
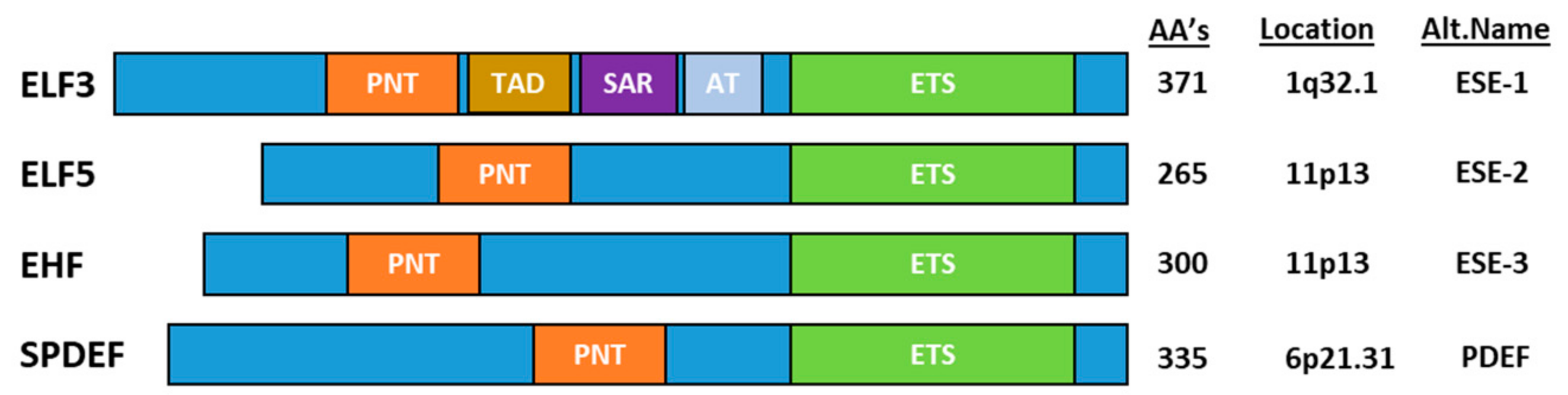
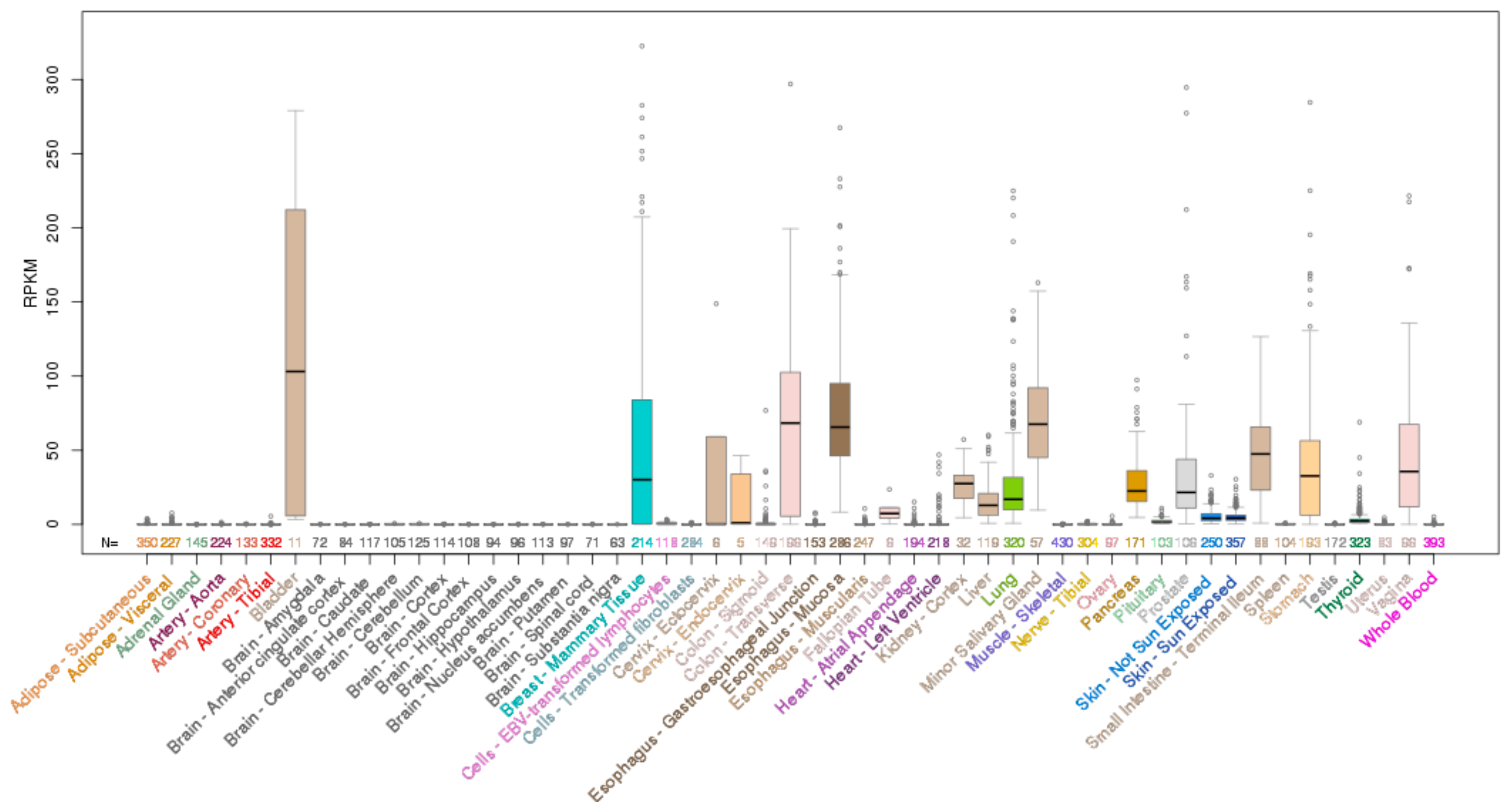
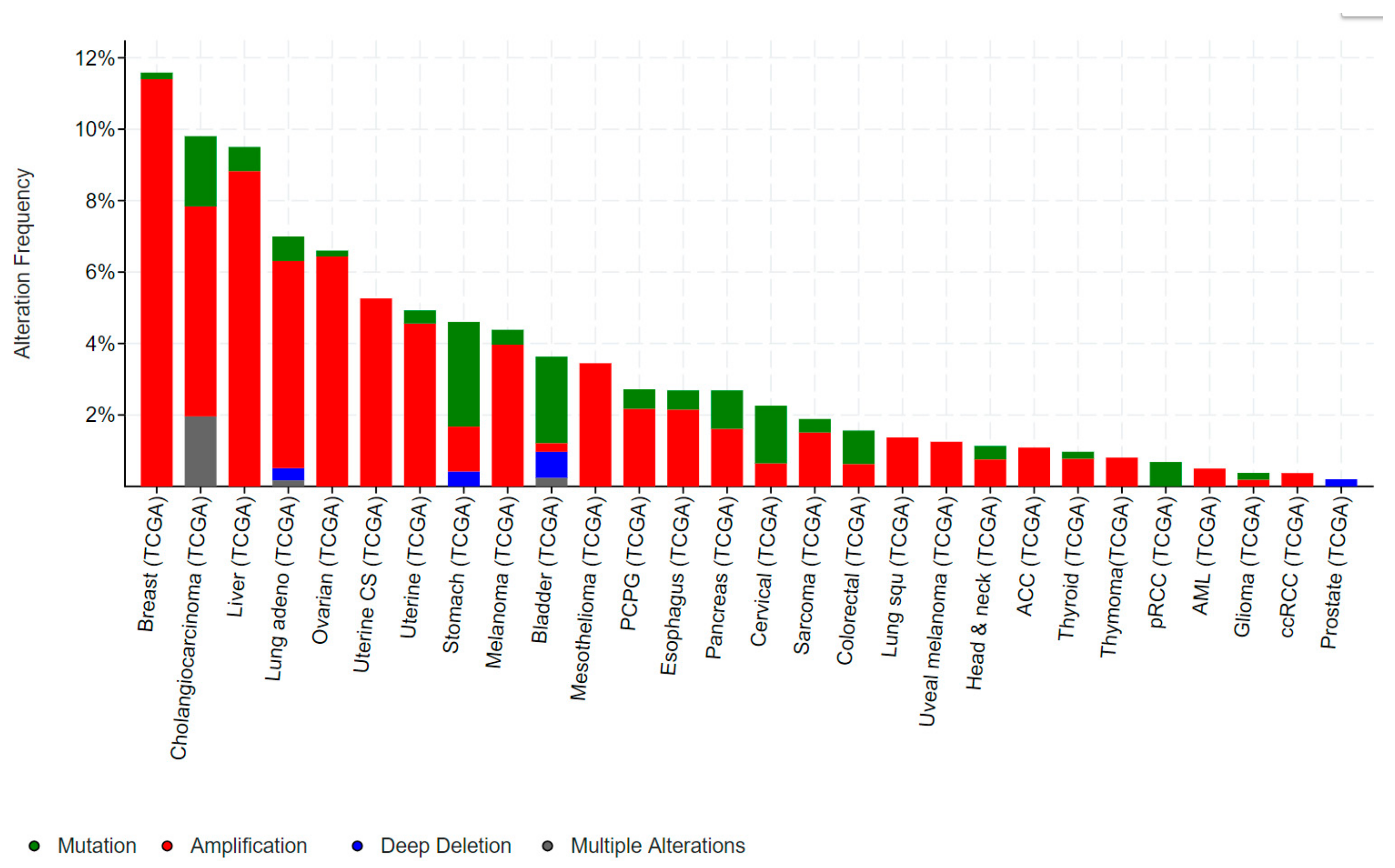
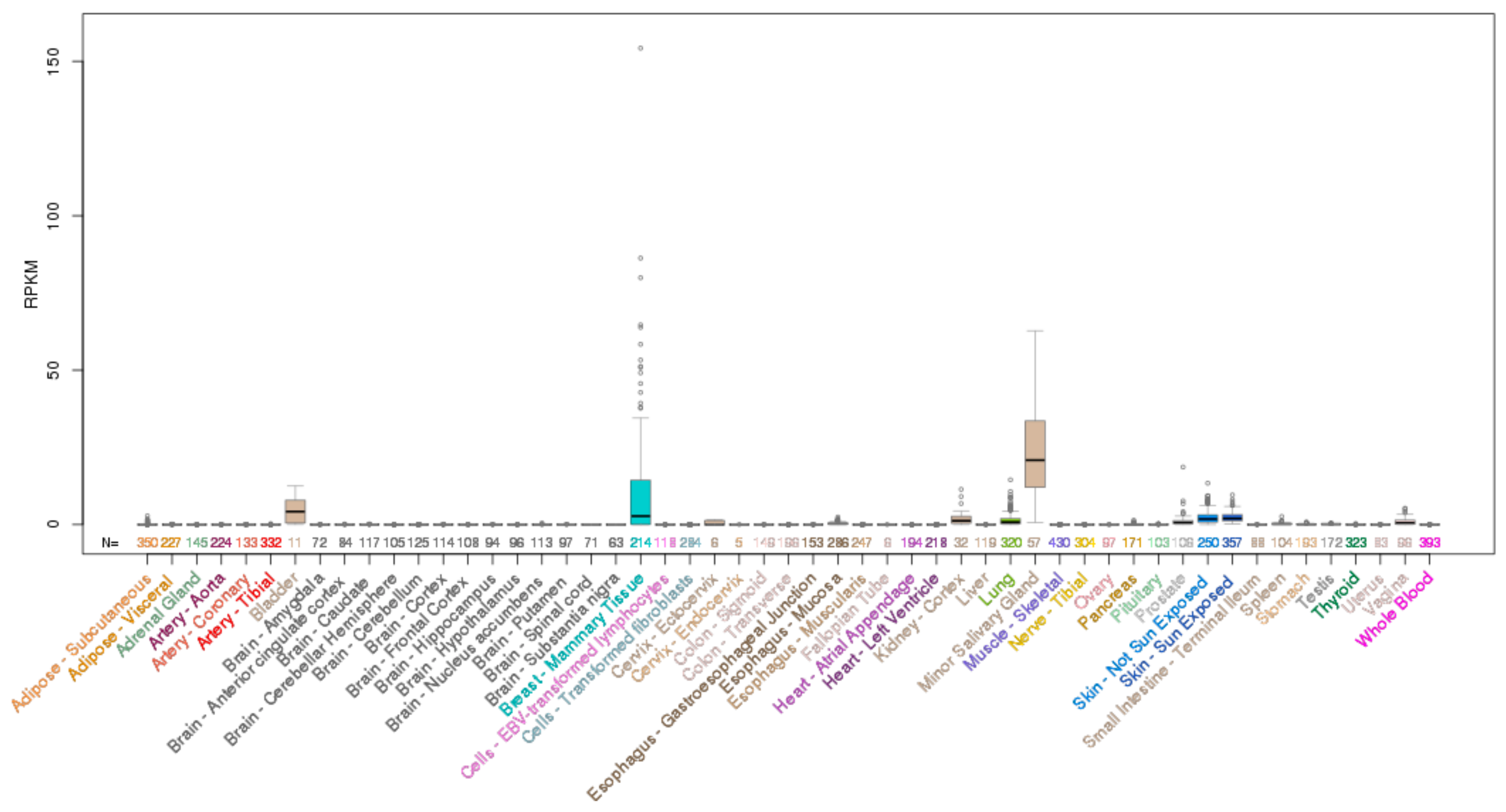
ELF3, ELF5, EHF and SPDEF Transcription Factors in Tissue Homeostasis and Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Role of ELF3 in Normal Tissue Homeostasis
