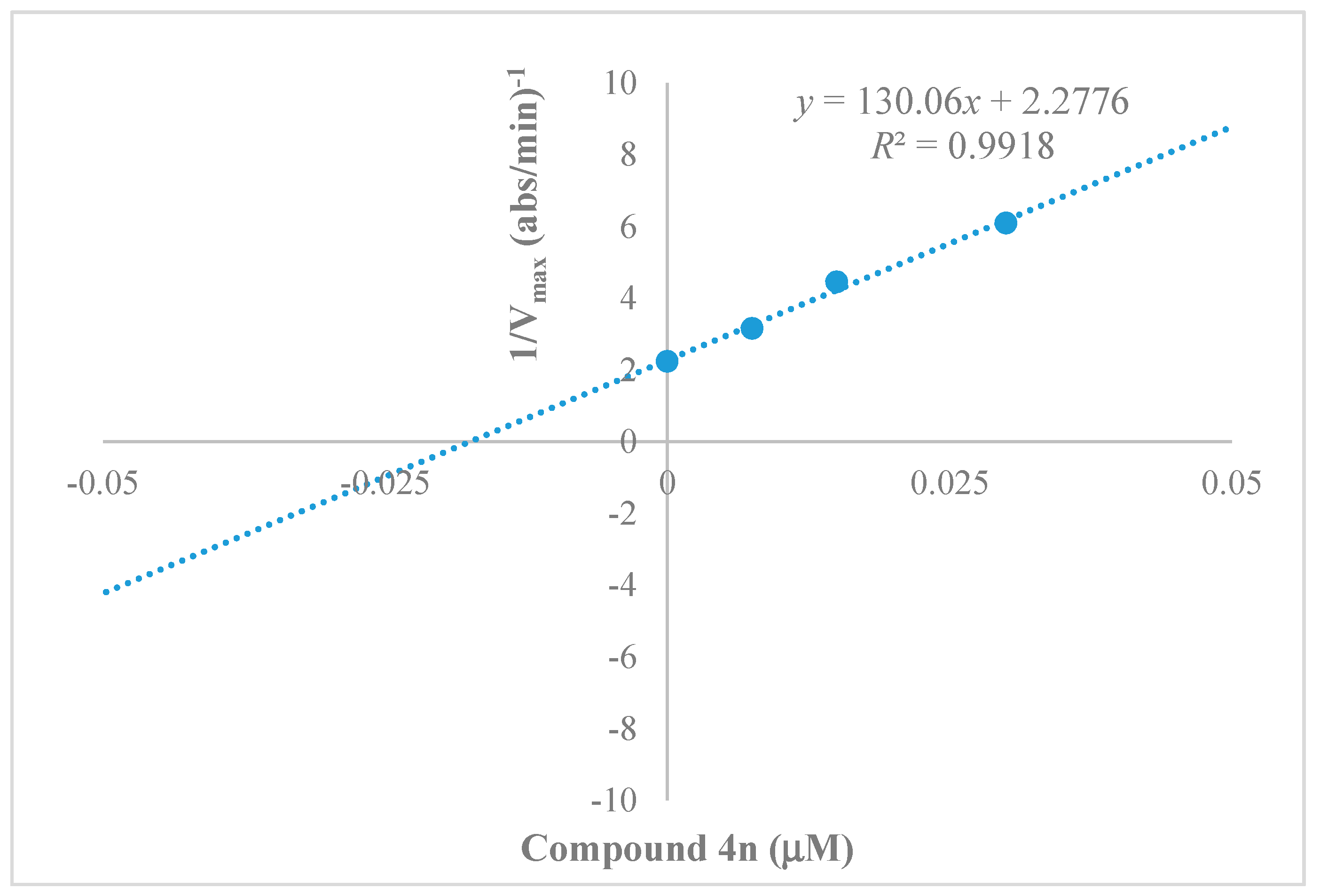
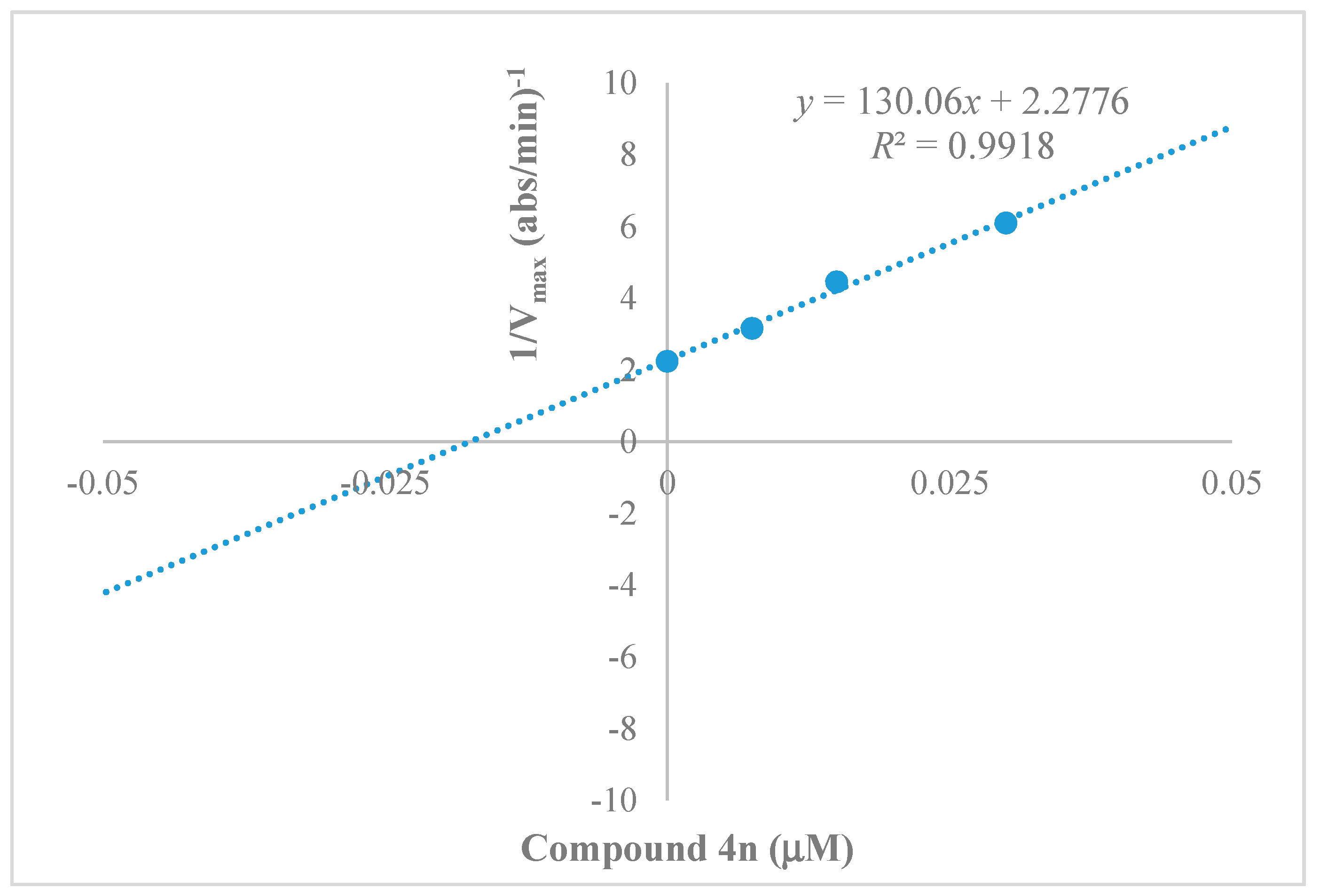
2.3. Kinetics Characterization of BChE Inhibition
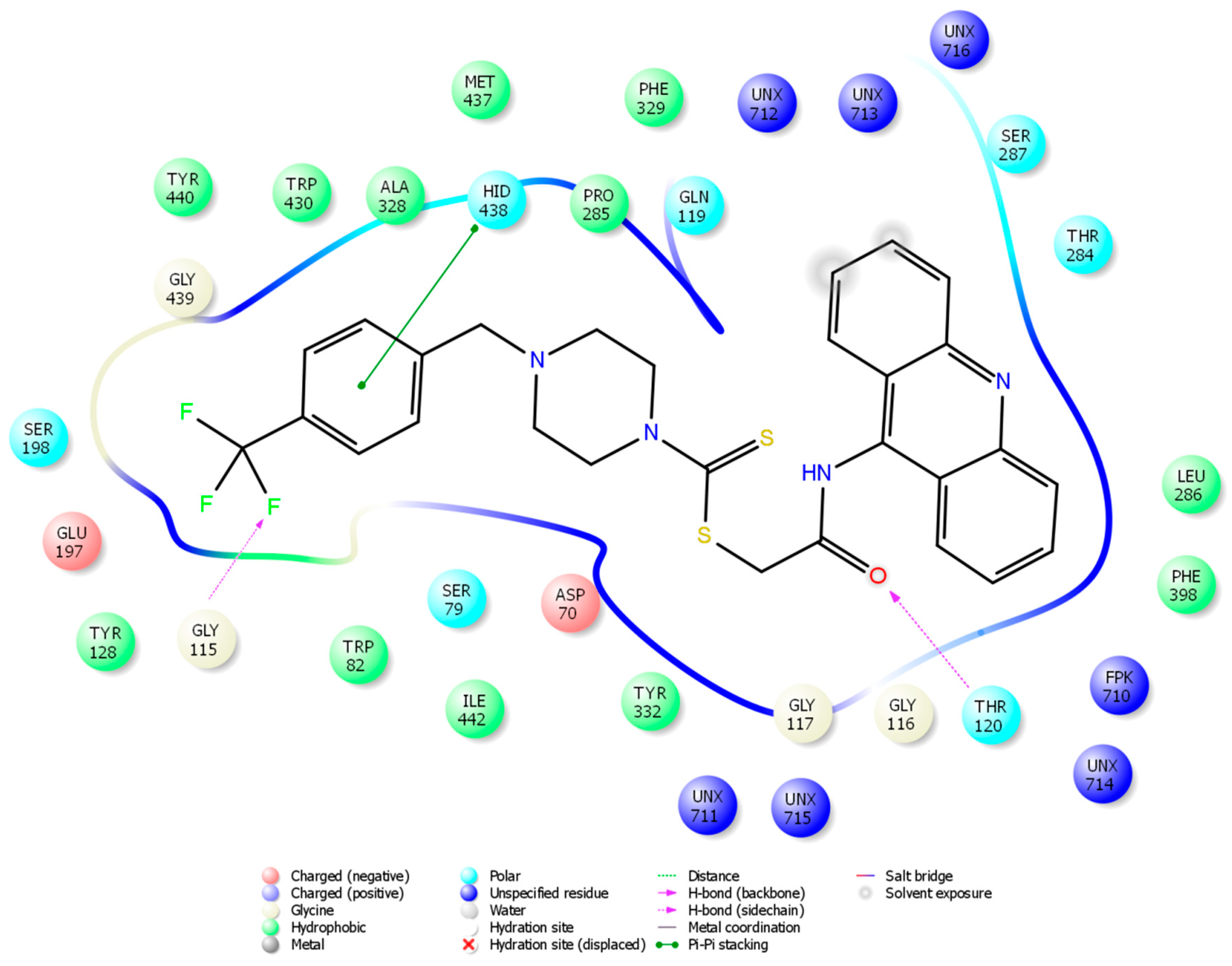
2.4. Molecular Docking
2.5. MTT Cell Viability Assay and Selectivity Indexes
2.6. BBB Permeability and Drug-Likeness Score (DLS)
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure for the Synthesis of N-(9-Acridinyl)-2-Chloroacetamide Derivatives (2)
3.1.2. General Procedure for the Synthesis of Sodium N-Substituted Piperazine/Morpholine/Piperidine Dithiocarbamates (3a–3u)
3.1.3. General Procedure for the Synthesis of 2-(9-Acridinylamino)-2-Oxoethyl 4-Piperazinyl/Morpholinyl/Piperidinylcarbodithioate Derivatives (4a–4u)
3.2. AChE/BChE Activity Assay
- Blank (B): the well in which the inhibitor compound and substrate were not added;
- Control (C): the well where the inhibitor compound was not added;
- A(B): the difference in absorbance reading for the blank;
- A(C): the difference in absorbance reading for the control;
- A(I): the difference in absorbance reading for the inhibitor compounds.
3.3. Enzyme Kinetics
3.4. Molecular Docking
3.5. Cell Viability Assay and Selectivity Indexes
3.6. BBB Permeability and Drug-Likeness Score (DLS)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- World Alzheimer Report 2015. Available online: https://www.alz.co.uk/research/worldalzheimerreport2015summary.pdf (accessed on 26 January 2017).
- Dementia: A Public Health Priority. Available online: http://www.who.int/mental_health/publications/dementia_report_2012/en/ (accessed on 26 January 2017).
- Melnikova, I. Therapies for Alzheimer's disease. Nat. Rev. Drug Discov. 2007, 6, 341–342. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, S. Advances in the pharmacotherapy of Alzheimer’s disease. Can. Med. Assoc. J. 2002, 166, 616–623. [Google Scholar]
- Darvesh, S.; Grantham, D.L.; Hopkins, D.A. Distribution of butyrylcholinesterase in the human amygdala and hippocampal formation. J. Comp. Neurol. 1998, 393, 374–390. [Google Scholar] [CrossRef]
