Direct Asymmetric Reductive Amination for the Synthesis of (S)-Rivastigmine

Abstract
:1. Introduction
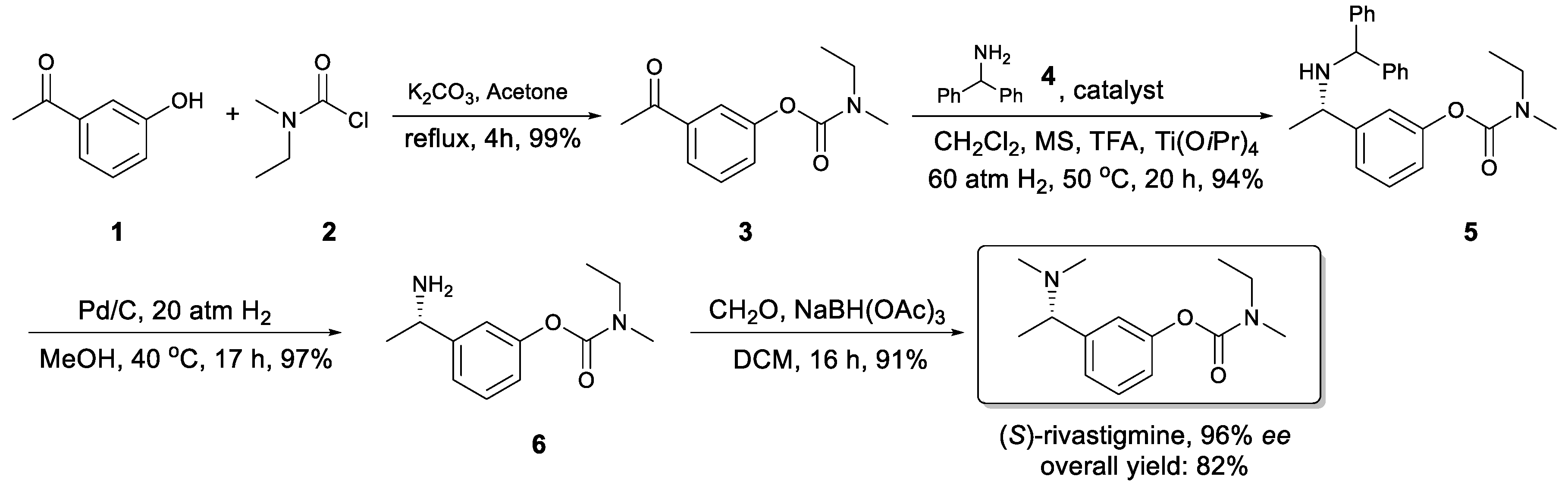
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Characterization
3.3. Preparation of Compound 3
3.4. Preparation of Compound 5
3.5. Preparation of Compound 6
3.6. Preparation of (S)-Rivastigmine
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Huang, Y.; Mucke, L. Alzheimer mechanisms and therapeutic strategies. Cell 2012, 148, 1204–1222. [Google Scholar] [CrossRef] [PubMed]
- Scarpini, E.; Scheltens, P.; Feldman, H. Treatment of Alzheimer’s disease: Current status and new perspectives. Lancet Neurol. 2003, 2, 539–547. [Google Scholar] [CrossRef]
- Frisoni, G.B.; Fox, N.C.; Jack, C.R., Jr.; Scheltens, P.; Thompson, P.M. The clinical use of structural MRI in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 67–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berchtold, N.C.; Cotman, C.W. Evolution in the conceptualization of dementia and Alzheimer’s disease: Greco-roman period to the 1960s. Neurobiol. Aging 1998, 19, 173–189. [Google Scholar] [CrossRef]
- Scheltens, P.; Blennow, K.; Breteler, M.M.; Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Panek, D.; Więckowska, A.; Wichur, T.; Bajda, M.; Godyń, J.; Jończyk, J.; Mika, K.; Janockova, J.; Soukup, O.; Knez, D.; et al. Design, synthesis and biological evaluation of new phthalimide and saccharin derivatives with alicyclic amines targeting cholinesterases, beta-secretase and amyloid beta aggregation. Eur. J. Med. Chem. 2017, 125, 676–695. [Google Scholar] [CrossRef] [PubMed]
- Farlow, M.R.; Cummings, J.L. Effective Pharmacologic Management of Alzheimer’s Disease. Am. J. Med. 2007, 120, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Polinsky, R.J. Clinical pharmacology of rivastigmine: A new-generation acetylcholinesterase inhibitor for the treatment of Alzheimer’s disease. Clin. Ther. 1998, 20, 634–647. [Google Scholar] [CrossRef]
- Rosler, M.; Anand, R.; Cicin-Sain, A.; Gauthier, S.; Agid, Y.; Dal-Bianco, P.; Stahelin, H.B.; Hartman, R.; Gharabawi, M. Efficacy and safety of rivastigmine in patients with Alzheimer’s disease: International randomised controlled trial commentary: Another piece of the Alzheimer’s jigsaw. Br. Med. J. 1999, 318, 633–638. [Google Scholar] [CrossRef]