1.1.1. Elf3 Knockout Mice
1.1.2. ELF3 in the Intestinal Epithelium
1.1.3. ELF3 in the Lung Epithelium
1.1.4. ELF3 in the Urothelium
1.1.5. ELF3 in Squamous Epithelium
1.1.6. ELF3 in Non-Epithelial Cells
1.2. Role of ELF3 in Cancer
1.2.1. ELF3 in Bladder Cancer
1.2.2. ELF3 in Ovarian Cancer (OC)
1.2.3. ELF3 in Biliary Tract Cancers
1.2.4. ELF3 in Gastric and Colorectal Cancer
1.2.5. ELF3 in Cervical Cancer
1.2.6. ELF3 in Breast Cancer
1.2.7. ELF3 in Prostate Cancer
1.2.8. ELF3 in Lung Cancer
1.2.9. ELF3 in Hepatocellular Cancer (HCC)
1.3. Role of ELF5 in Normal Tissue Homeostasis
1.3.1. Elf5 Knockout Mice
1.3.2. ELF5 in the Mammary Epithelium
1.3.3. ELF5 in the Kidney
1.3.4. ELF5 in the Skin
1.4. Role of ELF5 in Cancer
1.4.1. ELF5 in Breast Cancer
1.4.2. ELF5 in Prostate, Urothelial, Ovarian and Renal Cancer
1.5. Role of EHF in Normal Tissue Homeostasis
1.5.1. EHF in the Airway Epithelium
1.5.2. EHF in the Skin
1.5.3. EHF in the Intestinal Epithelium
1.5.4. EHF in Non-Epithelial Cells
1.6. Role of EHF in Cancer
1.6.1. EHF in Gastric and Colorectal Cancer.
1.6.2. EHF in Thyroid Cancer
1.6.3. EHF in Ovarian Cancer
1.6.4. EHF in Prostate Cancer
1.6.5. EHF in Pancreatic and Oesophageal Cancer
1.7. Role of SPDEF in Normal Tissue Homeostasis
1.7.1. Spdef Knockout Mice
1.7.2. SPDEF in the Prostate Epithelium
1.7.3. Role of SPDEF in Cancer
1.7.4. SPDEF in Colorectal Cancer
1.7.5. SPDEF in Hepatocellular Cancer
1.7.6. SPDEF in Bladder Cancer
1.7.7. SPDEF in Prostate Cancer
1.7.8. SPDEF in Ovarian Cancer
1.7.9. SPDEF in Breast Cancer
1.8. ESE Transcription Factors as Therapeutic Targets in Cancer
1.8.1. Direct Targeting
1.8.2. Indirect Targeting
1.8.3. Targeting Pathways Altered by ESE Factors in Cancer
1.8.4. Re-inducing Expression of ESE Factors
2. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sizemore, G.M.; Pitarresi, J.R.; Balakrishnan, S.; Ostrowski, M.C. The ETS family of oncogenic transcription factors in solid tumours. Nat. Rev. Cancer 2017, 17, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Sharrocks, A.D. The ETS-domain transcription factor family. Nat. Rev. Mol. Cell. Biol. 2001, 2, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Seidel, J.J.; Graves, B.J. An ERK2 docking site in the Pointed domain distinguishes a subset of ETS transcription factors. Genes Dev. 2002, 16, 127–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.S.; Sinha, S. Determination of the consensus DNA-binding sequence and a transcriptional activation domain for ESE-2. Biochem. J. 2006, 398, 497–507. [Google Scholar] [CrossRef] [Green Version]
- Feldman, R.J.; Sementchenko, V.I.; Watson, D.K. The epithelial-specific Ets factors occupy a unique position in defining epithelial proliferation, differentiation and carcinogenesis. Anticancer Res. 2003, 23, 2125–2131. [Google Scholar] [PubMed]
- Archer, L.K.; Frame, F.M.; Maitland, N.J. Stem cells and the role of ETS transcription factors in the differentiation hierarchy of normal and malignant prostate epithelium. J. Steroid Biochem. Mol. Biol. 2017, 166, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Oliver, J.R.; Kushwah, R.; Hu, J. Multiple roles of the epithelium-specific ETS transcription factor, ESE-1, in development and disease. Lab. Invest. 2012, 92, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.Y.; Waring, P.; Ristevski, S.; Wang, C.; Wilson, T.; Pritchard, M.; Hertzog, P.; Kola, I. Inactivation of the transcription factor Elf3 in mice results in dysmorphogenesis and altered differentiation of intestinal epithelium. Gastroenterology 2002, 122, 1455–1466. [Google Scholar] [CrossRef] [PubMed]
- Flentjar, N.; Chu, P.Y.; Ng, A.Y.; Johnstone, C.N.; Heath, J.K.; Ernst, M.; Hertzog, P.J.; Pritchard, M.A. TGF-betaRII rescues development of small intestinal epithelial cells in Elf3-deficient mice. Gastroenterology 2007, 132, 1410–1419. [Google Scholar] [CrossRef] [PubMed]
- Oliver, J.R.; Kushwah, R.; Wu, J.; Pan, J.; Cutz, E.; Yeger, H.; Waddell, T.K.; Hu, J. Elf3 plays a role in regulating bronchiolar epithelial repair kinetics following Clara cell-specific injury. Lab. Invest. 2011, 91, 1514–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, M.; Hinley, J.; Schmitt, C.; Wahlicht, T.; Kramer, S.; Southgate, J. Identification of ELF3 as an early transcriptional regulator of human urothelium. Dev. Biol. 2014, 386, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Oettgen, P.; Alani, R.M.; Barcinski, M.A.; Brown, L.; Akbarali, Y.; Boltax, J.; Kunsch, C.; Munger, K.; Libermann, T.A. Isolation and characterization of a novel epithelium-specific transcription factor, ESE-1, a member of the ets family. Mol. Cell. Biol. 1997, 17, 4419–4433. [Google Scholar] [CrossRef] [PubMed]
- Brembeck, F.H.; Opitz, O.G.; Libermann, T.A.; Rustgi, A.K. Dual function of the epithelial specific ets transcription factor, ELF3, in modulating differentiation. Oncogene 2000, 19, 1941–1949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, N.; Yoshida, S.; Araie, M.; Handa, H.; Nabeshima, Y. Ets family transcription factor ESE-1 is expressed in corneal epithelial cells and is involved in their differentiation. Mech. Dev. 2000, 97, 27–34. [Google Scholar] [CrossRef]
- Zhan, Y.; Yuan, L.; Kondo, M.; Oettgen, P. The counter-regulatory effects of ESE-1 during angiotensin II-mediated vascular inflammation and remodeling. Am. J. Hypertens. 2010, 23, 1312–1317. [Google Scholar] [CrossRef] [PubMed]
- Otero, M.; Plumb, D.A.; Tsuchimochi, K.; Dragomir, C.L.; Hashimoto, K.; Peng, H.; Olivotto, E.; Bevilacqua, M.; Tan, L.; Yang, Z.; et al. E74-like factor 3 (ELF3) impacts on matrix metalloproteinase 13 (MMP13) transcriptional control in articular chondrocytes under proinflammatory stress. J. Biol. Chem. 2012, 287, 3559–3572. [Google Scholar] [CrossRef] [PubMed]
- Wondimu, E.B.; Culley, K.L.; Quinn, J.; Chang, J.; Dragomir, C.L.; Plumb, D.A.; Goldring, M.B.; Otero, M. Elf3 Contributes to Cartilage Degradation in vivo in a Surgical Model of Post-Traumatic Osteoarthritis. Sci. Rep. 2018, 8, 6438. [Google Scholar] [CrossRef] [PubMed]