- Decker, M.; Kraus, B.; Heilmann, J. Design, synthesis and pharmacological evaluation of hybrid molecules out of quinazolinimines and lipoic acid lead to highly potent and selective butyrylcholinesterase inhibitors with antioxidant properties. Bioorg. Med. Chem. 2008, 16, 4252–4261. [Google Scholar] [CrossRef] [PubMed]
- Carolan, C.G.; Dillon, G.P.; Khan, D.; Ryder, S.A.; Gaynor, J.M.; Reidy, S.; Marquez, J.F.; Jones, M.; Holland, V.; Gilmer, J.F. Isosorbide-2-benzyl carbamate-5-salicylate, a peripheral anionic site binding subnanomolar selective butyrylcholinesterase inhibitor. J. Med. Chem. 2010, 53, 1190–1199. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, S.A.; Ayaz, M.; Brandt, W.; Wessjohann, L.A.; Westermann, B. Cation-π and π-π stacking interactions allow selective inhibition of butyrylcholinesterase by modified quinine and cinchonidine alkaloids. Biochem. Biophys. Res. Commun. 2011, 404, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Guillozet, A.L.; Smiley, J.F.; Mash, D.C.; Mesulam, M.M. Butyrylcholinesterase in the life cycle of amyloid plaques. Ann. Neurol. 1997, 42, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Podoly, E.; Shalev, D.E.; Shenhar-Tsarfaty, S.; Bennett, E.R.; Ben Assayag, E.; Wilgus, H.; Livnah, O.; Soreq, H. The butyrylcholinesterase K variant confers structurally derived risks for Alzheimer pathology. J. Biol. Chem. 2009, 284, 17170–17179. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.X. Future perspectives of AD research and clinical practice. Med. Hypotheses 2004, 63, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Lemke, T.L.; Williams, D.A.; Roche, V.F.; Zito, S.W. Foye’s Principles of Medicinal Chemistry, 6th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2008. [Google Scholar]
- Madalageri, P.M.; Kotresh, O. Synthesis, DNA protection and antimicrobial activity of some novel chloromethyl benzimidazole derivatives bearing dithiocarbamates. J. Chem. Pharm. Res. 2012, 4, 2697–2703. [Google Scholar]
- Levent, S.; Acar-Çevik, U.A.; Sağlık, B.N.; Özkay, Y.; Can, O.D.; Özkay, U.D.; Uçucu, Ü. Anticholinesterase activity screening of some novel dithiocarbamate derivatives including piperidine and piperazine moieties. Phosphorus Sulfur Silicon Relat. Elem. 2017, 192, 469–474. [Google Scholar] [CrossRef]
