Recent Advances in Mitochondria-Targeted Gene Delivery


Abstract
:1. Introduction
2. Physical Approaches
3. Chemical Approaches
4. Biological Approaches
5. Combinatorial Approaches
6. Cargo DNAs
7. Applications as Disease Therapies
8. Concluding Remarks
Funding
Conflicts of Interest
References
- Robin, E.D.; Wong, R. Mitochondrial DNA molecules and virtual number of mitochondria per cell in mammalian cells. J. Cell. Physiol. 1988, 136, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Legros, F.; Malka, F.; Frachon, P.; Lombes, A.; Rojo, M. Organization and dynamics of human mitochondrial DNA. J. Cell Sci. 2004, 117 Pt 13, 2653–2662. [Google Scholar] [CrossRef]
- D’Erchia, A.M.; Atlante, A.; Gadaleta, G.; Pavesi, G.; Chiara, M.; De Virgilio, C.; Manzari, C.; Mastropasqua, F.; Prazzoli, G.M.; Picardi, E.; et al. Tissue-specific mtDNA abundance from exome data and its correlation with mitochondrial transcription, mass and respiratory activity. Mitochondrion 2015, 20, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.B.; Chinnery, P.F. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat. Rev. Genet. 2015, 16, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Yasuzaki, Y.; Yamada, Y.; Kanefuji, T.; Harashima, H. Localization of exogenous DNA to mitochondria in skeletal muscle following hydrodynamic limb vein injection. J. Control. Release 2013, 172, 805–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuzaki, Y.; Yamada, Y.; Fukuda, Y.; Harashima, H. Condensation of plasmid DNA enhances mitochondrial association in skeletal muscle following hydrodynamic limb vein injection. Pharmaceuticals 2014, 7, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Yasuzaki, Y.; Yamada, Y.; Ishikawa, T.; Harashima, H. Validation of Mitochondrial Gene Delivery in Liver and Skeletal Muscle via Hydrodynamic Injection Using an Artificial Mitochondrial Reporter DNA Vector. Mol. Pharm. 2015, 12, 4311–4320. [Google Scholar] [CrossRef] [PubMed]
- Bonnefoy, N.; Fox, T.D. Directed alteration of Saccharomyces cerevisiae mitochondrial DNA by biolistic transformation and homologous recombination. Methods Mol. Biol. 2007, 372, 153–166. [Google Scholar] [PubMed]
- Cardoso, A.M.; Morais, C.M.; Cruz, A.R.; Cardoso, A.L.; Silva, S.G.; do Vale, M.L.; Marques, E.F.; de Lima, M.C.P.; Jurado, A.S. Gemini Surfactants Mediate Efficient Mitochondrial Gene Delivery and Expression. Mol. Pharm. 2015, 12, 716–730. [Google Scholar] [CrossRef] [PubMed]
- Salvado, R.; Sousa, F.; Queiroz, J.; Costa, D. Development of mitochondrial targeting plasmid DNA nanoparticles: Characterization and in vitro studies. Colloid Surf. A 2015, 480, 287–295. [Google Scholar] [CrossRef]
