Abstract
The ketones was successfully prepared from secondary alcohols using 9-azabicyclo[3.3.1]nonane-N-oxyl (ABNO) as the catalyst and 2,6-lutidine as the base in acetonitrile solution. The electrochemical activity of ABNO for oxidation of 1-phenylethanol was investigated by cyclic voltammetry, in situ Fourier transform infrared spectroscopy (FTIR) and constant current electrolysis experiments. The resulting cyclic voltammetry indicated that ABNO exhibited much higher electrochemical activity when compared with 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) under the similar conditions. A reasonable reaction mechanism of the electrocatalytic oxidation of 1-phenylethanol to acetophenone was proposed. In addition, a series of secondary alcohols could be converted to the corresponding ketones at room temperature in 80–95% isolated yields.
1. Introduction
The carbonyl compounds are important intermediates for synthesis of fine chemicals, such as fragrances, pharmaceuticals, and food additives, and the worldwide demand for carbonyl compounds is increasing every year [1,2,3]. Presently, the preparation of aldehydes and ketones by selective oxidation of the corresponding primary and secondary alcohols is one of the most fundamental functional group transformations in organic chemistry [4,5,6,7]. The stoichiometric or excess amounts of oxidants containing chromium reagents, manganese reagents, or hypervalent iodine are often required among the traditional protocols [8,9,10]. Unfortunately, these oxidants would generate equivalent wastes and pose a risk to the environment [11,12]. It is well known that green chemistry is a crucial principle of chemical research and its application in organic synthesis has attracted more and more attention [13]. From both environmentally benign and sustainable viewpoints, it is a great desire to develop a green and efficient catalyst for the synthesis of the corresponding carbonyl compounds.
Recently, a stable nitroxyl radical 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) has been successfully applied to synthesize aldehydes, nitriles, and imines because of its cleaness and inexpensiveness [14,15,16]. Several combinations of TEMPO and transition metals, especially Fe and Cu, have been widely used for the synthesis of aldehydes from alcohols [17,18,19,20,21,22,23,24,25,26,27,28,29]. Despite of using molecular oxygen as the terminal oxidant, the transition-metal catalyst still exists in these processes. Subsequently, our group developed several catalytic oxidation systems for synthesizing the carbonyl compounds without transition metals, such as TEMPO/HBr/tert-butyl nitrite (TBN)/O2, TEMPO/TBN/O2, TEMPO/2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ)/TBN/O2, etc [30,31,32]. Although a lot of meaningful works about the preparation of aldehydes from primary alcohols catalyzed by TEMPO were studied both in homogeneous and heterogeneous conditions, due to the high steric hindrance arising from four methyl group in TEMPO, the synthesis of ketones from secondary alcohols is still a serious challenge [33,34,35,36].
Subsequently, several less hindered nitroxyl radicals, such as 2-azaadamantan-N-oxyl (AZADO), 1-Me-AZADO, and 5-F-AZADO, have been utilized by Iwabuchi and co-workers [37,38,39]. Owing to their less steric hindrance around the reaction center, AZADO and its derivatives exhibited high activity at a low loading for the preparation of carbonyl compounds from selective oxidation of high hindered alcohols as compared with TEMPO [40]. However, AZADOs should be synthesized through long and complicated processes. Another nitroxyl radical, 9-azabicyclo[3.3.1]nonane-N-oxyl (ABNO) has the similar activity with AZADOs and it could be prepared via only four steps [41]. It put forward the possibility to prepare the corresponding carbonyl compounds from primary and secondary alcohols with ABNO as the catalyst under mild conditions. Previously, the ABNO/TBN/O2 system for the aerobic oxidation of secondary alcohols in water has been developed [42]. This reaction should be performed at 80 °C under 0.3–0.5 Mpa of oxygen.
As we all know, the electrochemical method in synthesis possesses the advantages of friendly environment, low cost, and high atom utilization, paving a new way for production of many fine chemicals [43,44,45,46]. In 2012, Raja et al. disclosed an electrochemical method to achieve the preparation of aromatic aldehydes with sodium nitrate as an effective redox mediator in biphasic medium [47]. However, the primary alcohols substituted with strong electron-withdrawing group, such as -nitro and some of secondary alcohols gave low product yields. Lately, we prepared a series of TEMPO-modified polymer electrodes, which could be used successfully for the selective oxidation of alcohols to aldehydes [48,49,50]. It is well known that the detailed information of the electrochemical behaviour of organic compounds is crucial for electrochemical synthesis. Stahl’s group reported that 4-acetamido-TEMPO (ACT) possessed of the superior activity than AZADO and ABNO at high pH with electrocatalytic studies about the nitroxyl/oxoammonium redox potential [51]. In continuation with our previous work about the synthesis of aldehydes from alcohols, we try to study the electrochemical performance of ABNO as the catalyst for the selective oxidation of secondary alcohols in the presence of 2,6-lutidine in acetonitrile solution. In this work, the electrochemical behaviour of ABNO for the oxidation of 1-phenylethanol to acetophenone on the Pt electrode was studied to a better understanding of the catalytic properties (Scheme 1).