- Demir Özkay, Ü.; Can, Ö.D.; Sağlık, B.N.; Acar Çevik, U.; Levent, S.; Özkay, Y.; Ilgın, S.; Atlı, Ö. Design, synthesis, and AChE inhibitory activity of new benzothiazole–piperazines. Bioorg. Med. Chem. Lett. 2016, 26, 5387–5394. [Google Scholar] [CrossRef] [PubMed]
- Korabecny, J.; Andrs, M.; Nepovimova, E.; Dolezal, R.; Babkova, K.; Horova, A.; Malinak, D.; Mezeiova, E.; Gorecki, L.; Sepsova, V.; et al. 7-Methoxytacrine-p-anisidine hybrids as novel dual binding site acetylcholinesterase inhibitors for Alzheimer’s disease treatment. Molecules 2015, 20, 22084–22101. [Google Scholar] [CrossRef] [PubMed]
- Khoury, R.; Patel, K.; Gold, J.; Hinds, S.; Grossberg, G.T. Recent progress in the pharmacotherapy of Alzheimer’s disease. Drugs Aging 2017, 34, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Korabecny, J.; Musilek, K.; Holas, O.; Nepovimova, E.; Jun, D.; Zemek, F.; Opletalova, V.; Patocka, J.; Dohnal, V.; Nachon, F.; et al. Synthesis and in vitro evaluation of N-(bromobut-3-en-2-yl)-7-methoxy-1,2,3,4-tetrahydroacridin-9-amine as a cholinesterase inhibitor with regard to Alzheimer’s disease treatment. Molecules 2010, 15, 8804–8812. [Google Scholar] [CrossRef] [PubMed]
- Panek, D.; Więckowska, A.; Wichur, T.; Bajda, M.; Godyń, J.; Jończyk, J.; Mika, K.; Janockova, J.; Soukup, O.; Knez, D.; et al. Design, synthesis and biological evaluation of new phthalimide and saccharin derivatives with alicyclic amines targeting cholinesterases, beta-secretase and amyloid beta aggregation. Eur. J. Med. Chem. 2017, 125, 676–695. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.Y.; Dai, Y.C.; Li, N.G.; Dong, Z.X.; Gu, T.; Shi, Z.H.; Xue, X.; Tang, Y.P.; Duan, J.A. Novel multitarget-directed tacrine derivatives as potential candidates for the treatment of Alzheimer's disease. J. Enzyme Inhib. Med. Chem. 2017, 32, 572–587. [Google Scholar] [CrossRef] [PubMed]
- Spilovska, K.; Korabecny, J.; Sepsova, V.; Jun, D.; Hrabinova, M.; Jost, P.; Muckova, L.; Soukup, O.; Janockova, J.; Kucera, T.; et al. Novel tacrine-scutellarin hybrids as multipotent anti-Alzheimer’s agents: Design, synthesis and biological evaluation. Molecules 2017, 22, 1006. [Google Scholar] [CrossRef] [PubMed]