- Santos, J.; Sousa, F.; Queiroz, J.; Costa, D. Rhodamine based plasmid DNA nanoparticles for mitochondrial gene therapy. Colloids Surf. B Biointerfaces 2014, 121, 129–140. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, G.G.M.; Rammohan, R.; Cheng, S.M.; Torchilin, V.P.; Weissig, V. DQAsome-mediated delivery of plasmid DNA toward mitochondria in living cells. J. Control. Release 2003, 92, 189–197. [Google Scholar] [CrossRef]
- Lyrawati, D.; Trounson, A.; Cram, D. Expression of GFP in the mitochondrial compartment using DQAsome-mediated delivery of an artificial mini-mitochondrial genome. Pharm. Res. 2011, 28, 2848–2862. [Google Scholar] [CrossRef] [PubMed]
- Weissig, V.; Lizano, C.; Torchilin, V.P. Selective DNA release from DQAsome/DNA complexes at mitochondria-like membranes. Drug Deliv. 2000, 7, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.; Jung, M.K.; Song, S.J.; Green, E.S.; Lee, S.; Park, H.S.; Jeong, S.H.; Han, J.; Mun, J.Y.; Ko, K.S.; et al. Functional nanosome for enhanced mitochondria-targeted gene delivery and expression. Mitochondrion 2017, 37, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Kogure, K.; Akita, H.; Yamada, Y.; Harashima, H. Multifunctional envelope-type nano device (MEND) as a non-viral gene delivery system. Adv. Drug Deliv. Rev. 2008, 60, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Fukuda, Y.; Harashima, H. An analysis of membrane fusion between mitochondrial double membranes and MITO-Porter, mitochondrial fusogenic vesicles. Mitochondrion 2015, 24, 50–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, Y.; Furukawa, R.; Yasuzaki, Y.; Harashima, H. Dual function MITO-Porter, a nano carrier integrating both efficient cytoplasmic delivery and mitochondrial macromolecule delivery. Mol. Ther. 2011, 19, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Ishikawa, T.; Harashima, H. Validation of the use of an artificial mitochondrial reporter DNA vector containing a Cytomegalovirus promoter for mitochondrial transgene expression. Biomaterials 2017, 136, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.C.; Wang, X.B.; Zhou, M.; Fei, H. A Mitochondria-Targeting Gold-Peptide Nanoassembly for Enhanced Cancer-Cell Killing. Adv. Healthc. Mater. 2013, 2, 1638–1643. [Google Scholar] [CrossRef] [PubMed]
