3.5. Experimental Procedures
Ethyl 2-bromo[13C3]propionate (
5): Thionyl chloride (4.63 g, 38.9 mmol, 2.82 mL, 3.00 eq.) and [
13C
3]propionic acid (1.00 g, 12.9 mmol, 1.00 eq.) were heated to reflux for 2.5 h. Bromine (1.56 g, 19.5 mmol, 0.50 mL, 1.50 eq.) was added over the course of 2.5 h and the heating continued overnight. After cooling to 0 °C, ethanol (1.49 g, 32.4 mmol, 1.90 mL, 2.50 eq.) was added during 30 min and stirring continued for 24 h at rt. 60 mL brine were added at 0 °C and the resulting suspension extracted with diethyl ether (3 × 50 mL). The collected organic phases were washed successively with sat. NaHCO
3 (1 × 50 mL), sat. Na
2S
2O
3 (1 × 50 mL) and brine (1 × 50 mL). After drying over Na
2SO
4 and evaporation of the solvent, the crude product was distilled under reduced pressure resulting in a colorless clear oil or directly used in the next reaction.
1H NMR (400 MHz, CDCl
3, 292 K):
δ [ppm] = 4.36 (m,
J = 154.9, 7.0 Hz, 1 H), 4.23 (qdd,
J = 7.2, 3.3, 1.9 Hz, 2 H), 1.82 (ddt,
J = 130.6, 6.9, 4.7 Hz, 3 H), 1.30 (m, 3 H);
13C NMR (101 MHz, CDCl
3, 292 K):
δ [ppm] = 170.4 (
13C, d,
J = 64.2 Hz), 62.1 (s), 40.4 (
13C, dd,
J = 64.2, 36.3 Hz), 21.8 (
13C, d,
J = 36.3 Hz), 14.0 (s). The obtained data of the unlabeled substances matched those reported in [
74].
Potassium phthalimide: Phthalimide (2.00 g, 13.6 mmol, 1.00 eq.) was heated to reflux in ethanol (50 mL) and poured into a solution of potassium hydroxide (0.76 g, 13.6 mmol, 1.00 eq.) in 0.75 mL water and 2.30 ml ethanol. The resulting suspension was cooled to 0 °C and filtered via Buchner funnel. After washing the greenish residuum with ethanol (1 × 25 mL) and aceton (2 × 25 mL), the remaining solvent was evaporated under reduced pressure to obtain the product as greenish crystals (1.96 g, 10.6 mmol, 71%). 1H NMR (400 MHz, D2O, 292 K): δ [ppm] = 7.58–7.31 (m, 4 H).
Ethyl 2-(1,3-dioxoisoindolin-2-yl)[13C3]propionate(6): Potassium phthalimide (4.99 g, 26.9 mmol, 2.50 eq.) was added to a solution of ethyl 2-bromo)[13C3]propionate (1.98 g, 10.7 mmol, 1.00 eq.) in 150 mL acetonitrile and heated to reflux for 24 h. The solvent was removed under reduced pressure and the residue diluted with dichloromethane (75 mL) and water (75 mL). After separation of the phases, the aqueous phase was extracted with dichloromethane (2 × 50 mL). The combined organic phases were washed with water (2 × 40 mL) and brine (1 × 30 mL), dried over Na2SO4 and the solvent evaporated under reduced pressure to obtain the crude product (2.28 mg, 9.11 mmol) as a white solid, which was directly used in the next reaction. 1H-NMR (400 MHz, CDCl3, 292 K): δ [ppm] = 7.87 (m, 2 H), 7.74 (m, 2 H), 5.00 (m, J = 136.6, 7.5, 5.4 Hz, 1 H), 4.21 (qdd, J = 7.1, 3.3, 2.5 Hz, 2 H), 1.69 (ddt, J = 130.5, 7.3, 4.6 Hz, 3 H), 1.23 (t, J = 7.1 Hz, 3 H); 13C NMR (101 MHz, CDCl3, 292 K): δ [ppm] = 169.8 (13C, d, J = 61.9 Hz), 167.5 (s), 134.2 (s), 132.1 (s), 123.6 (s), 62.0 (s), 47.7 (13C, dd, J = 61.8, 37.6 Hz), 15.4 (13C, d, J = 37.5 Hz), 14.2 (s); ESI-MS calcd: [(M + H)+] m/z 251.2, [(M + Na)+] m/z 273.2, found: [(M + H)+] m/z 251.3, [(M + Na)+] m/z 273.0.
[13C3]Alanine hydrochloride (7): Acetic acid (25 mL) and 6 N HCl (130 mL) were added to ethyl 2-(1,3-dioxoisoindolin-2-yl)[13C3]propionate (2.28 mg, 9.11 mmol, 1.00 eq.) and heated to reflux overnight. The solvent was evaporated under reduced pressure and the residue dissolved in water. The aqueous phase was washed with ethyl acetate (3 × 25 mL) and the solvent evaporated under reduced pressure to obtain the product as a white solid (1.33 g, 10.4 mmol, 78%). 1H NMR (400 MHz, D2O, 292 K): δ [ppm] = 4.10 (ddq, J = 146.3, 13.0, 6.9 Hz, 1 H), 1.56 (ddt, J = 130.9, 7.3, 4.5 Hz, 3 H); 13C NMR (101 MHz, D2O, 292 K): δ [ppm] = 172.9 (13C, dd, J = 58.9, 1.5 Hz), 48.8 (13C, dd, J = 59.0, 34.3 Hz), 15.3 (13C, d, J = 34.0 Hz); ESI-MS calcd: [(M − HCl + H)+] m/z 93.1,found: [(M − HCl + H)+] m/z 93.0.
Butyl [13C3]alaninate hydrochloride(8): Thionyl chloride (1.85 g, 15.5 mmol, 1.13 mL, 1.50 eq.) was added to a solution of [
13C
3]alanine hydrochloride (1.33 g, 10.4 mmol, 1.00 eq.) in
n-butanol (100 mL) at 0 °C and heated to reflux for 2 h. The solvent was evaporated under reduced pressure and the residue crystallized from diethyl ether/pentane to receive the product as an off-white solid (1.61 g, 8.70 mmol, 83%).