- Košak, U.; Brus, B.; Knez, D.; Šink, R.; Žakelj, S.; Trontelj, J.; Pišlar, A.; Šlenc, J.; Gobec, M.; Živin, M.; et al. Development of an in vivo active reversible butyrylcholinesterase inhibitor. Sci. Rep. 2016, 6, 39495. [Google Scholar] [CrossRef] [PubMed]
- Sawatzky, E.; Wehle, S.; Kling, B.; Wendrich, J.; Bringmann, G.; Sotriffer, C.A.; Heilmann, J.; Decker, M. Discovery of highly selective and nanomolar carbamate-based butyrylcholinesterase inhibitors by rational investigation into their inhibition mode. J. Med. Chem. 2016, 59, 2067–2082. [Google Scholar] [CrossRef] [PubMed]
- Krátký, M.; Štěpánková, S.; Vorčáková, K.; Švarcová, M.; Vinsova, J. Novel cholinesterase inhibitors based on o-aromatic N,N-disubstituted carbamates and thiocarbamates. Molecules 2016, 21, 191. [Google Scholar] [CrossRef] [PubMed]
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 2003, 278, 41141–41147. [Google Scholar] [CrossRef] [PubMed]
- Lucier, G.W.; McDaniel, O.S.; Matthews, H.B. Microsomal rat liver UDP glucuronyltransferase. Effects of piperonyl butoxide and other factors on enzyme activity. Arch. Biochem. Biophys. 1971, 145, 520–530. [Google Scholar] [CrossRef]
- Schneider, R.W. Effects of nonpathogenic strains of fusarium oxysporum on celery root infection by F. oxysporum f. sp. apii and a novel use of the Limeweaver-Burk double reciprocal plot technique. Phytopathology 1984, 74, 646. [Google Scholar] [CrossRef]
- Ahmed, M.; Rocha, J.B.; Correa, M.; Mazzanti, C.M.; Zanin, R.F.; Morsch, A.L.; Morsch, V.M.; Schetinger, M.R. Inhibition of two different cholinesterases by tacrine. Chem. Biol. Interact. 2006, 162, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Mai, Y.C.; Li, Y.; Hou, J.Q.; Huang, S.L.; Ou, T.M.; Tan, J.H.; An, L.K.; Li, D.; Gu, L.Q.; et al. Synthesis and evaluation of novel rutaecarpine derivatives and related alkaloids derivatives as selective acetylcholinesterase inhibitors. Eur. J. Med. Chem. 2010, 45, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Mu, C.; Wang, B.; Jin, J. Graveoline analogs exhibiting selective acetylcholinesterase inhibitory activity as potential lead compounds for the treatment of alzheimer’s disease. Molecules 2016, 21, 132. [Google Scholar] [CrossRef] [PubMed]