- Kushwah, R.; Oliver, J.R.; Wu, J.; Chang, Z.; Hu, J. Elf3 regulates allergic airway inflammation by controlling dendritic cell-driven T cell differentiation. J. Immunol. 2011, 187, 4639–4653. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, G.; Sun, X.; Chen, C.; Wu, S.; Huang, P.; Li, Z.; Dean, M.; Huang, Y.; Jia, W.; Zhou, Q.; et al. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat. Genet. 2013, 45, 1459–1463. [Google Scholar] [CrossRef] [PubMed]
- Dadhania, V.; Zhang, M.; Zhang, L.; Bondaruk, J.; Majewski, T.; Siefker-Radtke, A.; Guo, C.C.; Dinney, C.; Cogdell, D.E.; Zhang, S.; et al. Meta-Analysis of the Luminal and Basal Subtypes of Bladder Cancer and the Identification of Signature Immunohistochemical Markers for Clinical Use. EBioMedicine 2016, 12, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Kardos, J.; Chai, S.; Mose, L.E.; Selitsky, S.R.; Krishnan, B.; Saito, R.; Iglesia, M.D.; Milowsky, M.I.; Parker, J.S.; Kim, W.Y.; et al. Claudin-low bladder tumors are immune infiltrated and actively immune suppressed. JCI Insight 2016, 1, e85902. [Google Scholar] [CrossRef] [PubMed]
- Ryland, G.L.; Hunter, S.M.; Doyle, M.A.; Caramia, F.; Li, J.; Rowley, S.M.; Christie, M.; Allan, P.E.; Stephens, A.N.; Bowtell, D.D.; et al. Mutational landscape of mucinous ovarian carcinoma and its neoplastic precursors. Genome Med. 2015, 7, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, T.L.; Leung, C.S.; Wong, K.K.; Gutierrez-Hartmann, A.; Kwong, J.; Gershenson, D.M.; Mok, S.C. ELF3 is a negative regulator of epithelial-mesenchymal transition in ovarian cancer cells. Oncotarget 2017, 8, 16951–16963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Farshidfar, F.; Zheng, S.; Gingras, M.C.; Newton, Y.; Shih, J.; Robertson, A.G.; Hinoue, T.; Hoadley, K.A.; Gibb, E.A.; Roszik, J.; et al. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell. Rep. 2017, 19, 2878–2880. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, Z.; Li, X.; Ye, J.; Wu, X.; Tan, Z.; Liu, C.; Shen, B.; Wang, X.A.; Wu, W.; et al. Whole-exome and targeted gene sequencing of gallbladder carcinoma identifies recurrent mutations in the ErbB pathway. Nat. Genet. 2014, 46, 872–876. [Google Scholar] [CrossRef] [PubMed]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Huang, M.N.; Padmanabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.W.T.; Ng, L.M.; et al. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma. Cancer Discov. 2017, 7, 1116–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yachida, S.; Wood, L.D.; Suzuki, M.; Takai, E.; Totoki, Y.; Kato, M.; Luchini, C.; Arai, Y.; Nakamura, H.; Hama, N.; et al. Genomic Sequencing Identifies ELF3 as a Driver of Ampullary Carcinoma. Cancer Cell 2016, 29, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Gingras, M.C.; Covington, K.R.; Chang, D.K.; Donehower, L.A.; Gill, A.J.; Ittmann, M.M.; Creighton, C.J.; Johns, A.L.; Shinbrot, E.; Dewal, N.; et al. Ampullary Cancers Harbor ELF3 Tumor Suppressor Gene Mutations and Exhibit Frequent WNT Dysregulation. Cell. Rep. 2016, 14, 907–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.L.; Chen, Z.F.; Chen, H.M.; Wang, M.Y.; Kong, X.; Wang, Y.C.; Sun, T.T.; Hong, J.; Zou, W.; Xu, J.; et al. Elf3 drives beta-catenin transactivation and associates with poor prognosis in colorectal cancer. Cell Death Dis. 2014, 5, e1263. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Lee, S.H. Identification of ESE1 as a beta-Catenin Binding Protein. Anticancer Res. 2016, 36, 2697–2703. [Google Scholar] [PubMed]
- Ojesina, A.I.; Lichtenstein, L.; Freeman, S.S.; Pedamallu, C.S.; Imaz-Rosshandler, I.; Pugh, T.J.; Cherniack, A.D.; Ambrogio, L.; Cibulskis, K.; Bertelsen, B.; et al. Landscape of genomic alterations in cervical carcinomas. Nature. 2014, 506, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Gajulapalli, V.N.; Samanthapudi, V.S.; Pulaganti, M.; Khumukcham, S.S.; Malisetty, V.L.; Guruprasad, L.; Chitta, S.K.; Manavathi, B. A transcriptional repressive role for epithelial-specific ETS factor ELF3 on oestrogen receptor alpha in breast cancer cells. Biochem. J. 2016, 473, 1047–1061. [Google Scholar] [CrossRef] [PubMed]
- Merino, V.F.; Nguyen, N.; Jin, K.; Sadik, H.; Cho, S.; Korangath, P.; Han, L.; Foster, Y.M.N.; Zhou, X.C.; Zhang, Z.; et al. Combined Treatment with Epigenetic, Differentiating, and Chemotherapeutic Agents Cooperatively Targets Tumor-Initiating Cells in Triple-Negative Breast Cancer. Cancer Res. 2016, 76, 2013–2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.H.; Scott, G.K.; Kuo, W.L.; Xiong, X.; Suzdaltseva, Y.; Park, J.W.; Sayre, P.; Erny, K.; Collins, C.; Gray, J.W.; et al. ESX: A structurally unique Ets overexpressed early during human breast tumorigenesis. Oncogene 1997, 14, 1617–1622. [Google Scholar] [CrossRef] [PubMed]
- Neve, R.M.; Ylstra, B.; Chang, C.H.; Albertson, D.G.; Benz, C.C. ErbB2 activation of ESX gene expression. Oncogene 2002, 21, 3934–3938. [Google Scholar] [CrossRef] [PubMed]
- Kar, A.; Gutierrez-Hartmann, A. ESE-1/ELF3 mRNA expression associates with poor survival outcomes in HER2(+) breast cancer patients and is critical for tumorigenesis in HER2(+) breast cancer cells. Oncotarget 2017, 8, 69622–69640. [Google Scholar] [CrossRef] [PubMed]
- Kar, A.; Liu, B.; Gutierrez-Hartmann, A. ESE-1 Knockdown Attenuates Growth in Trastuzumab-resistant HER2(+) Breast Cancer Cells. Anticancer Res. 2017, 37, 6583–6591. [Google Scholar] [PubMed]