1H NMR (400 MHz, CDCl
3, 292 K):
δ [ppm] = 8.74 (br, 3 H), 4.22 (m,
J = 146.2, 6.6 Hz, 1 H), 4.19 (m, 2 H), 1.72 (m,
J = 131.1, 7.0, 4.6 Hz, 3 H), 1.64 (dq,
J = 8.7, 6.7 Hz, 2 H), 1.38 (h,
J = 7.4 Hz, 2 H), 0.93 (t,
J = 7.4 Hz, 3 H);
13C NMR (101 MHz, CDCl
3, 292 K):
δ [ppm] = 170.1 (
13C, dd,
J = 62.0, 1.5 Hz), 66.4 (s), 49.4 (
13C, dd,
J = 62.1, 34.1 Hz), 30.5 (s), 19.1 (s), 16.3 (
13C, d,
J = 35.0 Hz), 13.8 (s); ESI-MS calcd: [(M – HCl + H)
+]
m/z 149.1,found: [(M − HCl + H)
+]
m/z 149.0. The obtained data of the unlabeled substances matched those reported in [
62].
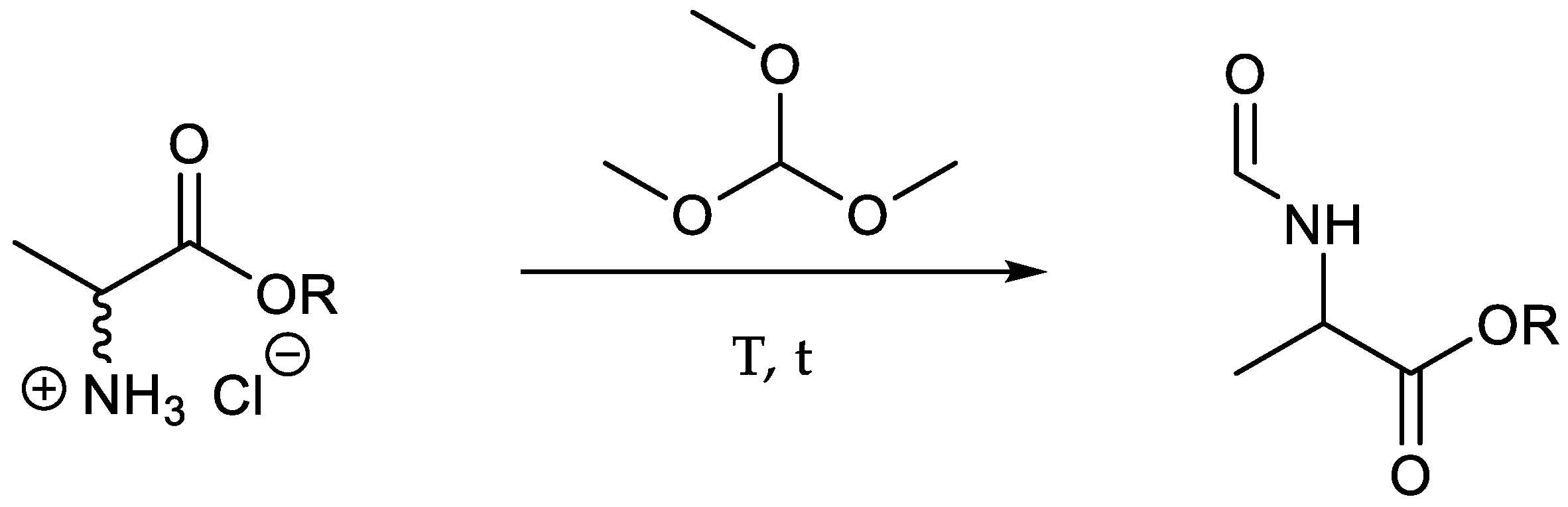
Butyl-N-formyl-[13C3]alaninate (
9): A mixture of butyl [
13C
3]alaninate hydrochloride (1.61 g, 8.70 mmol, 1.00 eq.) and trimethyl orthoformiate (4.62 g, 4.76 mL, 43.5 mmol, 5.00 eq.) was heated to 165 °C for 2 h. The volatile compounds were removed under reduced pressure to obtain the product as a yellow oil (1.24 g, 7.06 mmol, 84%).
1H-NMR (400 MHz, CDCl
3, 292 K):
δ [ppm] = 8.19 (d,
J = 5.1 Hz, 1 H), 6.20 (s, 1 H), 4.67 (ddt,
J = 143.1, 12.4, 7.1 Hz, 1 H), 4.17 (tt,
J = 6.6, 2.8 Hz, 2 H), 1.70–1.56 (m, 2 H + 1.5 H), 1.39 (dq,
J = 14.6, 7.3 Hz, 2 H), 1.28 (dt,
J = 7.1, 4.5 Hz, 1.5 H), 0.94 (t,
J = 7.4 Hz, 3 H);
13C-NMR (101 MHz, CDCl
3, 292 K):
δ [ppm] = 172.8 (
13C, dd,
J = 61.2, 1.2 Hz), 160.4 (s), 65.7 (s), 47.06 (
13C, dd,
J = 61.3, 34.8 Hz), 30.6 (s), 19.1 (s), 18.8 (
13C, dd,
J = 34.9, 1.3 Hz), 13.8 (s); ESI-MS: calcd: [(M + H)
+]
m/z 177.2, found: [(M + H)
+]
m/z 177.2.The obtained data of the unlabeled substances matched those reported in [
43].