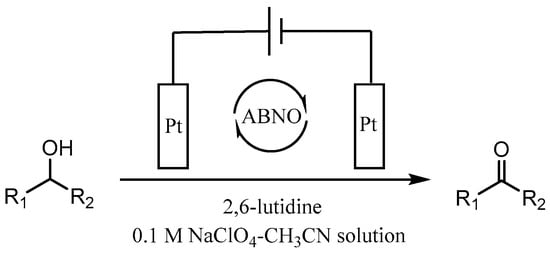
Scheme 1.
The electrochemical synthesis of ketones from secondary alcohols.
2. Results and Discussion
2.1. Cyclic Voltammetric Study for Oxidation of 1-Phenylethanol
The cyclic voltammograms for the oxidation of 1-phenylethanol in 0.1 M NaClO4-CH3CN solution using ABNO as the catalyst were shown in Figure 1. We first started out investigation in 0.1 M NaClO4-CH3CN solution containing 1-phenylethanol (1.0 mmol) and 2,6-lutidine (1.0 mmol) at Pt electrode (Figure 1a). No obvious reaction peaks appeared in the range of scanning potential between −0.1 and 0.8 V. As shown in Figure 1b, a pair of reversible redox peaks was well centered at about 0.25 V in the presence of ABNO (0.1 mmol). The oxidation peak at 0.3 V corresponded to the one-electron oxidation of nitroxyl radical to oxoammonium ion (ABNO+) rather than the response of 1-phenylethanol or 2,6-lutidine [51]. By the addition of 1-phenylethanol, the reaction was so weak that we could not detect the obvious change about the oxidation peak current (Figure 1c). However, when the base 2,6-lutidine was added to the reaction solution, the apparent increase of oxidation peak current from 1.37 mA to 1.51 mA was observed (Figure 1d). It certified that the active oxoammonium cations reacted with 1-phenylethanol on the surface of the working electrode and 2,6-lutidine was beneficial to facilitate the oxidation of 1-phenylethanol [52].
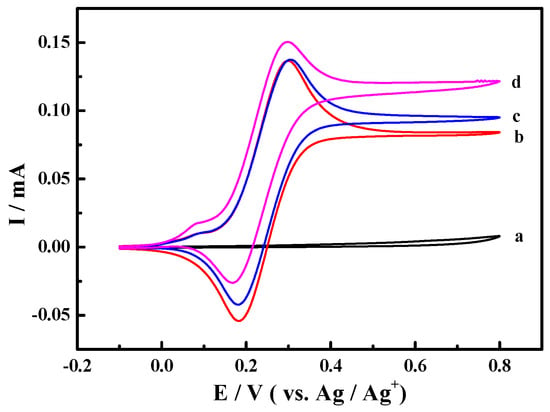
Figure 1.
Cyclic voltammograms of Pt electrode in 0.1 M NaClO4-CH3CN solution with (a) 1-phenylethanol (1.0 mmol) and 2,6-lutidine (1.0 mmol); (b) active oxoammonium cations (ABNO) (0.1 mmol); (c) ABNO (0.1 mmol) and 1-phenylethanol (1.0 mmol); and, (d) ABNO (0.1 mmol), 1-phenylethanol (1.0 mmol) and 2,6-lutidine (1.0 mmol) at the scan rate of 50 mV·s−1.
For comparison, other experiments for oxidation of 1-phenylethanol in 0.1 M NaClO4-CH3CN solution using TEMPO as the catalyst under the similar conditions have been conducted. As shown in Figure 2, the redox peaks at about 0.34 and 0.22 V could be seen in the presence of TEMPO (0.1 mmol), which were corresponded to one electron transfer from TEMPO to TEMPO+ [53]. When 1-phenylethanol and 2,6-lutidine were added to the above reaction solution sequentially, the corresponding cyclic voltammograms were almost overlapped and were omitted in Figure 2 hence.
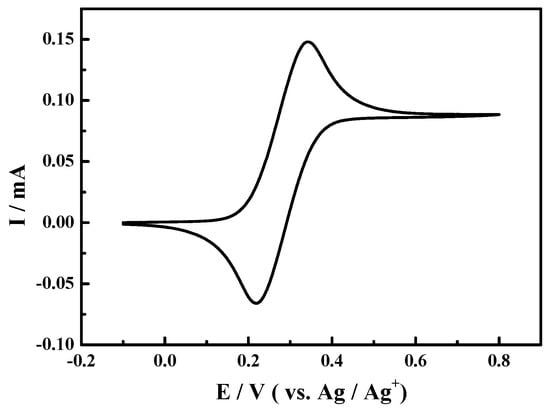
Figure 2.