- Longoni, N.; Sarti, M.; Albino, D.; Civenni, G.; Malek, A.; Ortelli, E.; Pinton, S.; Mello-Grand, M.; Ostano, P.; D’Ambrosio, G.; et al. ETS Transcription Factor ESE1/ELF3 Orchestrates a Positive Feedback Loop That Constitutively Activates NF-κB and Drives Prostate Cancer Progression. Cancer Res. 2013, 73, 4533–4547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shatnawi, A.; Norris, J.D.; Chaveroux, C.; Jasper, J.S.; Sherk, A.B.; McDonnell, D.P.; Giguere, V. ELF3 is a repressor of androgen receptor action in prostate cancer cells. Oncogene 2014, 33, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, Z.; Huo, S.; Chen, Z.; Ou, Z.; Mai, J.; Ding, S.; Zhang, J. Overexpression of ELF3 facilitates cell growth and metastasis through PI3K/Akt and ERK signaling pathways in non-small cell lung cancer. Int. J. Biochem. Cell. Biol. 2018, 94, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Xu, M.; Xu, J.; Wu, K.; Fang, Q.; Liang, Y.; Zhou, S.; Cen, D.; Ji, L.; Han, W.; et al. ELF3 promotes epithelial-mesenchymal transition by protecting ZEB1 from miR-141–3p-mediated silencing in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 387. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chehab, R.; Tkalcevic, J.; Naylor, M.J.; Harris, J.; Wilson, T.J.; Tsao, S.; Tellis, I.; Zavarsek, S.; Xu, D.; et al. Elf5 is essential for early embryogenesis and mammary gland development during pregnancy and lactation. EMBO J. 2005, 24, 635–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnison, M.; Beaton, A.; Davey, H.W.; Broadhurst, R.; L’Huillier, P.; Pfeffer, P.L. Loss of the extraembryonic ectoderm in Elf5 mutants leads to defects in embryonic patterning. Development 2005, 132, 2299–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearton, D.J.; Smith, C.S.; Redgate, E.; van Leeuwen, J.; Donnison, M.; Pfeffer, P.L. Elf5 counteracts precocious trophoblast differentiation by maintaining Sox2 and 3 and inhibiting Hand1 expression. Dev. Biol. 2014, 392, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.S.; Chakrabarti, R.; Escamilla-Hernandez, R.; Sinha, S. Elf5 conditional knockout mice reveal its role as a master regulator in mammary alveolar development: Failure of Stat5 activation and functional differentiation in the absence of Elf5. Dev. Biol. 2009, 329, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, K.; Shillingford, J.M.; Smith, G.H.; Grimm, S.L.; Wagner, K.U.; Oka, T.; Rosen, J.M.; Robinson, G.W.; Hennighausen, L. Signal transducer and activator of transcription (Stat) 5 controls the proliferation and differentiation of mammary alveolar epithelium. J. Cell. Biol. 2001, 155, 531–542. [Google Scholar] [CrossRef]
- Chakrabarti, R.; Wei, Y.; Romano, R.A.; DeCoste, C.; Kang, Y.; Sinha, S. Elf5 regulates mammary gland stem/progenitor cell fate by influencing notch signaling. Stem Cells 2012, 30, 1496–1508. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, R.; Hwang, J.; Blanco, M.A.; Wei, Y.; Lukacisin, M.; Romano, R.A.; Smalley, K.; Liu, S.; Yang, Q.; Ibrahim, T.; et al. Elf5 inhibits the epithelial-mesenchymal transition in mammary gland development and breast cancer metastasis by transcriptionally repressing Snail2. Nat. Cell. Biol. 2012, 14, 1212–1222. [Google Scholar] [CrossRef] [PubMed]
- Grassmeyer, J.; Mukherjee, M.; deRiso, J.; Hettinger, C.; Bailey, M.; Sinha, S.; Visvader, J.E.; Zhao, H.; Fogarty, E.; Surendran, K. Elf5 is a principal cell lineage specific transcription factor in the kidney that contributes to Aqp2 and Avpr2 gene expression. Dev. Biol. 2017, 424, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.S.; Cheng, J.; Segre, J.; Sinha, S. Generation and analysis of Elf5-LacZ mouse: Unique and dynamic expression of Elf5 (ESE-2) in the inner root sheath of cycling hair follicles. Histochem. Cell Biol. 2008, 129, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Tummala, R.; Sinha, S. Differentiation-specific transcriptional regulation of the ESE-2 gene by a novel keratinocyte-restricted factor. J. Cell. Biochem. 2006, 97, 766–781. [Google Scholar] [CrossRef] [PubMed]
- Omata, F.; McNamara, K.M.; Suzuki, K.; Abe, E.; Hirakawa, H.; Ishida, T.; Ohuchi, N.; Sasano, H. Effect of the normal mammary differentiation regulator ELF5 upon clinical outcomes of triple negative breast cancers patients. Breast Cancer 2018, 25, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Piggin, C.L.; Roden, D.L.; Gallego-Ortega, D.; Lee, H.J.; Oakes, S.R.; Ormandy, C.J. ELF5 isoform expression is tissue-specific and significantly altered in cancer. Breast Cancer Res. 2016, 18, 4. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Ortega, D.; Ledger, A.; Roden, D.L.; Law, A.M.; Magenau, A.; Kikhtyak, Z.; Cho, C.; Allerdice, S.L.; Lee, H.J.; Valdes-Mora, F.; et al. ELF5 Drives Lung Metastasis in Luminal Breast Cancer through Recruitment of Gr1+ CD11b+ Myeloid-Derived Suppressor Cells. PLoS Biol. 2015, 13, e1002330. [Google Scholar] [CrossRef] [PubMed]
- Kalyuga, M.; Gallego-Ortega, D.; Lee, H.J.; Roden, D.L.; Cowley, M.J.; Caldon, C.E.; Stone, A.; Allerdice, S.L.; Valdes-Mora, F.; Launchbury, R.; et al. Ormandy. ELF5 suppresses estrogen sensitivity and underpins the acquisition of antiestrogen resistance in luminal breast cancer. PLoS Biol. 2012, 10, e1001461. [Google Scholar]
- Yao, B.; Zhao, J.; Li, Y.; Li, H.; Hu, Z.; Pan, P.; Zhang, Y.; Du, E.; Liu, R.; Xu, Y. Elf5 inhibits TGF-beta-driven epithelial-mesenchymal transition in prostate cancer by repressing SMAD3 activation. Prostate 2015, 75, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Guo, Y.; Yang, X.; Zhang, Z.; Zhang, C.; Xu, Y. ELF5-Mediated AR Activation Regulates Prostate Cancer Progression. Sci. Rep. 2017, 7, 42759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, B.; Cao, X.; Liang, X.; Zhang, X.; Zhang, W.; Sun, G.; Wang, D. Epigenetic regulation of Elf5 is associated with epithelial-mesenchymal transition in urothelial cancer. PLoS ONE 2015, 10, e0117510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Qiu, L.; Xie, X.; Yang, H.; Liu, Y.; Lin, X.; Huang, H. ELF5 in epithelial ovarian carcinoma tissues and biological behavior in ovarian carcinoma cells. Oncol. Rep. 2017, 37, 1412–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapinskas, E.J.; Svobodova, S.; Davis, I.D.; Cebon, J.; Hertzog, P.J.; Pritchard, M.A. The Ets transcription factor ELF5 functions as a tumor suppressor in the kidney. Twin Res. Hum. Genet. 2011, 14, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Wright, F.A.; Strug, L.J.; Doshi, V.K.; Commander, C.W.; Blackman, S.M.; Sun, L.; Berthiaume, Y.; Cutler, D.; Cojocaru, A.; Collaco, J.M.; et al. Genome-wide association and linkage identify modifier loci of lung disease severity in cystic fibrosis at 11p13 and 20q13.2. Nat. Genet. 2011, 43, 539–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Duan, R.; Cao, H.; Field, D.; Newnham, C.M.; Koehler, D.R.; Zamel, N.; Pritchard, M.A.; Hertzog, P.; Post, M.; et al. Regulation of epithelium-specific Ets-like factors ESE-1 and ESE-3 in airway epithelial cells: Potential roles in airway inflammation. Cell Res. 2008, 18, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Fossum, S.L.; Mutolo, M.J.; Tugores, A.; Ghosh, S.; Randell, S.H.; Jones, L.C.; Leir, S.H.; Harris, A. Ets homologous factor (EHF) has critical roles in epithelial dysfunction in airway disease. J. Biol. Chem. 2017, 292, 10938–10949. [Google Scholar] [CrossRef] [PubMed]