- Boddapati, S.V.; D’Souza, G.G.; Erdogan, S.; Torchilin, V.P.; Weissig, V. Organelle-targeted nanocarriers: Specific delivery of liposomal ceramide to mitochondria enhances its cytotoxicity in vitro and in vivo. Nano Lett. 2008, 8, 2559–2563. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Dodwadkar, N.S.; Deshpande, P.P.; Torchilin, V.P. Liposomes loaded with paclitaxel and modified with novel triphenylphosphonium-PEG-PE conjugate possess low toxicity, target mitochondria and demonstrate enhanced antitumor effects in vitro and in vivo. J. Control. Release 2012, 159, 393–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.; Dodwadkar, N.S.; Piroyan, A.; Torchilin, V.P. Surface conjugation of triphenylphosphonium to target poly(amidoamine) dendrimers to mitochondria. Biomaterials 2012, 33, 4773–4782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinnery, P.F.; Taylor, R.W.; Diekert, K.; Lill, R.; Turnbull, D.M.; Lightowlers, R.N. Peptide nucleic acid delivery to human mitochondria. Gene Ther. 1999, 6, 1919–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flierl, A.; Jackson, C.; Cottrell, B.; Murdock, D.; Seibel, P.; Wallace, D.C. Targeted delivery of DNA to the mitochondrial compartment via import sequence-conjugated peptide nucleic acid. Mol. Ther. 2003, 7, 550–557. [Google Scholar] [CrossRef]
- Chuah, J.A.; Matsugami, A.; Hayashi, F.; Numata, K. Self-Assembled Peptide-Based System for Mitochondrial-Targeted Gene Delivery: Functional and Structural Insights. Biomacromolecules 2016, 17, 3547–3557. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Koilkonda, R.D.; Chou, T.H.; Porciatti, V.; Ozdemir, S.S.; Chiodo, V.; Boye, S.L.; Boye, S.E.; Hauswirth, W.W.; Lewin, A.S.; et al. Gene delivery to mitochondria by targeting modified adenoassociated virus suppresses Leber’s hereditary optic neuropathy in a mouse model. Proc. Natl. Acad. Sci. USA 2012, 109, E1238–E1247. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Ozdemir, S.S.; Koilkonda, R.D.; Chou, T.H.; Porciatti, V.; Chiodo, V.; Boye, S.L.; Hauswirth, W.W.; Lewin, A.S.; Guy, J. Mutant NADH dehydrogenase subunit 4 gene delivery to mitochondria by targeting sequence- modified adeno-associated virus induces visual loss and optic atrophy in mice. Mol. Vis. 2012, 18, 1668–1683. [Google Scholar] [PubMed]
- Suda, T.; Liu, D. Hydrodynamic gene delivery: Its principles and applications. Mol. Ther. 2007, 15, 2063–2069. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.X.; He, Y.F.; Shang, Y.Z.; Liu, H.L. Interaction between the Gemini Surfactant (12-6-12) and DNA. Acta Phys. Chim. Sin. 2011, 27, 156–162. [Google Scholar]