- Nachon, F.; Carletti, E.; Ronco, C.; Trovaslet, M.; Nicolet, Y.; Jean, L.; Renard, P.Y. Crystal structures of human cholinesterases in complex with huprine W and tacrine: Elements of specificity for anti-Alzheimer’s drugs targeting acetyl- and butyryl-cholinesterase. Biochem. J. 2013, 453, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.T.; Clark, D.E. In Silico Predictions of Blood-Brain Barrier Penetration: Considerations to “Keep in Mind”. J. Pharmacol. Exp. Ther. 2005, 315, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bhatia, P.A.; Daanen, J.F.; Latsaw, S.P.; Rohde, J.; Kolasa, T.; Hakeem, A.A.; Matulenko, M.A.; Nakane, M.; Uchic, M.E.; et al. Synthesis and evaluation of 3-aryl piperidine analogs as potent and efficacious dopamine D4 receptor agonists. Bioorg. Med. Chem. 2005, 13, 4667–4678. [Google Scholar] [CrossRef] [PubMed]
- Demir-Özkay, U.; Özkay, Y.; Can, Ö. Synthesis and analgesic effects of 2-(2-carboxyphenylsulfanyl)-N-(4-substitutedphenyl)acetamide derivatives. Med. Chem. Res. 2011, 20, 152–157. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.J.; Feather-Stone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef] [Green Version]
- Yurttaş, L.; Kaplancıklı, Z.A.; Ozkay, Y. Design, synthesis and evaluation of new thiazole-piperazines as acetylcholinesterase inhibitors. J. Enzyme Inhib. Med. Chem. 2013, 28, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Maestro, version 10.6; Schrödinger, LLC: New York, NY, USA, 2016.
- Schrödinger, version 2016-2; Schrödinger, LLC: New York, NY, USA, 2016.
- LigPrep, version 3.8; Schrödinger, LLC: New York, NY, USA, 2016.
- Glide, version 7.1; Schrödinger, LLC: New York, NY, USA, 2016.
- Berridge, M.V.; Herst, P.M.; Tan, A.S. Tetrazolium dyes as tools in cell biology: New insights into their cellular reduction. Biotechnol. Annu. Rev. 2005, 11, 127–152. [Google Scholar] [PubMed]
- The Blood-Brain Barrier (BBB) Prediction Server—CBLigand. Available online: http://www.cbligand.org/BBB/index.php (accessed on 27 February 2017).
- Drug-Likeness and Molecular Property Prediction. Available online: http://molsoft.com/mprop/ (accessed on 2 March 2017).