- Mutolo, M.J.; Leir, S.H.; Fossum, S.L.; Browne, J.A.; Harris, A. A transcription factor network represses CFTR gene expression in airway epithelial cells. Biochem. J. 2018, 475, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Rubin, A.J.; Barajas, B.C.; Furlan-Magaril, M.; Lopez-Pajares, V.; Mumbach, M.R.; Howard, I.; Kim, D.S.; Boxer, L.D.; Cairns, J.; Spivakov, M.; et al. Lineage-specific dynamic and pre-established enhancer-promoter contacts cooperate in terminal differentiation. Nat. Genet. 2017, 49, 1522–1528. [Google Scholar] [CrossRef] [PubMed]
- Tugores, A.; Le, J.; Sorokina, I.; Snijders, A.J.; Duyao, M.; Reddy, P.S.; Carlee, L.; Ronshaugen, M.; Mushegian, A.; Watanaskul, T.; et al. The epithelium-specific ETS protein EHF/ESE-3 is a context-dependent transcriptional repressor downstream of MAPK signaling cascades. J. Biol. Chem. 2001, 276, 20397–20406. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.; Stange, D.E.; Schepers, A.G.; van de Wetering, M.; Koo, B.K.; Itzkovitz, S.; Volckmann, R.; Kung, K.S.; Koster, J.; Radulescu, S.; et al. The Lgr5 intestinal stem cell signature: Robust expression of proposed quiescent ‘+4’ cell markers. EMBO J. 2012, 31, 3079–3091. [Google Scholar] [CrossRef] [PubMed]
- Terahara, K.; Yoshida, M.; Igarashi, O.; Nochi, T.; Pontes, G.S.; Hase, K.; Ohno, H.; Kurokawa, S.; Mejima, M.; Takayama, N.; et al. Comprehensive gene expression profiling of Peyer’s patch M cells, villous M-like cells, and intestinal epithelial cells. J. Immunol 2008, 180, 7840–7846. [Google Scholar] [CrossRef] [PubMed]
- Asai, T.; Morrison, S.L. The SRC family tyrosine kinase HCK and the ETS family transcription factors SPIB and EHF regulate transcytosis across a human follicle-associated epithelium model. J. Biol. Chem. 2013, 288, 10395–10405. [Google Scholar] [CrossRef] [PubMed]
- Appel, S.; Bringmann, A.; Grunebach, F.; Weck, M.M.; Bauer, J.; Brossart, P. Epithelial-specific transcription factor ESE-3 is involved in the development of monocyte-derived DCs. Blood 2006, 107, 3265–3270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprater, F.; Hovden, A.O.; Appel, S. Expression of ESE-3 isoforms in immunogenic and tolerogenic human monocyte-derived dendritic cells. PLoS ONE 2012, 7, e49577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, S.; Nakano, N.; Honjo, A.; Hara, M.; Maeda, K.; Nishiyama, C.; Kitaura, J.; Ohtsuka, Y.; Okumura, K.; Ogawa, H.; et al. The Transcription Factor Ehf Is Involved in TGF-beta-Induced Suppression of FcepsilonRI and c-Kit Expression and FcepsilonRI-Mediated Activation in Mast Cells. J. Immunol. 2015, 195, 3427–3435. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Qu, Y.; Li, X.; Sui, F.; Yao, D.; Yang, Q.; Shi, B.; Ji, M.; Hou, P. Increased expression of EHF via gene amplification contributes to the activation of HER family signaling and associates with poor survival in gastric cancer. Cell Death Dis. 2016, 7, e2442. [Google Scholar] [CrossRef] [PubMed]
- Taniue, K.; Oda, T.; Hayashi, T.; Okuno, M.; Akiyama, T. A member of the ETS family, EHF, and the ATPase RUVBL1 inhibit p53-mediated apoptosis. EMBO Rep. 2011, 12, 682–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, Y.; Sui, F.; Ma, J.; Ren, X.; Yang, Q.; Zhang, Y.; Guan, H.; Shi, B.; Hou, P.; Ji, M. Increased expression of EHF contributes to thyroid tumorigenesis through transcriptionally regulating HER2 and HER3. Oncotarget 2016, 7, 57978–57990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Z.; Guo, J.; Chen, L.; Luo, N.; Yang, W.; Qu, X. Knockdown of EHF inhibited the proliferation, invasion and tumorigenesis of ovarian cancer cells. Mol. Carcinog. 2016, 55, 1048–1059. [Google Scholar] [CrossRef] [PubMed]
- Cangemi, R.; Mensah, A.; Albertini, V.; Jain, A.; Mello-Grand, M.; Chiorino, G.; Catapano, C.V.; Carbone, G.M. Reduced expression and tumor suppressor function of the ETS transcription factor ESE-3 in prostate cancer. Oncogene 2008, 27, 2877–2885. [Google Scholar] [CrossRef] [PubMed]
- Albino, D.; Longoni, N.; Curti, L.; Mello-Grand, M.; Pinton, S.; Civenni, G.; Thalmann, G.; D’Ambrosio, G.; Sarti, M.; Sessa, F.; et al. ESE3/EHF controls epithelial cell differentiation and its loss leads to prostate tumors with mesenchymal and stem-like features. Cancer Res. 2012, 72, 2889–2900. [Google Scholar] [CrossRef] [PubMed]
- Albino, D.; Civenni, G.; Rossi, S.; Mitra, A.; Catapano, C.V.; Carbone, G.M. The ETS factor ESE3/EHF represses IL-6 preventing STAT3 activation and expansion of the prostate cancer stem-like compartment. Oncotarget 2016, 7, 76756–76768. [Google Scholar] [CrossRef] [PubMed]
- Kunderfranco, P.; Mello-Grand, M.; Cangemi, R.; Pellini, S.; Mensah, A.; Albertini, V.; Malek, A.; Chiorino, G.; Catapano, C.V.; Carbone, G.M. ETS transcription factors control transcription of EZH2 and epigenetic silencing of the tumor suppressor gene Nkx3.1 in prostate cancer. PLoS ONE 2010, 5, e10547. [Google Scholar]