- Cardoso, A.M.; Faneca, H.; Almeida, J.A.; Pais, A.A.; Marques, E.F.; de Lima, M.C.; Jurado, A.S. Gemini surfactant dimethylene-1,2-bis(tetradecyldimethylammonium bromide)-based gene vectors: A biophysical approach to transfection efficiency. Biochim. Biophys. Acta 2011, 1808, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Del Gaizo, V.; Payne, R.M. A novel TAT-mitochondrial signal sequence fusion protein is processed, stays in mitochondria, and crosses the placenta. Mol. Ther. 2003, 7, 720–730. [Google Scholar] [CrossRef]
- Futaki, S.; Ohashi, W.; Suzuki, T.; Niwa, M.; Tanaka, S.; Ueda, K.; Harashima, H.; Sugiura, Y. Stearylated arginine-rich peptides: A new class of transfection systems. Bioconjug. Chem. 2001, 12, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.K.; Myc, A.; Silpe, J.E.; Sumit, M.; Wong, P.T.; McCarthy, K.; Desai, A.M.; Thomas, T.P.; Kotlyar, A.; Holl, M.M.B.; et al. Dendrimer-Based Multivalent Vancomycin Nanoplatform for Targeting the Drug-Resistant Bacterial Surface. ACS Nano 2013, 7, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.L.; Pandita, D.; Rodrigues, J.; Pego, A.P.; Granja, P.L.; Balian, G.; Tomas, H. Receptor-Mediated Gene Delivery Using PAMAM Dendrimers Conjugated with Peptides Recognized by Mesenchymal Stem Cells. Mol. Pharm. 2010, 7, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, M.A.; Orr, B.G.; Banaszak Holl, M.M. Diffusion NMR study of generation-five PAMAM dendrimer materials. J. Phys. Chem. B 2014, 118, 7195–7202. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Shao, N.M.; Zhang, Q.; Cheng, Y.Y. Mitochondrial targeting dendrimer allows efficient and safe gene delivery. J. Mater. Chem. B 2014, 2, 2546–2553. [Google Scholar] [CrossRef]
- Chacinska, A.; Koehler, C.M.; Milenkovic, D.; Lithgow, T.; Pfanner, N. Importing Mitochondrial Proteins: Machineries and Mechanisms. Cell 2009, 138, 628–644. [Google Scholar] [CrossRef] [PubMed]
- Pfanner, N.; Meijer, M. The Tom and Tim machine. Curr. Biol. 1997, 7, R100–R103. [Google Scholar] [CrossRef]
- Van der Laan, M.; Meinecke, M.; Dudek, J.; Hutu, D.P.; Lind, M.; Perschil, I.; Guiard, B.; Wagner, R.; Pfanner, N.; Rehling, P. Motor-free mitochondrial presequence translocase drives membrane integration of preproteins. Nat. Cell Biol. 2007, 9, 1152–1159. [Google Scholar] [CrossRef] [PubMed]
- Esaki, M.; Kanamori, T.; Nishikawa, S.; Endo, T. Two distinct mechanisms drive protein translocation across the mitochondrial outer membrane in the late step of the cytochrome b(2) import pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 11770–11775. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Midoux, P.; Pichon, C.; Yaouanc, J.J.; Jaffres, P.A. Chemical vectors for gene delivery: A current review on polymers, peptides and lipids containing histidine or imidazole as nucleic acids carriers. Br. J. Pharmacol. 