Cyclic voltammogram of Pt electrode in 0.1 M NaClO4-CH3CN solution with 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) (0.1 mmol) at the scan rate of 50 mV·s−1.
To futher investigate the electrochemical behavior of ABNO for 1-phenylethanol oxidation, the cyclic voltammograms with various scan rates were studied and shown in Figure 3A. Due to the high solution resistance in CH3CN solution, the peak-to-peak separation between the oxidation and reduction peaks was about 100 mV at the scan rate of 50 mV·s−1, which was slightly more than the 59 mV separation [54]. Nevertheless, with the increasing of scanning rate, there was a less shift in the oxidation peak, suggesting that the electrochemical reaction of ABNO still had good reversibility [55]. The excellent linear correlation between the oxidation peak current (Ip) and the square root of scan rate (Figure 3B) exhibited that the electrode reaction was also a diffusion-controlled process, which provided further evidence for reacting on the surface of working electrode [56].
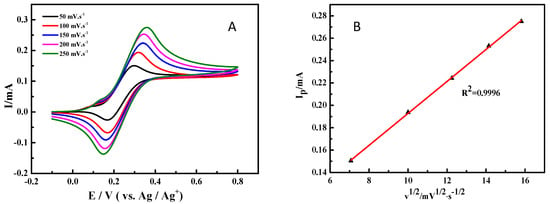
Figure 3.
(A) Cyclic voltammograms for the oxidation of 1-phenylethanol (1.0 mmol) with ABNO (0.1 mmol) and 2,6-lutidine (1.0 mmol) in 0.1 M NaClO4-CH3CN solution at various scan rates. (B) Linear plot between the oxidation peak current (Ip) and the square root of scan rate.
2.2. In situ FTIR Spectroscopic Analysis
In situ FTIR spectroscopy is one of the most useful techniques in electrochemistry today, and it can be used to study the organic reaction at the electrode surface. Figure 4 showed the in situ FTIR spectra that were obtained during the oxidation of 1-phenylethanol in the presence of ABNO and 2,6-lutidine in 0.1 M NaClO4-CH3CN solution at the scanning potentials varying from 0 to 800 mV. When the sample potential reached to 200 mV, some weak bands emerged gradually. With the increasing of potential, the corresponding infrared spectra signals could be seen clearly at approximately 400 mV. The downward bands at 1646, 1629, and 1587 cm−1 were contributed to ring stretching vibration of 2,6-lutidinium cation [57]. The band at 1176 cm−1 was related to C-H in-plane bending of 2,6-lutidinium cation [58,59]. It confirmed that 2,6-lutidine got a hydrogen proton to become 2,6-lutidinium cation during the electrocatalytic process [60,61]. Meanwhile, three negative-going bands at 1685, 1360, and 1267 cm−1 were detected, which were attributed to C=O stretching vibration, C-H deformation vibration in the methyl group, and skeleton vibration of acetophenone, respectively [62]. Moreover, a weak but important upward band of N-O stretching vibration, showing the participation of ABNO in this reaction, was observed at 1369 cm−1 [49]. The other band located at 1129 cm−1 was assigned to ClO4− ions of supporting electrolyte [63,64]. However, with the interference of the nearby signal peaks at about 1280 and 1627 cm−1, the ring stretching modes of 2,6-lutidinium cation and N+=O stretching vibration of oxoammonium ion could not be obviously found [48]. From the above results, we could get the conclusion that 1-phenylethanol was oxidized to acetophenone with ABNO as the catalyst and 2,6-lutidine as the base.
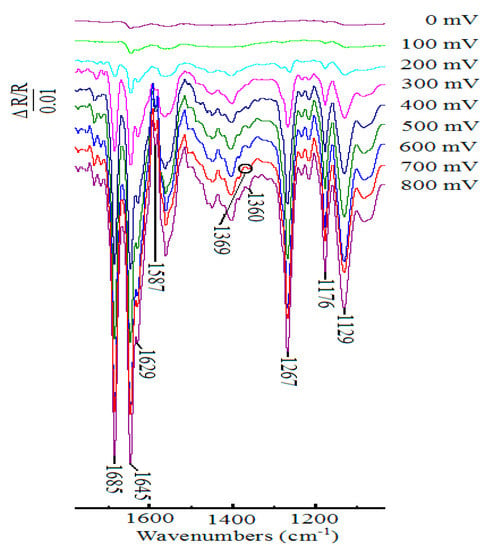
Figure 4.
In situ FTIR spectra collected on Pt disk electrode during the oxidation of 1-phenylethanol (1.0 mmol) in the presence of ABNO (0.1 mmol) and 2,6-lutidine (1.0 mmol) in 0.1 M NaClO4-CH3CN solution in a short time interval 10 s.