- Zhao, T.; Jiang, W.; Wang, X.; Wang, H.; Zheng, C.; Li, Y.; Sun, Y.; Huang, C.; Han, Z.B.; Yang, S.; et al. ESE3 Inhibits Pancreatic Cancer Metastasis by Upregulating E-Cadherin. Cancer Res. 2017, 77, 874–885. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xing, J.; Cheng, R.; Shao, Y.; Li, P.; Zhu, S.; Zhang, S. Abnormal Localization and Tumor Suppressor Function of Epithelial Tissue-Specific Transcription Factor ESE3 in Esophageal Squamous Cell Carcinoma. PLoS ONE 2015, 10, e0126319. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.H.; Badis, G.; Berger, M.F.; Kivioja, T.; Palin, K.; Enge, M.; Bonke, M.; Jolma, A.; Varjosalo, M.; Gehrke, A.R.; et al. Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J. 2010, 29, 2147–2160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregorieff, A.; Stange, D.E.; Kujala, P.; Begthel, H.; van den Born, M.; Korving, J.; Peters, P.J.; Clevers, H. The ets-domain transcription factor Spdef promotes maturation of goblet and paneth cells in the intestinal epithelium. Gastroenterology 2009, 137, 1333–1345. [Google Scholar] [CrossRef] [PubMed]
- Horst, D.; Gu, X.; Bhasin, M.; Yang, Q.; Verzi, M.; Lin, D.; Joseph, M.; Zhang, X.; Chen, W.; Li, Y.P.; et al. Requirement of the epithelium-specific Ets transcription factor Spdef for mucous gland cell function in the gastric antrum. J. Biol. Chem. 2010, 285, 35047–35055. [Google Scholar] [CrossRef] [PubMed]
- Marko, C.K.; Menon, B.B.; Chen, G.; Whitsett, J.A.; Clevers, H.; Gipson, I.K. Spdef null mice lack conjunctival goblet cells and provide a model of dry eye. Am. J. Pathol. 2013, 183, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, F.L.; Xiao, Y.; Bian, F.; Coursey, T.G.; Ko, B.Y.; Clevers, H.; de Paiva, C.S.; Pflugfelder, S.C. Goblet Cells Contribute to Ocular Surface Immune Tolerance-Implications for Dry Eye Disease. Int. J. Mol. Sci. 2017, 18, 978. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Korfhagen, T.R.; Xu, Y.; Kitzmiller, J.; Wert, S.E.; Maeda, Y.; Gregorieff, A.; Clevers, H.; Whitsett, J.A. SPDEF is required for mouse pulmonary goblet cell differentiation and regulates a network of genes associated with mucus production. J. Clin. Investig. 2009, 119, 2914–2924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajavelu, P.; Chen, G.; Xu, Y.; Kitzmiller, J.A.; Korfhagen, T.R.; Whitsett, J.A. Airway epithelial SPDEF integrates goblet cell differentiation and pulmonary Th2 inflammation. J. Clin. Investig. 2015, 125, 2021–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noah, T.K.; Kazanjian, A.; Whitsett, J.; Shroyer, N.F. SAM pointed domain ETS factor (SPDEF) regulates terminal differentiation and maturation of intestinal goblet cells. Exp. Cell Res. 2010, 316, 452–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oettgen, P.; Finger, E.; Sun, Z.; Akbarali, Y.; Thamrongsak, U.; Boltax, J.; Grall, F.; Dube, A.; Weiss, A.; Brown, L.; et al. PDEF, a novel prostate epithelium-specific ets transcription factor, interacts with the androgen receptor and activates prostate-specific antigen gene expression. J. Biol. Chem. 2000, 275, 1216–1225. [Google Scholar] [CrossRef]
- Noah, T.K.; Lo, Y.H.; Price, A.; Chen, G.; Washington, E.K.M.K.; Aronow, B.J.; Shroyer, N.F. SPDEF functions as a colorectal tumor suppressor by inhibiting beta-catenin activity. Gastroenterology 2013, 144, 1012–1023. [Google Scholar] [CrossRef] [PubMed]
- Moussa, O.; Turner, D.P.; Feldman, R.J.; Sementchenko, V.I.; McCarragher, B.D.; Desouki, M.M.; Fraig, M.; Watson, D.K. PDEF is a negative regulator of colon cancer cell growth and migration. J. Cell. Biochem. 2009, 108, 1389–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, Y.H.; Noah, T.K.; Chen, M.S.; Zou, W.; Borras, E.; Vilar, E.; Shroyer, N.F. SPDEF Induces Quiescence of Colorectal Cancer Cells by Changing the Transcriptional Targets of beta-catenin. Gastroenterology 2017, 153, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.B.; Zhou, S.L.; Pang, X.G.; Yin, D.; Miao, P.Z.; Yang, Y.; Chen, Q.; Zhu, K.; Gao, D.M.; Liu, T.S.; et al. Prostate-derived ETS factor improves prognosis and represses proliferation and invasion in hepatocellular carcinoma. Oncotarget 2017, 8, 52488–52500. [Google Scholar] [CrossRef] [PubMed]
- Tsui, K.H.; Lin, Y.H.; Chung, L.C.; Chuang, S.T.; Feng, T.H.; Chiang, K.C.; Chang, P.L.; Yeh, C.J.; Juang, H.H. Prostate-derived ets factor represses tumorigenesis and modulates epithelial-to-mesenchymal transition in bladder carcinoma cells. Cancer Lett. 2016, 375, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.R.; Koul, S.; Kumar, B.; Khandrika, L.; Venezia, S.; Maroni, P.D.; Meacham, R.B.; Koul, H.K. Loss of PDEF, a prostate-derived Ets factor is associated with aggressive phenotype of prostate cancer: Regulation of MMP 9 by PDEF. Mol. Cancer 2010, 9, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghadersohi, A.; Sharma, S.; Zhang, S.; Azrak, R.G.; Wilding, G.E.; Manjili, M.H.; Li, F. Prostate-derived Ets transcription factor (PDEF) is a potential prognostic marker in patients with prostate cancer. Prostate 2011, 71, 1178–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steffan, J.J.; Koul, S.; Meacham, R.B.; Koul, H.K. The transcription factor SPDEF suppresses prostate tumor metastasis. J. Biol. Chem. 2012, 287, 29968–29978. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.H.; Black, M.; Ustiyan, V.; Le, T.; Fulford, L.; Sridharan, A.; Medvedovic, M.; Kalinichenko, V.V.; Whitsett, J.A.; Kalin, T.V. SPDEF inhibits prostate carcinogenesis by disrupting a positive feedback loop in regulation of the Foxm1 oncogene. PLoS Genet. 2014, 10, e1004656. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Y.; Tsai, Y.C.; Yeh, H.L.; Suau, F.; Jiang, K.C.; Shao, A.N.; Huang, J.; Liu, Y.N. Loss of SPDEF and gain of TGFBI activity after androgen deprivation therapy promote EMT and bone metastasis of prostate cancer. Sci. Signal. 2017, 10, eaam6826. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.C.; Chen, W.Y.; Abou-Kheir, W.; Zeng, T.; Yin, J.J.; Bahmad, H.; Lee, Y.C.; Liu, Y.N. Androgen deprivation therapy-induced epithelial-mesenchymal transition of prostate cancer through downregulating SPDEF and activating CCL2. Biochim. Biophys. Acta 2018, 1864, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Rodabaugh, K.J.; Mhawech-Fauceglia, P.; Groth, J.; Lele, S.; Sood, A.K. Prostate-derived Ets factor is overexpressed in serous epithelial ovarian tumors. Int. J. Gynecol. Pathol. 