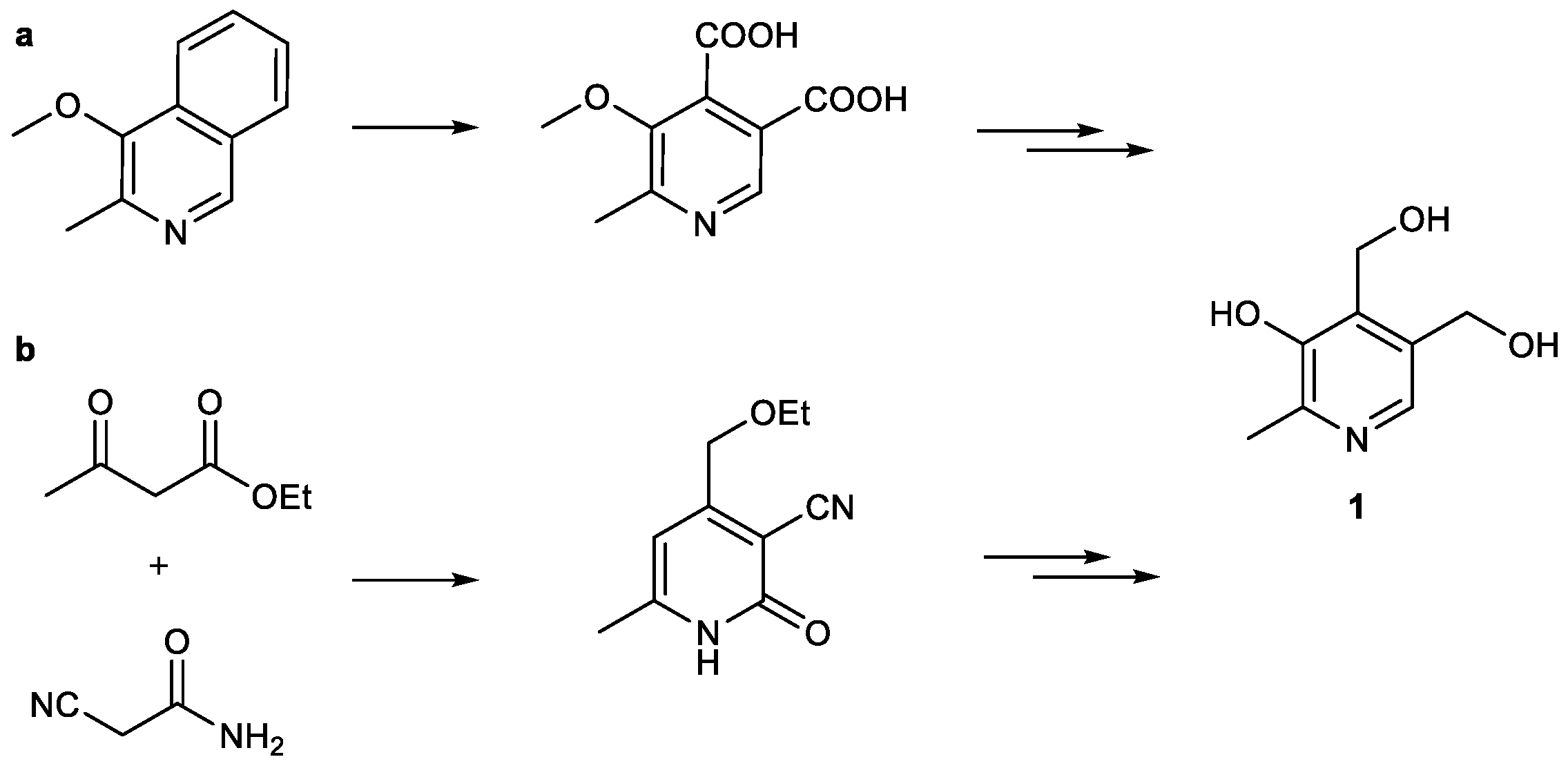
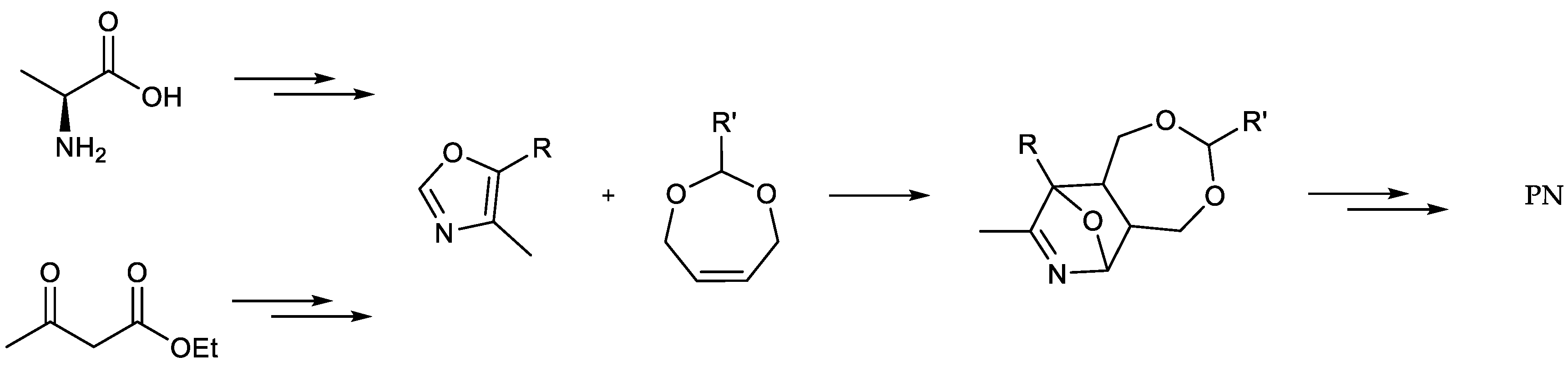
5-Butoxy-4-[13C1]methyl[4,5-13C2]oxazole (
10): A solution of butyl-
N-formyl-[
13C
3]alaninate (1.24 g, 7.06 mmol, 1.00 eq.) in dichloromethane was added to a homogenous mixture of celite (2 g), calcium oxide (2 g) and P
2O
5 (2.5 g, 17.6 mmol, 2.5 eq.) in dichloromethane (100 mL) under inert atmosphere. The mixture was stirred at room temperature for 30 min followed by heating to reflux for 48 h. After the first 24 h, additional 2.5 eq. P
2O
5 were added at rt and the heating was continued. A sat. Aqueous solution of NaHCO
3 was added at 0 °C, the mixture was filtered and the aqueous phase extracted with dichloromethane (3 × 50 mL). The combined organic phases were washed with brine (1 × 25 mL), dried over Na
2SO
4 and the solvent removed under reduced pressure. The crude product was purified
via column chromatography (pentane/diethyl ether = 3/1) to obtain the product (0.68 g, 4.33 mmol, 62%) as yellow oil.