2009, 157, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Daya, S.; Berns, K.I. Gene Therapy Using Adeno-Associated Virus Vectors. Clin. Microbiol. Rev. 2008, 21, 583–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, M.; Liu, F.; Chen, S.; Wang, M.; Cheng, A. Role of capsid proteins in parvoviruses infection. Virol. J. 2015, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Warrington, K.H.; Gorbatyuk, O.S.; Harrison, J.K.; Opie, S.R.; Zolotukhin, S.; Muzyczka, N. Adeno-associated virus type 2 VP2 capsid protein is nonessential and can tolerate large peptide insertions at its N terminus. J. Virol. 2004, 78, 6595–6609. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Mehta, A.; Wang, G.F.; Hauswirth, W.W.; Chiodo, V.; Boye, S.L.; Guy, J. Next-generation sequencing of mitochondrial targeted AAV transfer of human ND4 in mice. Mol. Vis. 2013, 19, 1482–1491. [Google Scholar] [PubMed]
- Coutinho, E.; Batista, C.; Sousa, F.; Queiroz, J.; Costa, D. Mitochondrial Gene Therapy: Advances in Mitochondrial Gene Cloning, Plasmid Production, and Nanosystems Targeted to Mitochondria. Mol. Pharm. 2017, 14, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Harashima, H. Enhancement in selective mitochondrial association by direct modification of a mitochondrial targeting signal peptide on a liposomal based nanocarrier. Mitochondrion 2013, 13, 526–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.; Mortensen, L.J.; Ravichandran, S.; Bentley, K.; DeLouise, L.A. Effect of Nanoparticle Surface Coating on Cell Toxicity and Mitochondria Uptake. J. Biomed. Nanotechnol. 2017, 13, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Mari, M.; Morales, A.; Colell, A.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Mitochondrial glutathione, a key survival antioxidant. Antioxid. Redox Signal. 2009, 11, 2685–2700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bibb, M.J.; Van Etten, R.A.; Wright, C.T.; Walberg, M.W.; Clayton, D.A. Sequence and gene organization of mouse mitochondrial DNA. Cell 1981, 26 Pt 2, 167–180. [Google Scholar] [CrossRef]
- Bonawitz, N.D.; Clayton, D.A.; Shadel, G.S. Initiation and beyond: Multiple functions of the human mitochondril transcription machinery. Mol. Cell 2006, 24, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta 1999, 1410, 103–123. [Google Scholar] [CrossRef]