To further observe the spectral changes with increasing time, the in situ time-resolved FTIR spectra experiment was carried out during the oxidation of 1-phenylethanol on Pt disk electrode with ABNO as the catalyst at 400 mV. The resulting spectra were displayed in Figure 5. The seven negative signals at 1685, 1360, and 1267 cm−1 and at 1645, 1629, 1587, and 1176 cm−1, illustrated to acetophenone and 2,6-lutidinium cation, respectively, could still be seen clearly [58,59,62]. The intensity of these bands significantly increased with the increasing of time. Meanwhile, no new bands emerged, which showed that no new reaction occurred on the electrode surface.
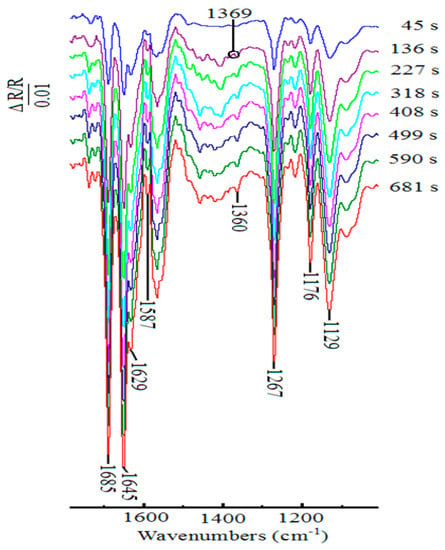
Figure 5.
In situ time-resolved FTIR spectra collected on Pt disk electrode during the oxidation of 1-phenylethanol (1.0 mmol) in the presence of ABNO (0.1 mmol) and 2,6-lutidine (1.0 mmol) in 0.1 M NaClO4-CH3CN solution at 400 mV.
For comparision, the in situ FTIR spectra collected during the oxidation of 1-phenylethanol in the presence of 2,6-lutidine with TEMPO as the catalyst and without any catalyst in 0.1 M NaClO4-CH3CN solution under the similar conditions were obtained. The spectra at 400 mV were put together and presented in Figure 6. As shown in Figure 6a, no additional bands were observed except for the band of supporting electrolyte at Pt electrode without any catalyst. In addition, with the addition of TEMPO as the catalyst in the reaction solution, all of the characteristic bands changed weakly (Figure 6b). Only with ABNO as the catalyst in the reaction solution, the intensity of the bands enhanced markedly, which was identify with cyclic voltammograms results (Figure 6c). Thus, ABNO could act as an efficient catalyst for the oxidation of secondary alcohols instead of TEMPO under the same conditions [65,66].
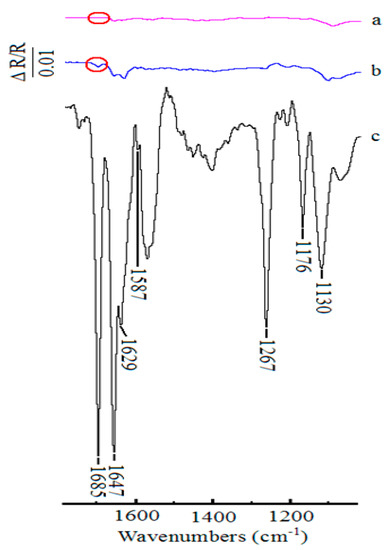
Figure 6.
Comparison of in situ FTIR spectra collected on Pt disk electrode during the oxidation of 1-phenylethanol (1.0 mmol) in the presence of 2,6-lutidine (1.0 mmol) at 400 mV. (a) without any catalyst, (b) with TEMPO (0.1 mmol) as the catalyst, (c) with ABNO (0.1 mmol) as the catalyst.
Based on the above observation and relevant literatures, a plausible mechanism for the oxidation of 1-phenylethanol was presented in Scheme 2. The catalyst ABNO could be oxidized to generate the active oxoammonium cations (ABNO+) via one electronic transfer process on the anode electrode [36,67]. Subsequently, 1-phenylethanol reacted with ABNO+ to give acetophenone and 9-hydroxy-9-azabicyclo[3.3.1]nonane (ABNOH), accompanied by one hydrogen proton generation [42,68]. Meanwhile, ABNOH was regenerated to ABNO [51,69]. The base 2,6-lutidine got the hydrogen proton to form 2,6-lutidinium cation during the oxidation reaction [48]. In addition, H2 was generated as the side product on the cathode electrode [54].
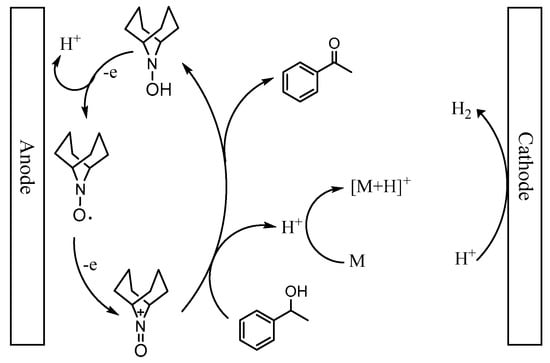
Scheme 2.