1H-NMR (400 MHz, CDCl
3, 292 K):
δ [ppm] = 7.37 (dd,
J = 6.9, 4.4 Hz, 1 H), 4.08 (td,
J = 6.6, 2.9 Hz, 2 H), 2.04 (ddd,
J = 128, 7.2, 4.6 Hz, 3 H), 1.76–1.64 (m, 2 H), 1.52–1.41 (m, 2 H), 0.96 (t,
J = 7.4 Hz, 3 H);
13C-NMR (101 MHz, CDCl
3, 292 K):
δ [ppm] = 154.6 (
13C, dd,
J = 97.5, 7.2 Hz), 142.4–142.0 (m), 112.0 (
13C, dd,
J = 97.5, 56.4 Hz), 74.4–74.2 (m), 31.5 (d,
J = 2.4 Hz), 18.9 (s), 13.8 (s), 10.1 (
13C, dd,
J = 56.4, 7.2 Hz); ESI-MS calcd: [(M + H)
+]
m/z 159.1,found: [(M + H)
+]
m/z 159.1; TLC:
Rf = 0.34 (pentane/diethyl ether = 3/1 [KMnO
4]).The obtained data of the unlabeled substances matched those reported in [
42].
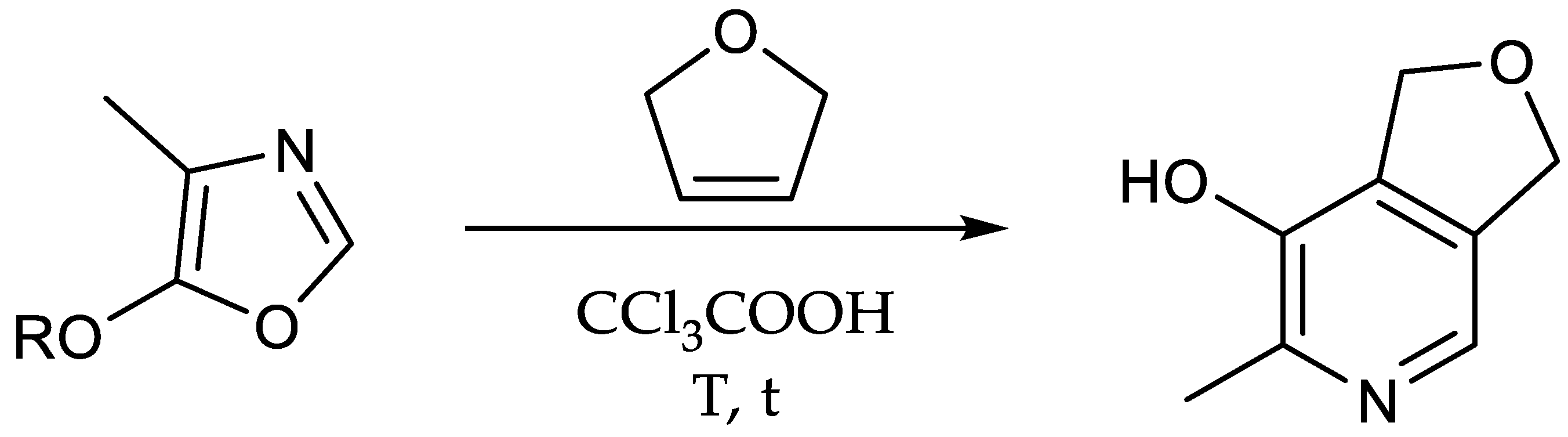
2-[13C1]Methyl-3-hydroxy-4,5-epoxydimethyl[2,3-13C2]pyridine (11): 5-Butoxy-4-[13C1]methyl[4,5-13C2]oxazole (0.68 g, 4.33 mmol, 1.00 eq.), 2,5-dihydrofuran (9.63 mL, 130 mmol, 30.0 eq.) and trichloroacetic acid (0.15 g, 0.95 mmol) were heated to 210 °C for 5 h in a pressure vial and thereafter allowed to cool to room temperature overnight. The reaction mixture was concentrated under reduced pressure and the crude product purified via column chromatography (diethyl ether/methanol = 10/0.5) to obtain the product (0.40 mg, 2.63 mmol, 61%) as brownish solid. 1H-NMR (400 MHz, MeOD, 292 K): δ [ppm] = 7.78 (d, J = 9.0 Hz, 1 H), 5.06 (d, J = 11.0 Hz, 4 H), 2.42 (ddd, J = 127.8, 6.5, 2.8 Hz, 3 H); 13C-NMR (101 MHz, MeOD, 292 K): δ [ppm] = 149.57 (13C, d, J = 66.6 Hz), 145.99 (13C, dd, J = 66.8, 50.6 Hz), 137.16 (s), 137.08 (s), 130.4–130.1 (m), 72.85 (d, J = 2.4 Hz), 72.33 (m), 17.64 (13C, dd, J = 50.6, 4.4 Hz); ESI-MS calcd: [(M + H)+] m/z 155.1, [(M − H)−] m/z 153.0,found: [(M + H)+] m/z 155.1, [(M − H)−] m/z 153.0; TLC: Rf = 0.27 (diethyl ether/methanol = 10/0.5 [KMnO4]).