Sample Availability: Samples of the compounds 4a–4u are available from the authors. |



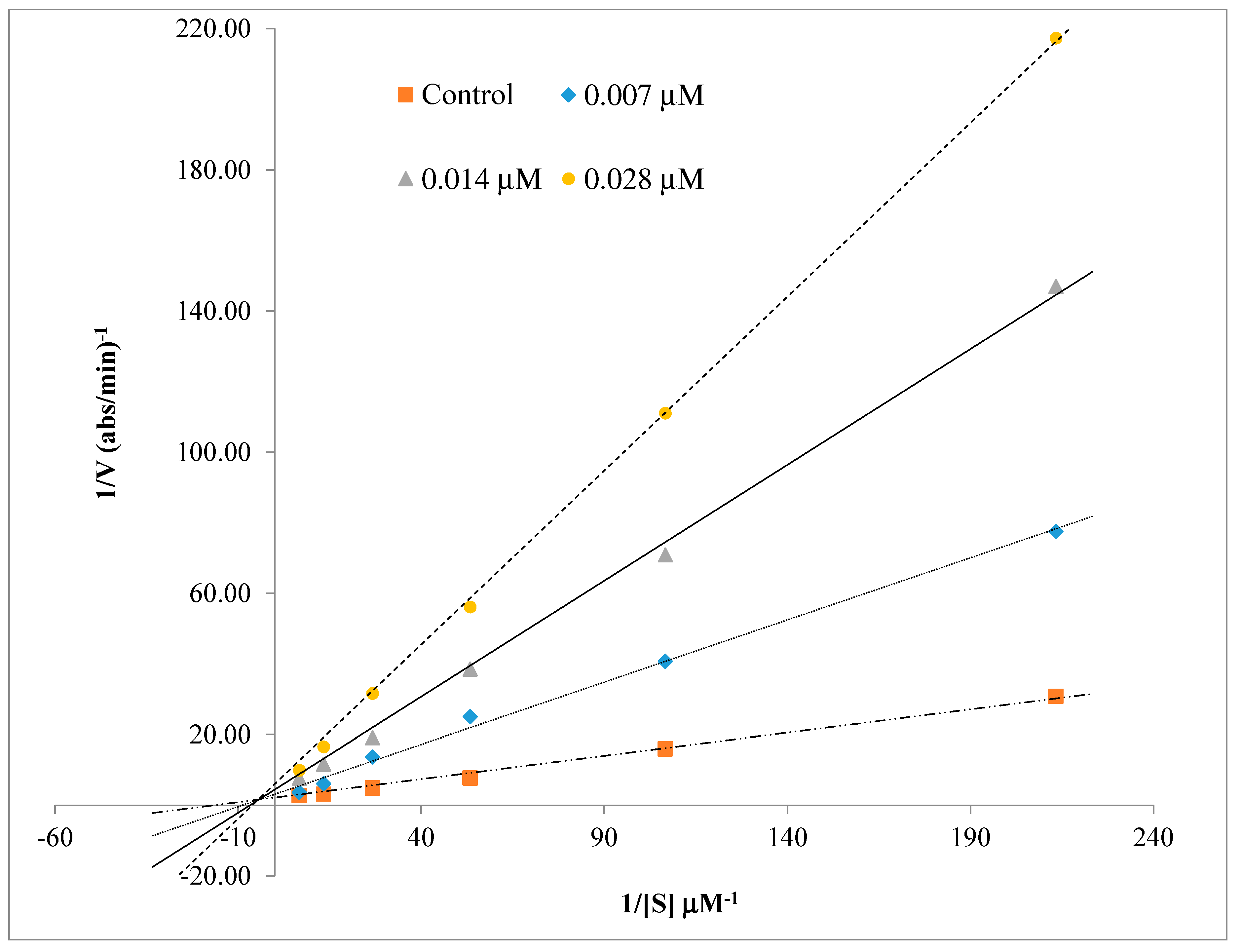



{kind=link}
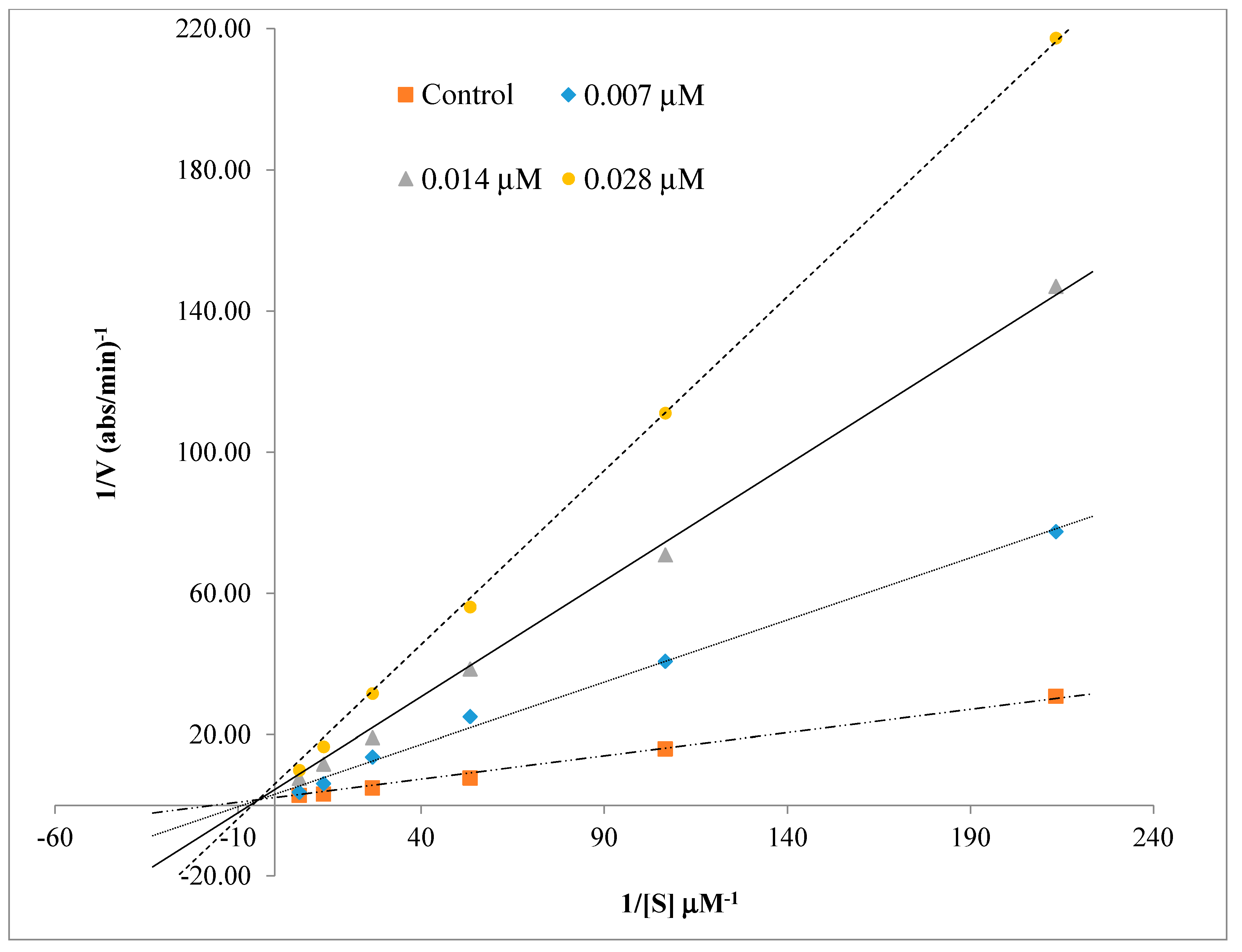{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AChE (Electrophorus electricus) (%) ± SD | AChE (Human) (%) ± SD | BChE (Equine) (%) ± SD | BChE (Human) (%) ± SD | |||||
|---|---|---|---|---|---|---|---|---|
| Compound | 10−4 M | IC50 (µM) | 10−4 M | IC50 (µM) | 10−4 M | IC50 (µM) | 10−4 M | IC50 (µM) |
| 4a | 17.03 ± 0.48 | - | 18.15 ± 0.51 | - | * 91.13 ± 2.94 | 0.226 ± 0.008 | * 93.25 ± 2.24 | 0.243 ± 0.009 |
| 4b | 32.41 ± 1.01 | - | 35.49 ± 1.00 | - | * 82.50 ± 2.07 | 0.264 ± 0.012 | * 84.18 ± 2.21 | 0.222 ± 0.010 |
| 4c | 5.11 ± 0.09 | - | 10.28 ± 0.22 | - | 22.70 ± 0.70 | - | 20.55 ± 0.61 | - |
| 4d | 31.64 ± 0.45 | - | 34.71 ± 0.77 | - | 40.19 ± 0.49 | - | 45.26 ± 0.60 | - |
| 4e | 7.09 ± 0.64 | - | 10.13 ± 0.51 | - | * 95.98 ± 2.88 | 1.420 ± 0.068 | * 96.57 ± 2.03 | 1.373 ± 0.057 |
| 4f | 4.70 ± 0.27 | - | 8.71 ± 0.62 | - | 12.80 ± 0.87 | - | 15.67 ± 0.79 | - |
| 4g | 10.44 ± 0.66 | - | 11.30 ± 0.58 | - | 33.65 ± 0.89 | - | 35.97 ± 0.80 | - |
| 4h | 15.23 ± 0.69 | - | 18.39 ± 0.57 | - | 12.64 ± 0.46 | - | 15.58 ± 0.67 | - |
| 4i | 35.91 ± 1.57 | - | 33.85 ± 1.15 | - | * 69.51 ± 1.16 | 2.038 ± 0.068 | * 72.90 ± 1.08 | 2.097 ± 0.085 |
| 4j | 2.88 ± 0.18 | - | 5.97 ± 0.22 | - | 27.86 ± 1.07 | - | 30.77 ± 1.20 | - |
| 4k | 8.62 ± 0.22 | - | 7.21 ± 0.18 | - | 25.14 ± 1.42 | - | 20.65 ± 1.20 | - |