A plausible mechanism for the oxidation of 1-phenylethanol in the presence of ABNO and 2,6-lutidine (M).
2.3. Preparation Electrolysis
Inspired by these results, the scope of this electrochemical method for the oxidation of secondary alcohols was explored. As shown in Table 1, 1-phenylethanol could be converted completely to acetophenone in 98% GC internal standard yield (entry 1). Due to the volatility of acetophenone during the process of purification and concentration, the isolated yield was relatively low (80%). Meanwhile, we tried to calculate the faradaic efficiency in this electrochemical process. The faradaic efficiencies with 1-phenylethanol were about 81.4% at 6 h, with 92% conversion and 99% selectivity. With the reaction proceeding, the concentration of the substrate was decreased and the reaction slowed down gradually. The faradaic efficiency was about 61.8% at 8.5 h. Similarly, a series of secondary benzyl alcohols with both electro-donating and electron-withdrawing substituents in phenyl group were provided in good to excellent isolated yields (entires 2–11). For example, four secondary benzylic alcohols with electro-donating group were converted into the corresponding ketones with good isolated yields under the standard conditions (entries 5–8). Meanwhile, halide substituents products such as chloro-acetophenone (o-, m-, and p-), 4-bromo-acetophenone and 4-fluoro-acetophenone were all obtained in excellent isolated yields (entries 2–4,10,11). Furthermore, 1-(4-nitrophenyl)ethanol could afford the target product efficiently (entry 9). When 1-phenylpropan-1-ol was chosen as the substrate, a full conversion with 88% isolated yield could be achieved (entry 12). Other excellent results were also obtained, including 1-phenyl-1-butanol and 1-(p-tolyl)propan-1-ol (entries 13 and 14). Notably, the conversion of 2-methyl-1-phenylpropan-1-ol could reach to 98% by prolonging the reaction time to 11 h (entry 15). The selectivity to 2-methylpropiophenone was 92% and the main by-product was benzaldehyde. Next, the polycyclic and heterocyclic substrates were smoothly converted to the corresponding ketones with satisfying isolated yields (entries 16–18). In order to explore the applicability of the protocol for unactivated secondary aliphatic alcohols, cycloheptanol and 2-octanol were tested. Fortunately, the high GC yields of their corresponding ketones were received in 98% and 90%, respectively (entries 19 and 20).

Table 1.
Electrochemical conversion of various secondary alcohols to ketones a.
3. Materials and Methods
3.1. Catalyst Preparation and Reagents
ABNO was synthesized in our laboratory according to the available procedures [41]. Other chemicals and solvents were purchased from the supplier and were used as received. In this work, the racemic secondary alcohols were used.
3.2. Cyclic Voltammetry Study
The cyclic voltammetric study was carried out by using CHI620B electrochemical workstation (CH Instrument Inc., Austin, TX, USA) with an “L” type Pt electrode as the working electrode. A big square platinum sheet (1.5 cm in length) and a Ag/Ag+ electrode (0.1 M AgNO3 in acetonitrile) were employed as the counter electrode and the reference one, respectively. The experiments were performed in 0.1 M NaClO4-CH3CN solution at the scan rate of 50 mV·s−1 at room temperature. All of the potentials in this article were referred to the Ag/Ag+ electrode (0.1 M AgNO3 in acetonitrile).
3.3. In Situ FTIR Spectroscopic Study
The in situ infrared spectroscopy experiments were performed on 263 A Potentiostat/Galvanostat and Nicolet 670 FTIR spectrometer (Thermo Fisher Nicolet, Waltham, MA, USA), equipped with a refrigerated MCT-A detector and KBr beam splitter. The disk electrode of Pt (0.6 cm in diameter) was used as the working electrode. A three-electrode spectro-electrochemical cell equipped with CaF2 window as the IR window was used for collecting the interferograms. Each single-beam spectrum was collected and two hundred interferograms were coadded at a spectral resolution of 8 cm−1. The sample potentials varied in a stepwise fashion from 0 to 800 mV and the reference potential was fixed at −100 mV [70,71,72].