- Spencer, C.M.; Noble, S. Rivastigmine: A review of its use in Alzheimer’s disease. Drugs Aging 1998, 13, 391–411. [Google Scholar] [CrossRef] [PubMed]
- Gaitonde, A.; Mangle, M.; Pawar, S. Novel Processes for the Preparation of Aminoalkyl Phenylcarbamates. International Patent WO 2005/061446, 7 July 2005. [Google Scholar]
- Boezio, A.A.; Pytkowicz, J.; Côté, A.; Charette, A.B. Asymmetric, catalytic synthesis of r-chiral amines using a novel bis(phosphine) monoxide chiral ligand. J. Am. Chem. Soc. 2003, 125, 14260–14261. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.L.; Buchwald, S.L. Copper-catalysed selective hydroamination reactions of alkynes. Nat. Chem. 2015, 7, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Wakchaure, V.N.; Kaib, P.S.J.; Leutzsch, M.; List, B. Disulfonimide-catalyzed asymmetric reduction of N-alkyl imines. Angew. Chem. Int. Ed. 2015, 54, 11852. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Zhang, F.L.; Xie, M.H. Novel convenient synthesis of rivastigmine. Synth. Commun. 2009, 39, 1527–1533. [Google Scholar] [CrossRef]
- Han, K.; Kim, C.; Park, J.; Kim, M.J. Chemoenzymatic synthesis of rivastigmine via dynamic kinetic resolution as a key step. J. Org. Chem. 2010, 75, 3105–3108. [Google Scholar] [CrossRef] [PubMed]
- Mangas-Sánchez, J.; Rodríguez-Mata, M.; Busto, E.; Gotor-Fernández, V.; Gotor, V. Chemoenzymatic synthesis of rivastigmine based on lipase-catalyzed processes. J. Org. Chem. 2009, 74, 5304–5310. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, M.; Koszelewski, D.; Tauber, K.; Sattler, J.; Banko, W.; Holzer, A.K.; Pickl, M.; Kroutil, W.; Faber, K. Improved chemoenzymatic asymmetric synthesis of (S)-rivastigmine. Tetrahedron 2012, 68, 7691–7694. [Google Scholar] [CrossRef]
- Fuchs, M.; Koszelewski, D.; Tauber, K.; Kroutil, W.; Faber, K. Chemoenzymatic asymmetric total synthesis of (S)-rivastigmine using ω-transaminases. Chem. Commun. 2010, 46, 5500–5502. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.C.; Zhu, G.L.; Xie, J.H.; Zhang, X.D.; Zhou, Q.L.; Li, Y.Q.; Shen, W.H.; Che, D.Q. Industrial scale-up of enantioselective hydrogenation for the asymmetric synthesis of rivastigmine. Org. Process. Res. Dev. 2013, 17, 307–312. [Google Scholar] [CrossRef]