| 4l | 3.90 ± 0.09 | - | 7.29 ± 0.18 | - | 45.88 ± 1.62 | - | 42.60 ± 1.53 | - |
| 4m | 25.43 ± 1.21 | - | 27.38 ± 1.10 | - | * 98.96 ± 3.47 | 0.092 ± 0.001 | * 97.50 ± 2.06 | 0.093 ± 0.002 |
| 4n | * 63.21 ± 1.76 | 45.945 ± 1.581 | * 68.39 ± 1.99 | 41.129 ± 1.694 | * 99.03 ± 3.26 | 0.015 ± 0.0006 | * 98.25 ± 2.63 | 0.014 ± 0.0005 |
| 4o | 25.30 ± 1.01 | - | 30.45 ± 1.15 | - | * 74.83 ± 2.17 | 1.071 ± 0.051 | * 75.25 ± 2.08 | 1.069 ± 0.039 |
| 4p | 48.40 ± 1.18 | - | 49.51 ± 1.50 | - | 48.65 ± 1.14 | - | 40.71 ± 1.08 | - |
| 4q | 16.35 ± 0.39 | - | 20.28 ± 0.51 | - | 39.90 ± 1.36 | - | 42.48 ± 1.22 | - |
| 4r | 37.33 ± 1.03 | - | 40.70 ± 1.00 | - | 45.30 ± 1.88 | - | 43.29 ± 1.17 | - |
| 4s | 32.17 ± 1.23 | - | 35.20 ± 1.12 | - | 38.40 ± 0.74 | - | 34.99 ± 0.63 | - |
| 4t | 15.94 ± 0.42 | - | 14.51 ± 0.44 | - | * 55.00 ± 1.74 | 1.515 ± 0.072 | * 54.26 ± 1.27 | 1.607 ± 0.062 |
| 4u | 25.88 ± 0.74 | - | 30.71 ± 0.52 | - | 29.50 ± 0.48 | - | 31.21 ± 0.50 | - |
| Aminoacridine | 75.18 ± 1.15 | 1.752 ± 0.072 | 78.22 ± 0.89 | 1.339 ± 0.047 | 87.29 ± 1.07 | 0.233 ± 0.009 | 86.12 ± 1.22 | 0.221 ± 0.007 |
| Donepezil | 98.56 ± 2.87 | 0.0077 ± 0.0003 | 96.95 ± 2.14 | 0.0072 ± 0.0002 | 71.65 ± 2.16 | 1.683 ± 0.064 | 76.50 ± 2.08 | 1.419 ± 0.047 |
| Tacrine | 97.29 ± 3.24 | 0.147 ± 0.004 | 98.79 ± 2.63 | 0.151 ± 0.002 | 98.61 ± 3.71 | 0.0068 ± 0.0002 | 99.15 ± 2.07 | 0.0070 ± 0.0002 |
| Compound | IC50 (µM) |
|---|---|