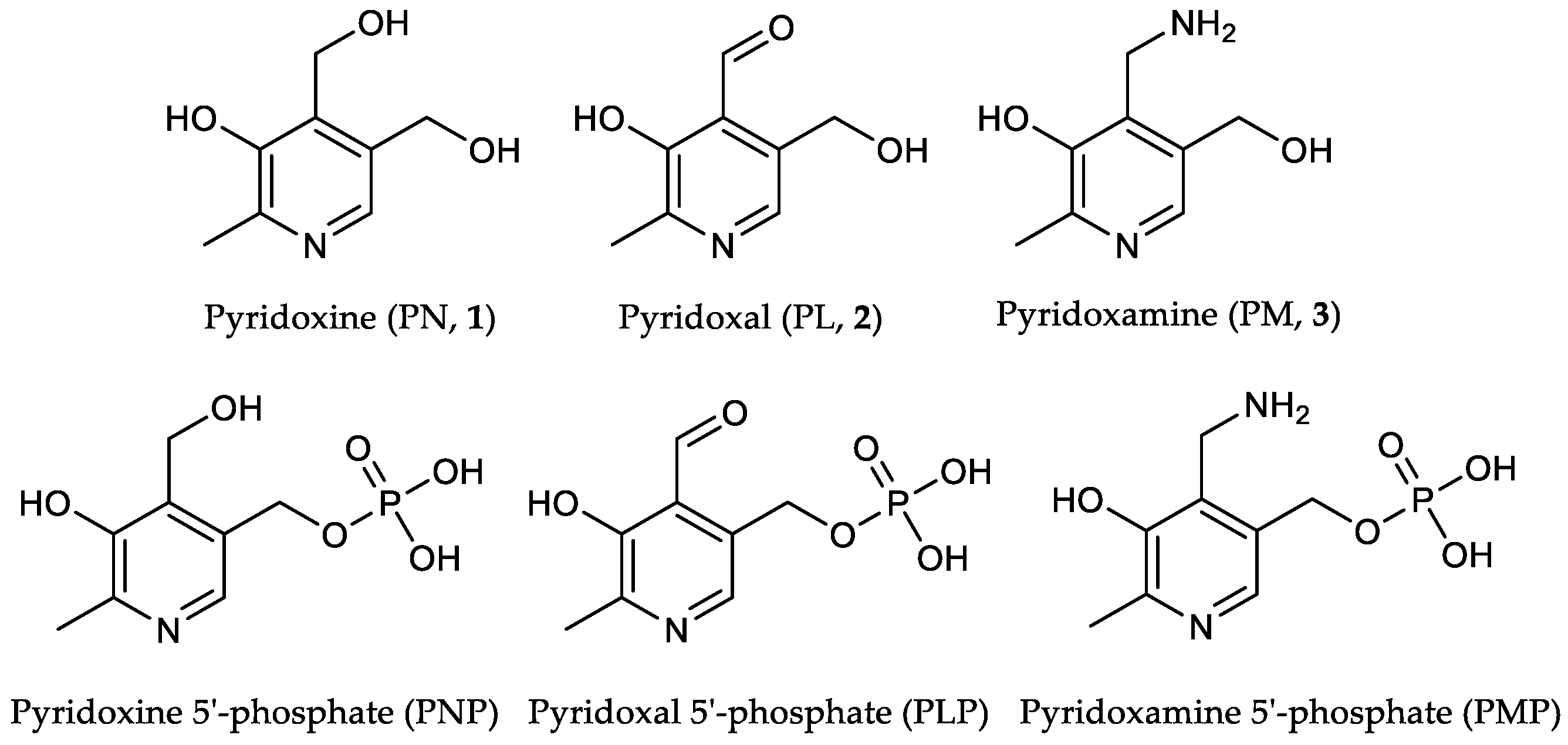
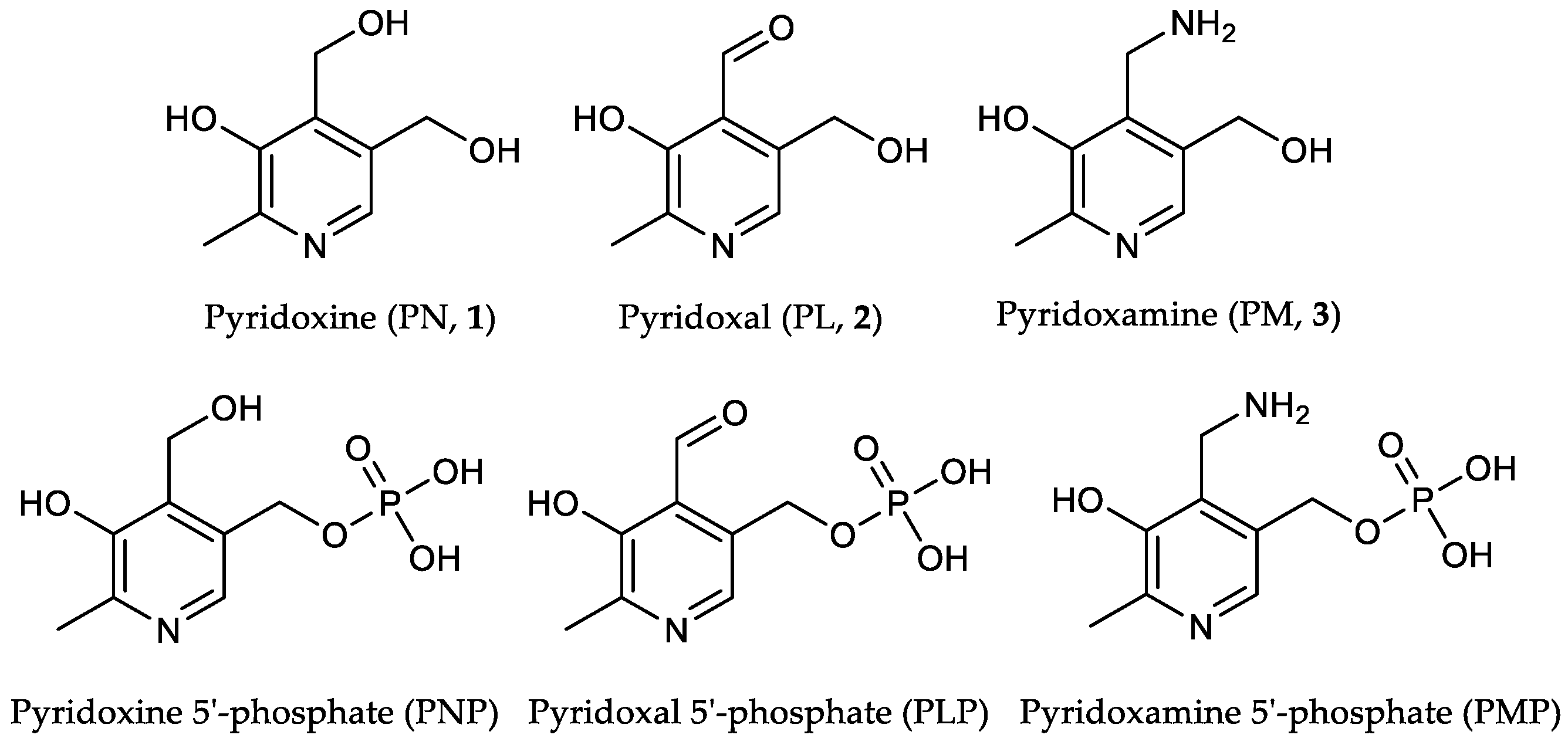
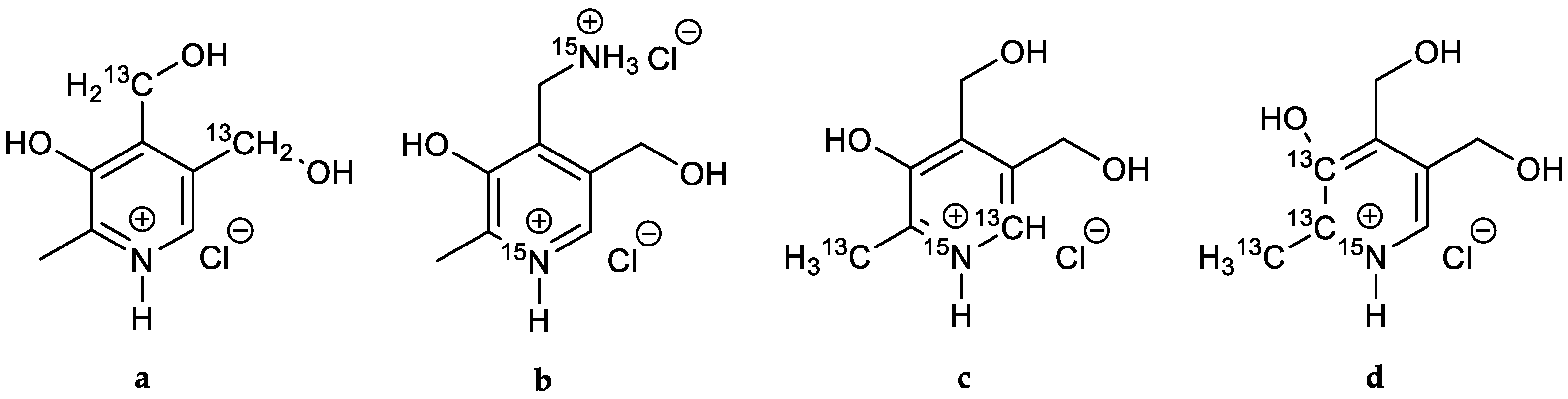
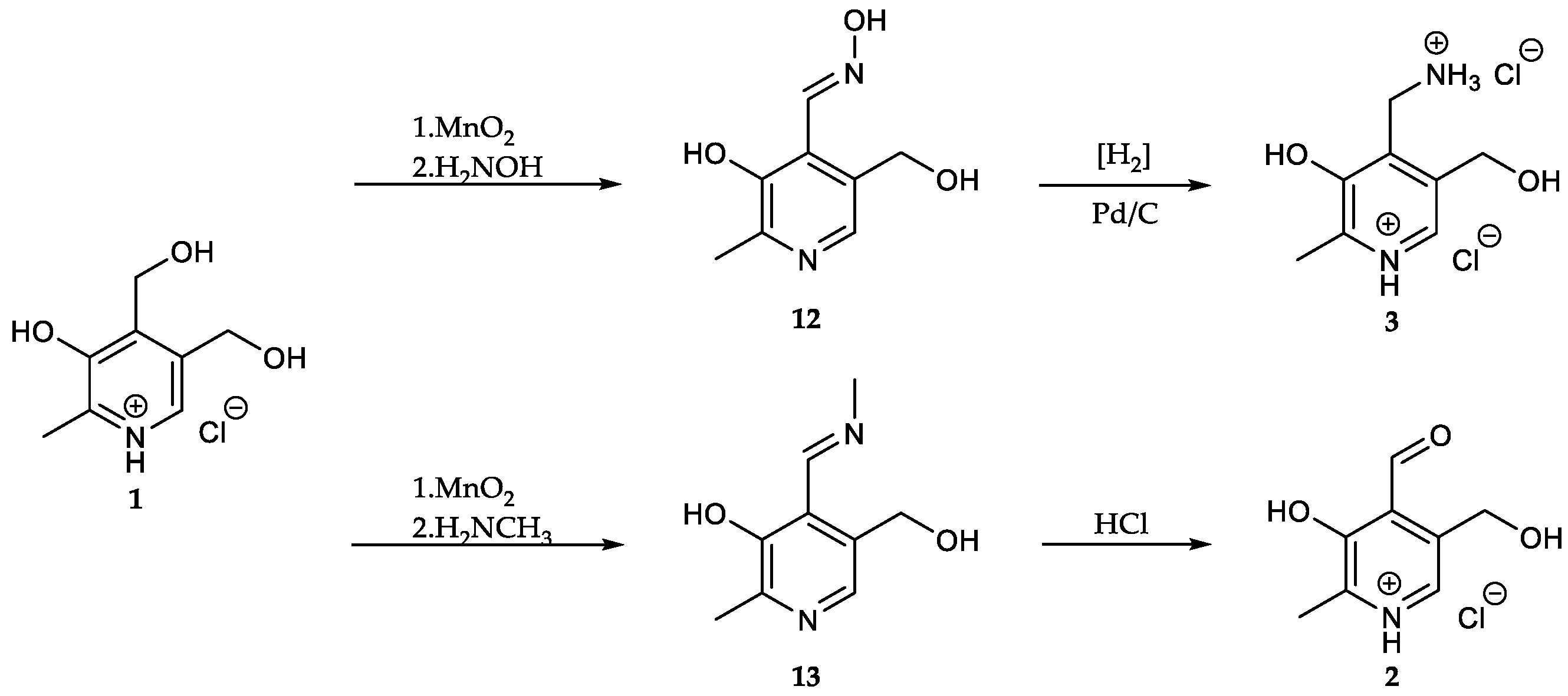
[13C3]Pyridoxine (PN, 1): A solution of 2-[13C1]methyl-3-hydroxy-4,5-epoxydimethyl[2,3-13C2]pyridine (0.40 mg, 2.63 mmol, 1.00 eq.) in 6 mL 48% HBraq. was heated to reflux for one hour. After cooling to rt, the solvent was evaporated under reduced pressure. Water (25 mL) and freshly prepared AgCl (8 g) were added and the reaction heated to reflux for one hour. After filtration over a Celite-Pad and washing with water, the solvent was removed under reduced pressure and the crude product purified via column chromatography (dichloromethane/methanol = 10/1) to obtain an off-white powder (0.45 g, 2.15 mmol, 82%). 1H-NMR (400 MHz, D2O, 292 K): δ [ppm] = 8.16 (d, J = 7.0 Hz, 1 H), 5.00 (d, J = 3.9 Hz, 2 H), 4.80 (s, 2 H), 2.64 (ddd, J = 131.4, 6.8, 3.1 Hz, 3 H); 13C-NMR: (101 MHz, D2O, 292 K): δ [ppm] = 150.5 (13C, dd, J = 72.7, 1.5 Hz), 140.5 (13C, dd, J = 72.9, 46.4 Hz), 138.5, 134.5, 127.4, 55.8 (d, J = 3.5 Hz), 54.5 (d), 12.1 (13C, dd, J = 46.5, 1.6 Hz); ESI-MS calcd: [(M − HCl + H)+] m/z 173.1, [(M − HCl − H)−] m/z 171.1,found: [(M − HCl + H)+] m/z 173.0, [(M – HCl − H)−] m/z 171.5; TLC: Rf = 0.28 (dichloromethane/methanol = 10/1 [KMnO4]).