- Huang, H.; Liu, X.; Zhou, L.; Chang, M.; Zhang, X. Direct asymmetric reductive amination for the synthesis of chiral β-arylamines. Angew. Chem. Int. Ed. 2016, 55, 5309–5312. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhao, Y.; Yang, Y.; Zhou, L.; Chang, M. Direct catalytic asymmetric reductive amination of aliphatic ketones utilizing diphenylmethanamine as coupling partner. Org. Lett. 2017, 19, 1942–1945. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Wu, Z.; Gao, G.; Zhou, L.; Chang, M. Iridium-catalyzed direct asymmetric reductive amination of aromatic ketones. Org. Chem. Front. 2017, 4, 1976–1980. [Google Scholar] [CrossRef]
- Claver, C.; Fernandez, E.; Gillon, A.; Heslop, K.; Hyett, D.J.; Martorell, A.; Orpen, A.G.; Pringle, P.G. Biarylphosphonites: A class of monodentatephosphorus(III) ligands that outperform their chelatinganalogues in asymmetric hydrogenation catalysis. Chem. Commun. 2000, 46, 961–962. [Google Scholar] [CrossRef]
- Berg van den, M.; Minnaard, A.J.; Schudde, E.P.; Esch, J.V.; de Vries, A.H.M.; de Vries, J.G.; Feringa, B.L. Highly enantioselective rhodium-catalyzed hydrogenation with monodentate ligands. J. Am. Chem. Soc. 2000, 122, 11539–11540. [Google Scholar] [CrossRef]
- Minnaard, A.J.; Feringa, B.L.; Lefort, L.; de Vries, J.G. Asymmetric hydrogenation using monodentate phosphoramidite ligands. Acc. Chem. Res. 2007, 40, 1267–1277. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Tang, W. Chiral monophosphorus ligands for asymmetric catalytic reactions. ACS Catal. 2016, 6, 4814–4858. [Google Scholar] [CrossRef]
- Hong, L.; Sun, W.; Yang, D.; Li, G.; Wang, R. Additive effects on asymmetric catalysis. Chem. Rev. 2016, 116, 4006–4123. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Villa-Marcos, B.; Xiao, J. Metal-brønsted acid cooperative catalysis for asymmetric reductive amination. J. Am. Chem. Soc. 2009, 131, 6967–6969. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Liu, S.; Zhang, X. Direct catalytic asymmetric reductive amination of simple aromatic ketones. Org. Lett. 2013, 15, 4354–4357. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.; Zhou, Y.; Zhang, X. Highly enantioselective reductive amination of simple aryl ketones catalyzed by Ir-f-binaphane in the presence of titanium(IV) isopropoxide and iodine. J. Org. Chem. 2003, 68, 4120–4122. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds (3–6 and rivastigmine) are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