| 4a | 26.81 |
| 4b | 25.64 |
| 4e | 324.90 |
| 4i | >1000 |
| 4m | 214.18 |
| 4n | >1000 |
| 4o | >1000 |
| Comp. | DLS | BBB Permeability |
|---|---|---|
| 4a | 2.21 | + |
| 4b | 2.16 | + |
| 4e | 1.73 | + |
| 4i | 1.80 | + |
| 4m | 1.88 | + |
| 4n | 1.40 | + |
| 4o | 1.54 | + |
| Tacrine | 0.97 | + |
| Donepezil | 0.91 | + |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussein, W.; Sağlık, B.N.; Levent, S.; Korkut, B.; Ilgın, S.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis and Biological Evaluation of New Cholinesterase Inhibitors for Alzheimer’s Disease. Molecules 2018, 23, 2033. https://doi.org/10.3390/molecules23082033
Hussein W, Sağlık BN, Levent S, Korkut B, Ilgın S, Özkay Y, Kaplancıklı ZA. Synthesis and Biological Evaluation of New Cholinesterase Inhibitors for Alzheimer’s Disease. Molecules. 2018; 23(8):2033. https://doi.org/10.3390/molecules23082033
Chicago/Turabian StyleHussein, Weiam, Begüm Nurpelin Sağlık, Serkan Levent, Büşra Korkut, Sinem Ilgın, Yusuf Özkay, and Zafer Asım Kaplancıklı. 2018. "Synthesis and Biological Evaluation of New Cholinesterase Inhibitors for Alzheimer’s Disease" Molecules 23, no. 8: 2033. https://doi.org/10.3390/molecules23082033
APA StyleHussein, W., Sağlık, B. N., Levent, S., Korkut, B., Ilgın, S., Özkay, Y., & Kaplancıklı, Z. A. (2018). Synthesis and Biological Evaluation of New Cholinesterase Inhibitors for Alzheimer’s Disease. Molecules, 23(8), 2033. https://doi.org/10.3390/molecules23082033