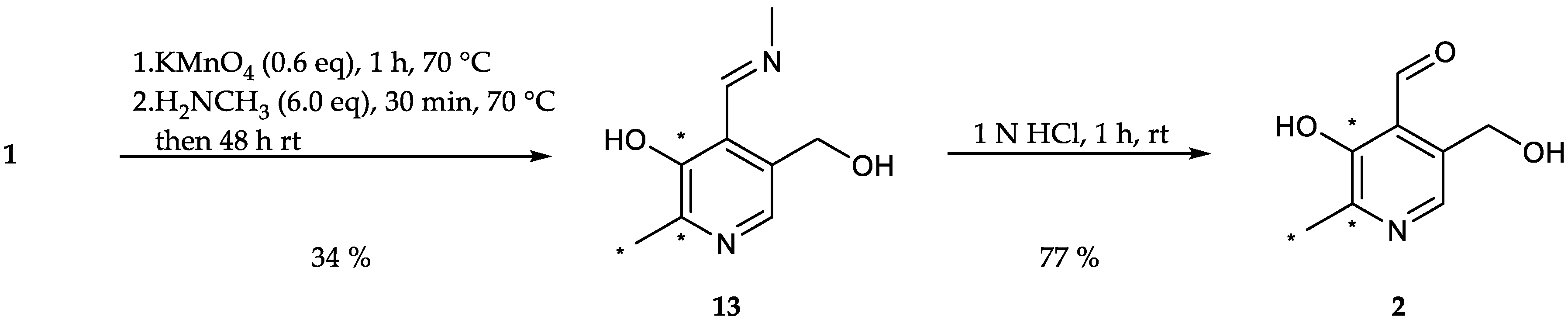
[13C3]N-(Pyridoxylidene)methylamine (13): A solution of potassium permanganate (0.18 g, 1.16 mmol, 0.60 eq.) in distilled water (20 mL) was added portion wise to a solution of [13C3]pyridoxine (0.40 g, 1.93 mmol, 1.00 eq.) in distilled water (10 mL) over the course of 1 h followed by heating to 70 °C for 1 h. Afterwards, the reaction mixture was reduced to 1/3 of its volume under reduced pressure. Methylamine hydrochloride (0.78 g, 11.5 mmol, 6.00 eq.) was added to the suspension and the pH adjusted to 8. The resulting mixture was heated to 70 °C for 30 min and afterwards stirred at room temperature for another 48 hours. The aqueous phase was extracted with dichloromethane (3 × 50 mL). The collected organic phases were dried over Na2SO4 and the solvent evaporated under reduced pressure to obtain the product as yellow-brownish crystals (0.12 g, 0.66 mmol, 34%). 1H-NMR (400 MHz, CDCl3, 292 K): δ [ppm] = 8.86 (dt, J = 5.6, 1.6 Hz, 1 H), 7.83 (d, J = 10.6 Hz, 1 H), 4.78 (s, 2 H), 3.57 (d, J = 1.5 Hz, 3 H), 2.51 (ddd, J = 127.9, 6.6, 2.7 Hz, 3 H); 13C-NMR (101 MHz, CDCl3, 292 K): δ [ppm] = 164.0 (s), 155.4 (13C, dd, J = 65.0, 4.8 Hz), 151.1 (13C, dd, J = 65.0, 51.6 Hz), 137.7 (s), 131.01 (s), 121.1 (s), 60.8 (d, J = 2.0 Hz), 46.3 (s), 19.1 (13C, dd, J = 51.6, 4.7 Hz); ESI-MS calcd: [(M + H)+] m/z 184.1, [(M − H)−] m/z 182.2,found: [(M + H)+] m/z 184.0, [(M − H)−] m/z 182.5.
[13C3]Pyridoxal (PL, 2): [13C3]N-(pyridoxylidene)methylamine (0.11 g, 0.64 mmol, 1.00 eq.) was dissolved in 10 mL 1 N HCl and stirred for 1 h at rt. The crude product was purified via column chromatography (dichloromethane/methanol = 8/1) to obtain 13C3-PL as white solid (0.08 g, 0.49 mmol, 77%). 1H-NMR (400 MHz, D2O, 292 K): δ [ppm] = 8.19 (d, J = 6.5 Hz, 1 H), 6.77 (s, 1 H), 5.36 (d, J = 13.9 Hz, 1 H), 5.22 (d, J = 13.9 Hz, 1 H), 2.68 (ddd, J = 131.6, 6.7, 3.2 Hz, 3 H); 13C-NMR (101 MHz, D2O, 292 K): δ [ppm] = 149.0 (13C, dd, J = 73.1, 1.6 Hz), 143.9 (13C, dd, J = 73.1, 46.0 Hz), 14.1 (13C, dd, J = 46.0, 4.8 Hz); ESI-MS: calcd: [(M+H)+] m/z 171.1, [(M−H)−] m/z 169.1,found: [(M+H)+] m/z 171.0, [(M−H)−] m/z 169.4; TLC: Rf = 0.3 (dichloromethane/methanol = 8/1 [KMnO4]).