2007, 26, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Ghadersohi, A.; Odunsi, K.; Lele, S.; Collins, Y.; Greco, W.R.; Winston, J.; Liang, P.; Sood, A.K. Prostate derived Ets transcription factor shows better tumor-association than other cancer-associated molecules. Oncol. Rep. 2004, 11, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Ghadersohi, A.; Odunsi, K.; Zhang, S.; Azrak, R.G.; Bundy, B.N.; Manjili, M.H.; Li, F. Prostate-derived Ets transcription factor as a favorable prognostic marker in ovarian cancer patients. Int. J. Cancer 2008, 123, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Feldman, R.J.; Sementchenko, V.I.; Gayed, M.; Fraig, M.M.; Watson, D.K. Pdef expression in human breast cancer is correlated with invasive potential and altered gene expression. Cancer Res. 2003, 63, 4626–4631. [Google Scholar] [PubMed]
- Turner, D.P.; Moussa, O.; Sauane, M.; Fisher, P.B.; Watson, D.K. Prostate-derived ETS factor is a mediator of metastatic potential through the inhibition of migration and invasion in breast cancer. Cancer Res. 2007, 67, 1618–1625. [Google Scholar] [CrossRef] [PubMed]
- Findlay, V.J.; Turner, D.P.; Moussa, O.; Watson, D.K. MicroRNA-mediated inhibition of prostate-derived Ets factor messenger RNA translation affects prostate-derived Ets factor regulatory networks in human breast cancer. Cancer Res. 2008, 68, 8499–8506. [Google Scholar] [CrossRef] [PubMed]
- Sood, A.K.; Geradts, J.; Young, J. Prostate-derived Ets factor, an oncogenic driver in breast cancer. Tumour Biol. 2017, 39, 1010428317691688. [Google Scholar] [CrossRef] [PubMed]
- Sood, A.K.; Wang, J.; Mhawech-Fauceglia, P.; Jana, B.; Liang, P.; Geradts, J. Sam-pointed domain containing Ets transcription factor in luminal breast cancer pathogenesis. Cancer Epidemiol. Biomark. Prev. 2009, 18, 1899–1903. [Google Scholar] [CrossRef] [PubMed]
- Buchwalter, G.; Hickey, M.M.; Cromer, A.; Selfors, L.M.; Gunawardane, R.N.; Frishman, J.; Jeselsohn, R.; Lim, E.; Chi, D.; Fu, X.; et al. PDEF promotes luminal differentiation and acts as a survival factor for ER-positive breast cancer cells. Cancer Cell 2013, 23, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Marcotte, R.; Sayad, A.; Brown, K.R.; Sanchez-Garcia, F.; Reimand, J.; Haider, M.; Virtanen, C.; Bradner, J.E.; Bader, G.D.; Mills, G.B.; et al. Functional Genomic Landscape of Human Breast Cancer Drivers, Vulnerabilities, and Resistance. Cell 2016, 164, 293–309. [Google Scholar] [CrossRef] [PubMed]
- Bhagwat, A.S.; Vakoc, C.R. Targeting Transcription Factors in Cancer. Trends Cancer 2015, 1, 53–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, H.; Fujiwara, Y.; Doki, Y.; Sugita, Y.; Sohma, I.; Miyata, H.; Takiguchi, S.; Yasuda, T.; Tomita, N.; Morishita, R.; et al. Gene therapy using ets-1 transcription factor decoy for peritoneal dissemination of gastric cancer. Int. J. Cancer 2007, 121, 1609–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dyke, M.W.; Dervan, P.B. Chromomycin, mithramycin, and olivomycin binding sites on heterogeneous deoxyribonucleic acid. Footprinting with (methidiumpropyl-EDTA)iron(II). Biochemistry 1983, 22, 2373–2377. [Google Scholar] [CrossRef] [PubMed]
- Raskatov, J.A.; Meier, J.L.; Puckett, J.W.; Yang, F.; Ramakrishnan, P.; Dervan, P.B. Modulation of NF-kappaB-dependent gene transcription using programmable DNA minor groove binders. Proc. Natl. Acad. Sci. USA 2012, 109, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sicot, G.; Cui, X.; Vogel, M.; Wuertzer, C.A.; Lezon-Geyda, K.; Wheeler, J.; Harki, D.A.; Muzikar, K.A.; Stolper, D.A.; et al. Targeting a DNA binding motif of the EVI1 protein by a pyrrole-imidazole polyamide. Biochemistry 2011, 50, 10431–10441. [Google Scholar] [CrossRef] [PubMed]
- Nhili, R.; Peixoto, P.; Depauw, S.; Flajollet, S.; Dezitter, X.; Munde, M.M.; Ismail, M.A.; Kumar, A.; Farahat, A.A.; Stephens, C.E.; et al. Targeting the DNA-binding activity of the human ERG transcription factor using new heterocyclic dithiophene diamidines. Nucleic Acids Res. 2013, 41, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Fang, R.; Cho, J.Y.; Libermann, T.A.; Oettgen, P. Positive and negative modulation of the transcriptional activity of the ETS factor ESE-1 through interaction with p300, CREB-binding protein, and Ku 70/86. J. Biol. Chem. 2004, 279, 25241–25250. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Cho, J.Y.; Park, Y.B.; Park, J.W.; Kwon, T.K. ESE-3 transcription factor is involved in the expression of death receptor (DR)-5 through putative Ets sites. Biochem. Biophys Res. Commun. 2006, 350, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Ceccacci, E.; Minucci, S. Inhibition of histone deacetylases in cancer therapy: Lessons from leukaemia. Br. J. Cancer 2016, 114, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Tse, J.W.T.; Jenkins, L.J.; Chionh, F.; Mariadason, J.M. Aberrant DNA Methylation in Colorectal Cancer: What Should We Target? Trends Cancer 2017, 3, 698–712. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luk, I.Y.; Reehorst, C.M.; Mariadason, J.M. ELF3, ELF5, EHF and SPDEF Transcription Factors in Tissue Homeostasis and Cancer. Molecules 2018, 23, 2191. https://doi.org/10.3390/molecules23092191
Luk IY, Reehorst CM, Mariadason JM. ELF3, ELF5, EHF and SPDEF Transcription Factors in Tissue Homeostasis and Cancer. Molecules. 2018; 23(9):2191. https://doi.org/10.3390/molecules23092191
Chicago/Turabian StyleLuk, Ian Y., Camilla M. Reehorst, and John M. Mariadason. 2018. "ELF3, ELF5, EHF and SPDEF Transcription Factors in Tissue Homeostasis and Cancer" Molecules 23, no. 9: 2191. https://doi.org/10.3390/molecules23092191
APA StyleLuk, I. Y., Reehorst, C. M., & Mariadason, J. M. (2018). ELF3, ELF5, EHF and SPDEF Transcription Factors in Tissue Homeostasis and Cancer. Molecules, 23(9), 2191. https://doi.org/10.3390/molecules23092191