- Walberg, M.W.; Clayton, D.A. Sequence and properties of the human KB cell and mouse L cell D-loop regions of mitochondrial DNA. Nucleic Acids Res. 1981, 9, 5411–5421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Holt, I.J.; Reyes, A. Human mitochondrial DNA replication. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Zollo, O.; Tiranti, V.; Sondheimer, N. Transcriptional requirements of the distal heavy-strand promoter of mtDNA. Proc. Natl. Acad. Sci. USA 2012, 109, 6508–6512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boshart, M.; Weber, F.; Jahn, G.; Dorsch-Hasler, K.; Fleckenstein, B.; Schaffner, W. A very strong enhancer is located upstream of an immediate early gene of human cytomegalovirus. Cell 1985, 41, 521–530. [Google Scholar] [CrossRef] [Green Version]
- Gammage, P.A.; Gaude, E.; Van Haute, L.; Rebelo-Guiomar, P.; Jackson, C.B.; Rorbach, J.; Pekalski, M.L.; Robinson, A.J.; Charpentier, M.; Concordet, J.P.; et al. Near-complete elimination of mutant mtDNA by iterative or dynamic dose-controlled treatment with mtZFNs. Nucleic Acids Res. 2016, 44, 7804–7816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, M.; Bacman, S.R.; Peralta, S.; Falk, M.J.; Chomyn, A.; Chan, D.C.; Williams, S.L.; Moraes, C.T. MitoTALEN: A General Approach to Reduce Mutant mtDNA Loads and Restore Oxidative Phosphorylation Function in Mitochondrial Diseases. Mol. Ther. 2015, 23, 1592–1599. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.V.; Bacman, S.R.; Arguello, T.; Zekonyte, U.; Williams, S.L.; Edgell, D.R.; Moraes, C.T. mitoTev-TALE: A monomeric DNA editing enzyme to reduce mutant mitochondrial DNA levels. EMBO Mol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Borgeld, H.J.; Zhang, J.; Muramatsu, S.; Gong, J.S.; Yoneda, M.; Maruyama, W.; Naoi, M.; Ibi, T.; Sahashi, K.; et al. Gene therapy for mitochondrial disease by delivering restriction endonuclease SmaI into mitochondria. J. Biomed. Sci. 2002, 9 Pt 1, 534–541. [Google Scholar]
- Yang, Y.; Wu, H.; Kang, X.; Liang, Y.; Lan, T.; Li, T.; Tan, T.; Peng, J.; Zhang, Q.; An, G.; et al. Targeted elimination of mutant mitochondrial DNA in MELAS-iPSCs by mitoTALENs. Protein Cell 2018, 9, 283–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuppen, H.A.; Blakely, E.L.; Turnbull, D.M.; Taylor, R.W. Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta 2010, 1797, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Menezes, M.P.; Ouvrier, R.A. Peripheral neuropathy associated with mitochondrial disease in children. Dev. Med. Child Neurol. 2012, 54, 407–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cwerman-Thibault, H.; Augustin, S.; Ellouze, S.; Sahel, J.A.; Corral-Debrinski, M. Gene therapy for mitochondrial diseases: Leber Hereditary Optic Neuropathy as the first candidate for a clinical trial. C. R. Biol. 2014, 337, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Man, P.Y.W.; Turnbull, D.M.; Chinnery, P.F. Leber hereditary optic neuropathy. J. Med. Genet. 2002, 39, 162–169. [Google Scholar] [CrossRef] [Green Version]