3.4. Preparation Electrolysis Experiments
The preparative electrolysis experiments were conducted with in an undivided cell containing 0.1 M NaClO4-CH3CN solution (15 mL), alcohol substrate (1.0 mmol), ABNO (0.1 mmol), and 2,6-lutidine (1.0 mmol) at a constant current of 10.0 mA with moderate magnetic stirring for 8.5 h in the atmosphere. Two square platinum sheets were employed as the anode and cathode, respectively. The electrolytic reaction was monitored by gas chromatography (GC) on a GC-2010 system (Shimadzu, Kyoto, Japan) equipped with a SH-Rtx-Was polar column and a flame ionization detector (FID). Both the injector and detector were maintained at 220 °C, the carrier gas is nitrogen, and the flow rate is 1.2 mL/min. The initial oven temperature of 100 °C was held for 2 min and then ramped up at 15 °C per min to 220 °C. This final temperature was held for 8 min. After the reaction was finished, the resulting mixture was concentrated in a rotary evaporator (Heidolph, Schwabach, Germany) and purified by column chromatography on silica gel using petroleum and ethyl acetate 15:1) as eluent to afford the products. The products were confirmed by GC-MS, 1H-NMR, and 13C-NMR. NMR spectroscopy was carried out on a Bruker Avance III spectrometer (Bruker, Fällanden, Switzerland). The GC-MS analysis was measured on Thermo Trace ISQ instrument (Thermo Fisher Nicolet, Waltham, MA, USA) with TG 5MS capillary column.
Acetophenone (colorless oil, yield 80%): 1H-NMR (500 MHz, CDCl3) δ 7.94–7.92 (m, 2H), 7.55–7.51 (m,1H), 7.44–7.41 (m, 2H), 2.57 (s, 3H). 13C-NMR (125 MHz, CDCl3) δ 198.0, 136.9, 132.9, 128.4, 128.1, 26.4. GC-MS (EI): m/z: 120.14 [M+].
1-(4-Chlorophenyl)ethanone (colorless oil, yield 94%): 1H-NMR (500 MHz, CDCl3) δ 7.89–7.87 (m, 2H), 7.42–7.41 (m, 2H), 2.57 (s, 3H). 13C-NMR (125 MHz, CDCl3) δ 196.7, 139.5, 135.4, 129.7, 128.8, 26.5. GC-MS (EI): m/z: 154.03 [M+].
1-(3-Chlorophenyl)ethanone (colorless oil, yield 88%): 1H-NMR (500 MHz, CDCl3) δ 7.92–7.91 (m, 1H), 7.83–7.81 (m, 1H), 7.54–7.51 (m, 1H), 7.42–7.39 (m, 1H), 2.59 (s, 3H). 13C-NMR (125 MHz, CDCl3) δ 196.6, 138.6, 134.9, 133.0, 129.9, 128.3, 126.4, 26.6. GC-MS (EI): m/z: 154.17 [M+].
1-(2-Chlorophenyl)ethanone (colorless oil, yield 87%): 1H-NMR (500 MHz, CDCl3) δ 7.56–7.54 (m, 1H), 7.43–7.37 (m, 2H), 7.34–7.31 (m, 1H), 2.65 (s, 3H). 13C-NMR (125 MHz, CDCl3) δ 200.4, 139.1, 132.0, 131.3, 130.6, 129.4, 126.9, 30.7. GC-MS (EI): m/z: 154.11 [M+].
1-(4-Fluorophenyl)ethanone (colorless oil, yield 85%): 1H-NMR (500 MHz, CDCl3) δ 8.00–7.96 (m, 2H), 7.15–7.10 (m, 2H), 2.58 (s, 3H). 13C-NMR (125 MHz, CDCl3) δ 196.4, 165.8 (d, J = 254.8 Hz), 133.6 (d, J = 2.4 Hz), 130.9 (d, J = 9.3 Hz), 115.6 (d, J = 21.9 Hz), 26.5. GC-MS (EI): m/z: 138.06 [M+].
1-(4-Bromophenyl)ethanone (white solid, yield 92%): 1H-NMR (500 MHz, CDCl3) δ 7.83–7.80 (m, 2H), 7.61–7.59 (m, 2H), 2.58 (s, 3H). 13C-NMR (125 MHz, CDCl3) δ 196.9, 135.8, 131.9, 129.8, 128.3, 26.5. GC-MS (EI): m/z: 197.98, 200.04 [M+].
1-(4-Methylphenyl)ethanone (colorless oil, yield 92%): 1H-NMR (500 MHz, CDCl3) δ 7.86–7.84(m, 2H), 7.28–7.24 (m, 2H), 2.56 (s, 3H), 2.40 (s, 3H). 13C-NMR (125 MHz, CDCl3) δ 197.7, 143.8, 134.7, 129.2, 128.4, 26.4, 21.5. GC-MS (EI): m/z: 134.15 [M+].
1-(3-Methylphenyl)ethanone (colorless oil, yield 93%): 1H-NMR (500 MHz, CDCl3) δ 7.77–7.74(m, 2H), 7.38–7.33 (m, 2H), 2.59 (s, 3H), 2.41 (s, 3H). 13C-NMR (125 MHz, CDCl3) δ 198.3, 138.3, 137.1, 133.8, 128.7, 128.4, 125.5, 26.6, 21.2. GC-MS (EI): m/z: 134.10 [M+].