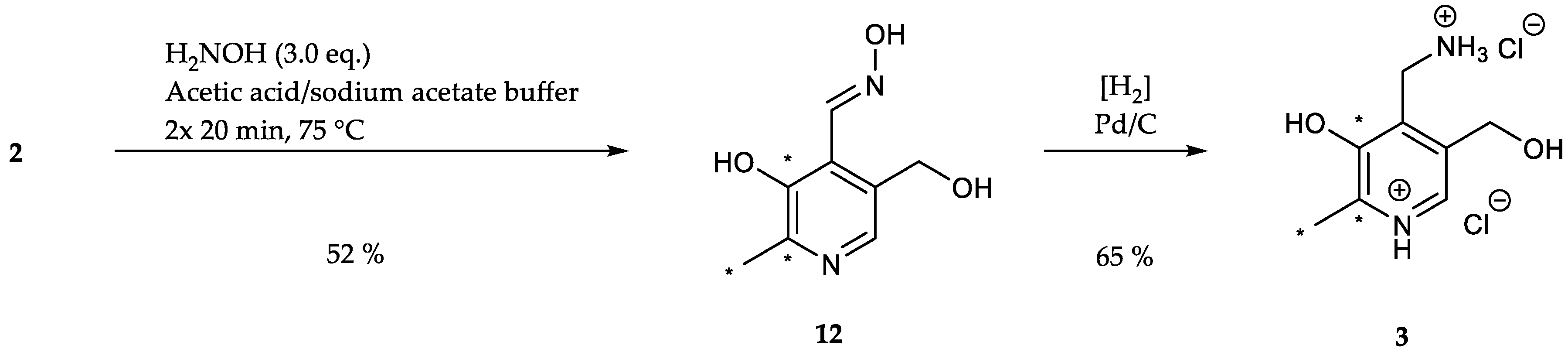
[13C3]N-(Pyridoxylidene)hydroxylamine (12): Hydroxylamine hydrochloride (0.09 g, 1.30 mmol, 3.00 eq.) was added to a solution of [13C3]pyridoxal (74.9 mg, 0.44 mmol, 1.00 eq.) in 5 mL sodium acetate/acetic acid buffer (pH = 4.8). After heating the reaction mixture for 20 min at 75 °C, 1 mL buffer was added followed by a further heating period of 20 min at 75 °C. The crude product was purified via column chromatography (dichloromethane/methanol = 10/1) to obtain the oxime as white solid (42.6 mg, 0.23 mmol, 52%). 1H-NMR (400 MHz, MeOD, 292 K): δ [ppm] = 8.64 (dd, J = 5.8, 1.2 Hz, 1 H), 7.89 (dd, J = 10.7, 1.5 Hz, 1 H), 4.69 (s, 2 H), 2.45 (ddd, J = 127.9, 6.6, 2.8 Hz, 3 H); 13C-NMR (101 MHz, MeOD, 292 K): δ [ppm] = 152.4 (13C, dd, J = 67.9, 4.5 Hz), 148.9 (13C, dd, J = 67.9, 51.0 Hz), 139.3 (s), 133.6 (s), 122.5 (s), 60.4 (d, J = 2.5 Hz), 18.4 (13C, dd, J = 51.0, 4.4 Hz); ESI-MS calcd: [(M+H)+] m/z 186.1, [(M−H)−] m/z 184.5,found: [(M+H)+] m/z 186.1, [(M−H)−] m/z 184.1; TLC: Rf = 0.27 (dichloromethane/methanol = 10/1 [KMnO4]).
[13C3]Pyridoxamine (PM, 3): Palladium/coal (24.8 mg, 30% wt, 0.07 mmol, 0.60 eq.) was added to a solution of [13C3]N-(pyridoxylidene)hydroxylamine (21.4 mg, 0.11 mmol, 1.00 eq.) in 4 mL methanol. The reaction vessel was flushed with H2 until complete conversion of the starting material (indicated through TLC). 0.5 mL 4 N HCl were added and the mixture filtered. After evaporation of the solvent, the crude product was crystallized from methanol/diethyl ether to obtain [13C3]-PM dihydrochloride as white solid (18.2 mg, 0.07 mmol, 65%). 1H-NMR (400 MHz, D2O, 292 K): δ [ppm] = 8.24 (d, J = 7.0 Hz, 1 H), 4.87 (s, 2 H), 4.44 (d, J = 4.3 Hz, 2 H), 2.72 (ddd, J = 131.5, 6.5, 3.3 Hz, 3 H); 13C-NMR (101 MHz, D2O, 292 K): δ [ppm] = 153.8 (13C, d, J = 69.9 Hz), 142.9 (13C, dd, J = 70.1, 46.3 Hz), 58.6 (d, J = 3.2 Hz), 34.6 (s), 15.0 (13C, dd, J = 46.5, 1.9 Hz); ESI-MS calcd: [(M+H)+] m/z 172.1, [(M−H)−] m/z 170.1,found: [(M+H)+] m/z 172.1, [(M−H)−] m/z 170.2.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
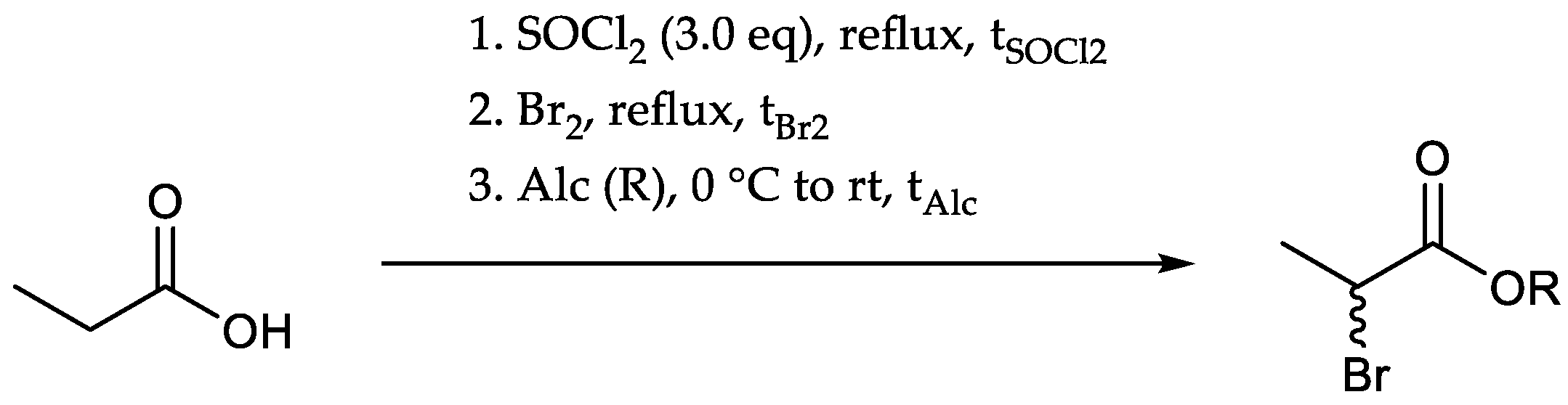{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}