- Tzen, C.Y.; Thajeb, P.; Wu, T.Y.; Chen, S.C. Melas with point mutations involving tRNALeu (A3243G) and tRNAGlu(A14693g). Muscle Nerve 2003, 28, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Hajek, P.; Chomyn, A.; Chan, E.; Seo, B.B.; Matsuno-Yagi, A.; Yagi, T.; Attardi, G. Lack of complex I activity in human cells carrying a mutation in MtDNA-encoded ND4 subunit is corrected by the Saccharomyces cerevisiae NADH-quinone oxidoreductase (NDI1) gene. J. Biol. Chem. 2001, 276, 38808–38813. [Google Scholar] [CrossRef] [PubMed]
- Seo, B.B.; Kitajima-Ihara, T.; Chan, E.K.; Scheffler, I.E.; Matsuno-Yagi, A.; Yagi, T. Molecular remedy of complex I defects: Rotenone-insensitive internal NADH-quinone oxidoreductase of Saccharomyces cerevisiae mitochondria restores the NADH oxidase activity of complex I-deficient mammalian cells. Proc. Natl. Acad. Sci. USA 1998, 95, 9167–9171. [Google Scholar] [CrossRef] [PubMed] [Green Version]

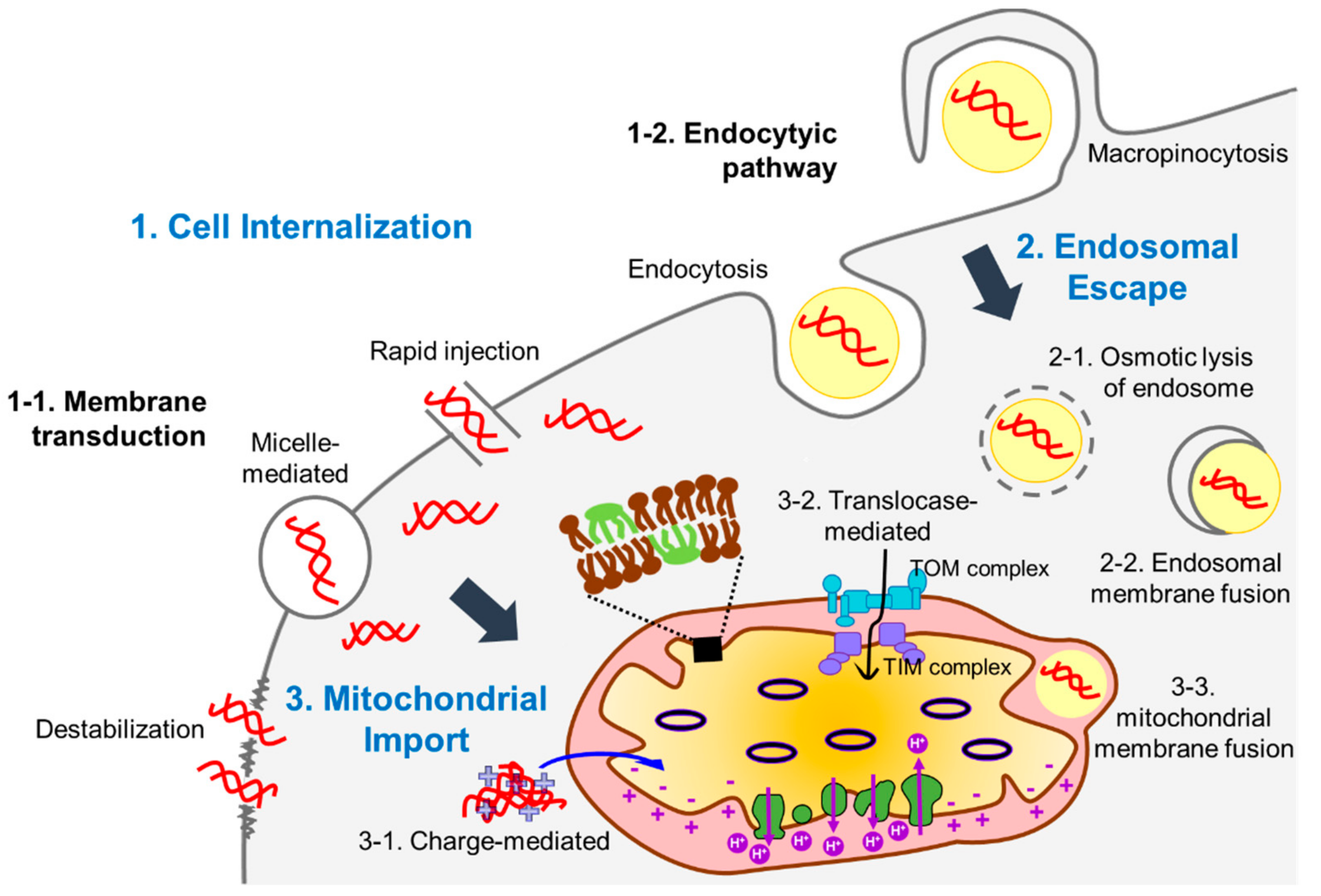

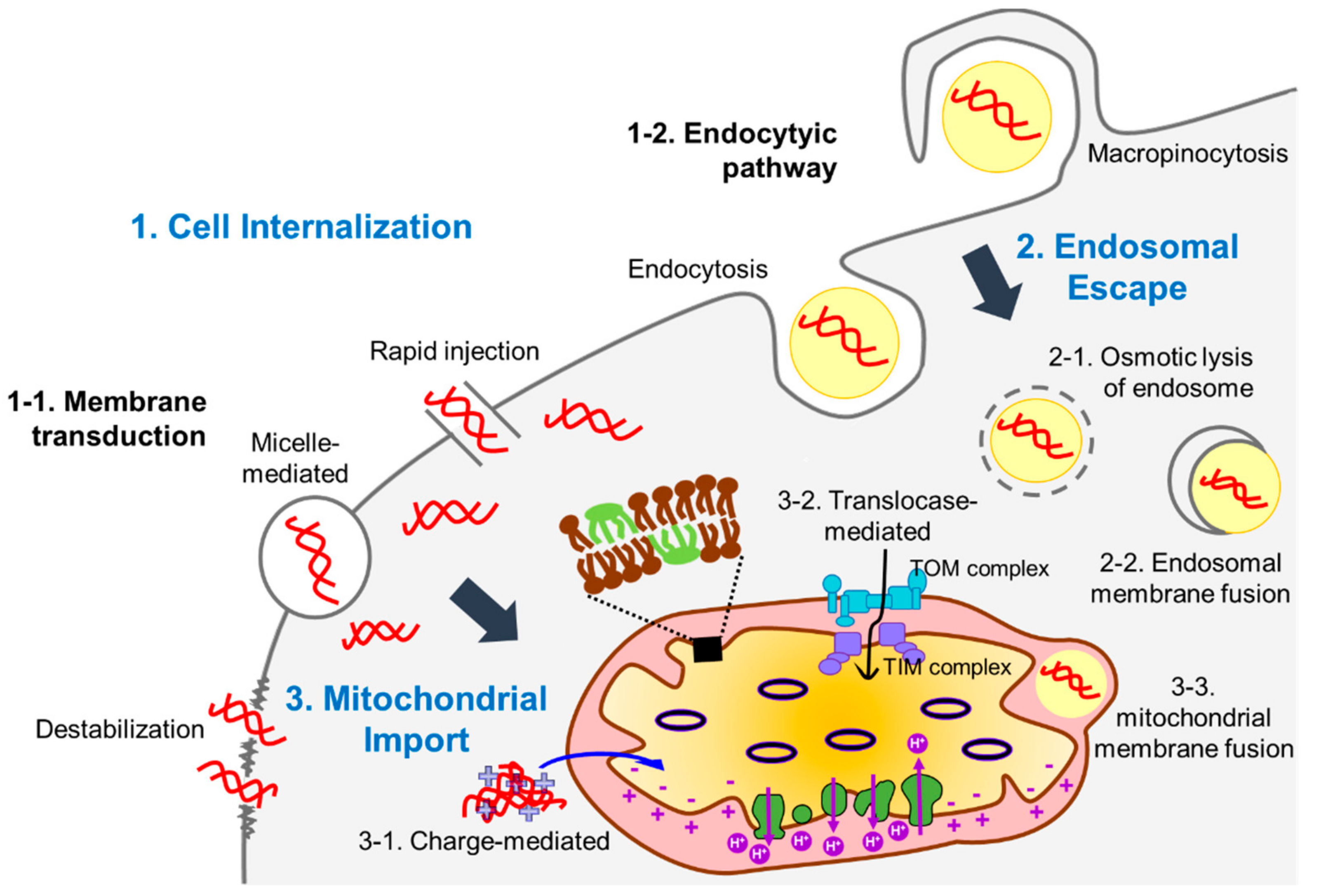
{kind=link}
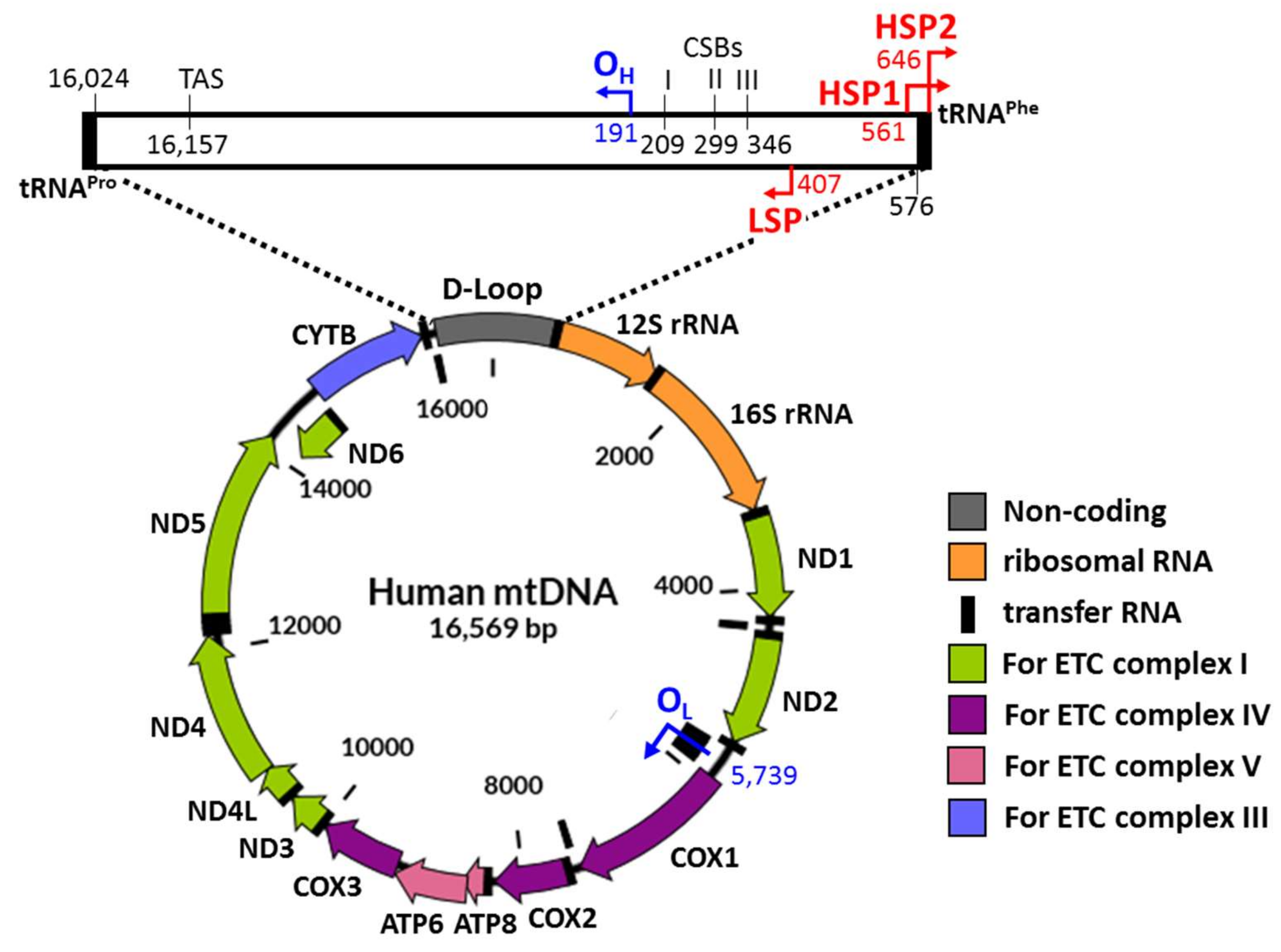
{kind=link}
| Classification | Key Acting Component | Delivery-Target Systems | Strategy | Advantages | Limitations | Ref. |
|---|---|---|---|---|---|---|
| Physical | Hydrodynamic injection | Rat, Mice | Cell penetration by hydrodynamic force | Simplicity | No mitochondria-targeting | [5,6,7] |
| Biolistics | Yeast | Cell penetration by bombardment | Cell type-independent | Potential cell damage, No mitochondria-targeting | [8] | |
| Chemical | Gemini surfactants | HeLa cell | Formation of cationic micelle-like structure with DNA | Works in small dose | Weak specificity to mitochondria | [9] |
| Rhodamine 123 | Normal human dermal fibroblast (NHDF) adult donor cell | Precipitation of DNA using lipophilic molecule with delocalized positive charge | Can be traced due to fluorescence | Expression from transferred DNA has not been confirmed | [10,11] | |
| DQAsomes | BT 20 cell | Transport of DNA by ampiphilic and cationic lipid-based vesicle | High specificity to mitochondria | Low transfection efficiency, Cytotoxicity | [12,13,14] | |
| DQA80s | Primary human dermal fibroblasts, HeLa cell | Lipid incorporation into DQAsomes | Improved transfection efficiency | Delivery of DNA around mitochondria not into the matrix | [15] | |