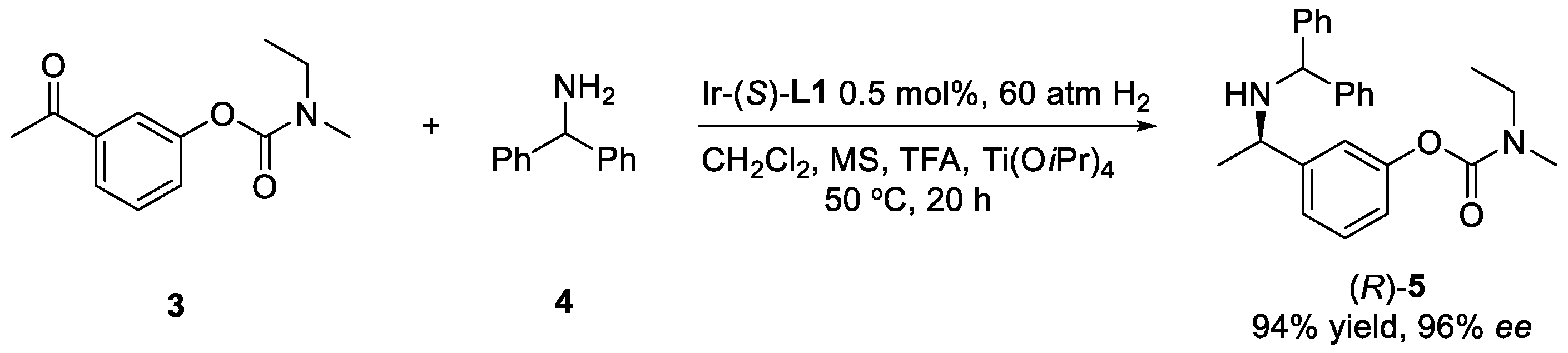{kind=link}
{kind=link}

| Entry | Ligand | Acid (Equiv.) | Yield (%) | ee (%) |
|---|---|---|---|---|
| 1 | L1 | TFA (0.5) | 84 | 96 |
| 2 b | L1 | TFA (0.5) | 76 | 96 |
| 3 c | L1 | TFA (0.5) | 45 | 77 |
| 4 | L1 | TFA (1.0) | 93 | 96 |
| 5 | L1 | 4-Cl-C6H4SO3H (1.0) | <15 | - |
| 6 | L1 | TsOH (1.0) | <15 | - |
| 7 | L1 | 4-NO2-C6H4CO2H (1.0) | 65 | 5 |
| 8 | L1 | CCl3CO2H (1.0) | 77 | 92 |
| 9 | L2 | TFA (1.0) | 60 | 90 |
| 10 | L3 | TFA (1.0) | 16 | 58 |
| 11 | L4 | TFA (1.0) | <10 | - |
| 12 | L5 | TFA (1.0) | <10 | - |
| 13 | L6 | TFA (1.0) | 94 | 95 |
| 14 | L7 | TFA (1.0) | 78 | 82 |

| Entry | Catalyst Loading (mol%) | Ti(OiPr)4 (mol%) | Acid (mol%) | Yield (%) | ee (%) |
|---|---|---|---|---|---|
| 1 | 1 | 20 | 50 | 79 | 93 |
| 2 | 1 | 30 | 50 | 85 | 96 |
| 3 | 1 | 40 | 50 | 87 | 94 |
| 4 | 1 | 30 | 70 | 91 | 96 |
| 5 | 1 | 30 | 80 | 94 | 97 |
| 6 b | 1 | 30 | 80 | 90 | 97 |
| 7 c | 1 | 30 | 80 | 75 | 98 |
| 8 | 0.5 | 30 | 80 | 94 | 96 |
| 9 | 0.2 | 30 | 80 | 88 | 96 |
| 10 d | 0.1 | 30 | 80 | 70 | 94 |
| 11 e | 0.1 | 30 | 80 | 94 | 95 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, G.; Du, S.; Yang, Y.; Lei, X.; Huang, H.; Chang, M. Direct Asymmetric Reductive Amination for the Synthesis of (S)-Rivastigmine. Molecules 2018, 23, 2207. https://doi.org/10.3390/molecules23092207
Gao G, Du S, Yang Y, Lei X, Huang H, Chang M. Direct Asymmetric Reductive Amination for the Synthesis of (S)-Rivastigmine. Molecules. 2018; 23(9):2207. https://doi.org/10.3390/molecules23092207
Chicago/Turabian StyleGao, Guorui, Shaozhi Du, Yang Yang, Xue Lei, Haizhou Huang, and Mingxin Chang. 2018. "Direct Asymmetric Reductive Amination for the Synthesis of (S)-Rivastigmine" Molecules 23, no. 9: 2207. https://doi.org/10.3390/molecules23092207
APA StyleGao, G., Du, S., Yang, Y., Lei, X., Huang, H., & Chang, M. (2018). Direct Asymmetric Reductive Amination for the Synthesis of (S)-Rivastigmine. Molecules, 23(9), 2207. https://doi.org/10.3390/molecules23092207