1-(2-Methylphenyl)ethanone (colorless oil, yield 95%): 1H-NMR (500 MHz, CDCl3) δ 7.71–7.69(m, 1H), 7.40–7.36 (m, 1H), 7.28–7.24 (m, 2H), 2.58 (s, 3H), 2.54 (s, 3H). 13C-NMR (125 MHz, CDCl3) δ 201.7, 138.3, 137.6, 132.0, 131.5, 129.3, 125.6, 29.5, 21.5. GC-MS (EI): m/z: 134.09 [M+].
1-(4-Methoxyphenyl)ethanone (colorless oil, yield 89%): 1H-NMR (500 MHz, CDCl3) δ 7.94–7.91 (m, 2H), 6.94–6.91 (m, 2H), 3.86 (s, 3H), 2.55 (s, 3H). 13C-NMR (125 MHz, CDCl3) δ 196.7, 163.5, 130.5, 130.3, 113.6, 55.4, 26.3. GC-MS (EI): m/z: 150.14 [M+].
1-(4-Nitrophenyl)ethanone (yellow crystal powder, yield 90%): 1H-NMR (500 MHz, CDCl3) δ 8.28–8.27 (m, 2H), δ 8.11–80.08 (m, 2H), δ 2.66 (s, 3H); 13C-NMR (125 MHz, CDCl3) δ 196.3, 150.3, 141.3, 129.2, 123.7, 26.9. GC-MS (EI): m/z: 165.05 [M+].
Propiophenone (colorless oil, yield 88%): 1H-NMR (500 MHz, CDCl3) δ 7.99–7.97 (m, 2H), 7.58–7.55(m, 1H), 7.49–7.45 (m, 2H), 3.04–3.00 (m, 2H), 1.24 (t, J = 7.2 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ 200.9, 137.0, 132.9, 128.6, 128.0, 31.8, 8.3. GC-MS (EI): m/z: 134.16 [M+].
Phenylbutanone (colorless oil, yield 92%): 1H-NMR (500 MHz, CDCl3) δ 7.95–7.93 (m, 2H), 7.54–7.50(m, 1H), 7.44–7.41 (m, 2H), 2.94–2.91 (m, 2H), 1.79–1.72 (m, 2H), 0.99 (t, J = 7.4 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ 200.2, 137.0, 132.7, 128.4, 127.9, 40.3, 17.6, 13.7. GC-MS (EI): m/z: 148.17 [M+].
2-Methylpropiophenone (colorless oil, yield 89%): 1H-NMR (500 MHz, CDCl3) δ 7.98–7.96 (m, 2H), 7.58–7.54 (m, 1H), 7.49–7.46 (m, 2H), 3.60–3.54 (m, 1H), 1.23 (d, J = 6.9 Hz, 6H). 13C-NMR (125 MHz, CDCl3) δ 204.5, 136.3, 132.8, 128.6, 128.3, 35.3, 19.1. GC-MS (EI): m/z: 148.18 [M+].
1-(4-Methylphenyl)propan-2-one (colorless oil, yield 93%): 1H-NMR (500 MHz, CDCl3) δ 7.87 (d, J = 8.2 Hz, 2H), 7.28–7.25 (m, 2H), 3.00–2.96 (m, 2H), 2.41 (s, 3H), 1.22 (t, J = 7.3 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ 200.5, 143.5, 134.4, 129.2, 128.1, 31.6, 21.5, 8.3. GC-MS (EI): m/z: 148.16 [M+].
1-Tetralone (white solid, yield 90%): 1H-NMR (500 MHz, CDCl3) δ 8.04–8.03 (m, 1H), 7.48–7.45 (m, 1H), 7.30 (t, J = 7.5 Hz, 1H), 7.25 (d, J = 7.6 Hz, 1H), 2.97 (t, J = 6.1 Hz, 2H), 2.67–2.64 (m, 2H), 2.17–2.12 (m, 2H). 13C-NMR (125 MHz, CDCl3) δ 198.3, 144.4, 133.3, 132.6, 128.7, 127.1, 126.6, 39.1, 29.7, 23.2. GC-MS (EI): m/z: 146.17 [M+].
Benzophenone (white solid, yield 95%): 1H-NMR (500 MHz, CDCl3) δ 7.82 (d, J = 7.4 Hz, 4H), 7.60 (t, J = 7.4 Hz, 2H), 7.49 (t, J = 7.6 Hz, 4H). 13C-NMR (125 MHz, CDCl3) δ 196.7, 137.6, 132.4, 130.0, 128.2. GC-MS (EI): m/z: 182.00 [M+].
1-(Thiophen-2-yl)ethanone (yellow oil, yield 80%): 1H-NMR (500 MHz, CDCl3) δ 7.69–7.68 (m, 1H), 7.63–7.62 (m, 1H), 7.12–7.10 (m, 1H), 2.55 (s, 3H). 13C-NMR (125 MHz, CDCl3) δ 190.6, 144.5, 133.7, 132.4, 128.0, 26.8. GC-MS (EI): m/z: 125.97 [M+].