| MITO-Porter | HeLa cell, Rat | Membrane fusion by lipid-based nano carrier | Easy surface modification | [16,17,18,19] | ||
| R8-MITO-Porter | Rat | Enhancing cellular uptake by functionalization with cationic peptide | High fusogenic activity with mitochondrial outer membrane | Low fusogenic activity with mitochondrial inner membrane, Moderate cytotoxicity | [16,17] | |
| KALA-MITO-Porter | HeLa cell, Mice | Enhancing cellular uptake by functionalization with membrane destabilizing peptide | Improved transfection efficiency | High cytotoxicity | [19,20] | |
| STPP-liposome | 4T1 cell, Mice | Conjugation of stearyl residue to lipophilic and cationic TPP for ampiphilic property | Selective accumulation in mitochondria | Cytotoxicity | [21] | |
| TPP-PEG-PE liposome | HeLa, 4T1 cell, Mice | Substitution of stearyl moiety with biocompatible PEG-PE polymer | Decreased cytotoxicity | Transgene expression not confirmed | [22] | |
| TPP-PAMAM dendrimer | HeLa, MCF-7, 4T1, NIH 3T3 cell | DNA condensation by high positive surface charge, endosomal escape by free tertiary amine groups | Efficient endosomal escape and high serum resistance | Transgene expression not confirmed | [23] | |
| Biological | MTS-PNA | Myoblasts, Fibroblasts, NT 2, IMR 32, HeLa, HepG2, C2C12 cell | MTS-guided localization of DNA hybridized with PNA to mitochondria | High specificity to mitochondria via actions of translocase | Only can transfer short nucleic acids | [24,25] |
| MTS-KH peptide | HEK 293 cell | Mitochondrial localization of MTS-conjugated DNA-binding peptide and exogenous DNA complex | Can transfer large DNA with high specificity to mitochondria | [26] | ||
| MTS-AAV | Neuronal G11778A NT 2 cybrid, HEK 293T cell, Mice | Mitochondrial localization of DNA by inserting MTS into the AAV capsid | Proven effects of transgene expression | Inability to carry large DNA | [27,28] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, Y.-h.; Lim, K.-i. Recent Advances in Mitochondria-Targeted Gene Delivery. Molecules 2018, 23, 2316. https://doi.org/10.3390/molecules23092316
Jang Y-h, Lim K-i. Recent Advances in Mitochondria-Targeted Gene Delivery. Molecules. 2018; 23(9):2316. https://doi.org/10.3390/molecules23092316
Chicago/Turabian StyleJang, Yoon-ha, and Kwang-il Lim. 2018. "Recent Advances in Mitochondria-Targeted Gene Delivery" Molecules 23, no. 9: 2316. https://doi.org/10.3390/molecules23092316
APA StyleJang, Y.-h., & Lim, K.-i. (2018). Recent Advances in Mitochondria-Targeted Gene Delivery. Molecules, 23(9), 2316. https://doi.org/10.3390/molecules23092316