Cycloheptanone (colorless oil, yield 57%): 1H-NMR (500 MHz, CDCl3) δ 2.49–2.47 (m, 4H), 1.70–1.64 (m, 8H); 13C-NMR (125 MHz, CDCl3) δ 215.3, 43.8, 30.4, 24.3. GC-MS (EI): m/z: 112.11 [M+].
4. Conclusions
In conclusion, the electrochemical behaviour of the nitroxyl radical ABNO for the oxidation of 1-phenylethanol in acetonitrile solution have been studied. The electrochemical measurements revealed that ABNO showed reversible redox behavior and it had lower potential than TEMPO. The oxoammonium ion (ABNO+) was generated by single-electron oxidation. According to the in situ FTIR spectra results, the base 2,6-lutidine received a hydrogen proton to become 2,6-lutidinium cation, which was a crucial process during the synthesis of acetophenone from 1-phenylethanol. Under mild reaction conditions, a variety of substrates, including aromatic, heteroaromatic, and aliphatic secondary alcohols could be converted to the corresponding ketones with good to excellent isolated yields.
Author Contributions
Funding acquisition, M.L.; Investigation, P.N. and X.L.; Supervision, Z.S. and M.L.; Writing–original draft, P.N.; Writing–review & editing, Z.S. and M.L.
Funding
This research was funded by the National Natural Science Foundations of China (21773211 and 21776260), and the Natural Science Foundation of Zhejiang Province (LY17B060007).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Musawir, M.; Davey, P.N.; Kelly, G.; Kozhevnikov, I.V. Highly efficient liquid-phase oxidation of primary alcohols to aldehydes with oxygen catalysed by Ru-Co oxide. Chem. Commun. 2003, 34, 1414–1415. [Google Scholar] [CrossRef]
- Gilhespy, M.; Lok, M.; Baucherel, X. Polymer-supported nitroxyl radical catalyst for selective aerobic oxidation of primary alcohols to aldehydes. Chem. Commun. 2005, 8, 1085–1086. [Google Scholar] [CrossRef] [PubMed]
- Zhan, B.Z.; Thompson, A. Recent developments in the aerobic oxidation of alcohols. Tetrahedron 2004, 60, 2917–2935. [Google Scholar] [CrossRef]
- Demizu, Y.; Shiigi, H.; Oda, T.; Matsumura, Y.; Onomura, O. Efficient oxidation of alcohols electrochemically mediated by azabicyclo-N-oxyls. Tetrahedron Lett. 2008, 49, 48–52. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Arends, I.W.C.E.; Dijksman, A. New developments in catalytic alcohol oxidations for fine chemicals synthesis. Catal. Today 2000, 57, 157–166. [Google Scholar] [CrossRef]
- Lenze, M.; Bauer, E.B. Chemoselective, iron(II)-catalyzed oxidation of a variety of secondary alcohols over primary alcohols utilizing H2O2 as the oxidant. Chem. Commun. 2013, 49, 5889–5891. [Google Scholar] [CrossRef] [PubMed]
- Okimoto, M.; Yoshida, T.; Hoshi, M.; Chiba, T.; Maeo, K. Successful application of indirct electrooxidation for the transformation of biaryl methanols to the corresponding biaryl ketones. Synthetic Commun. 2011, 41, 3134–3139. [Google Scholar] [CrossRef]
- Jiang, N.; Ragauskas, A.J. TEMPO-catalyzed oxidation of benzylic alcohols to aldehydes with the H2O2/HBr/ionic liquid [bmim]PF6 system. Tetrahedron Lett. 2005, 46, 3323–3326. [Google Scholar] [CrossRef]
- Uyanik, M.; Ishihara, K. Hypervalent iodine-mediated oxidation of alcohols. Chem. Commun. 2009, 2086–2099. [Google Scholar] [CrossRef]
- Sharma, V.B.; Jain, S.L.; Sain, B. A New and Efficient Transition Metal-Free Oxidation of Secondary Alcohols to Ketones Using Aqueous HBr and H2O2. Synlett 2005, 1, 173–175. [Google Scholar] [CrossRef]
- Raju, T.; Manivasagan, S.; Revathy, B.; Kulangiappar, K.; Muthukumaran, A. A mild and efficient method for the oxidation of benzylic alcohols by two-phase electrolysis. Tetrahedron Lett. 2007, 48, 3681–3684. [Google Scholar] [CrossRef]
- Campestrini, S.; Carraro, M.; Ciriminna, R.; Pagliaro, M.; Tonellato, U. Alcohols oxidation with hydrogen peroxide promoted by TPAP-doped ormosils. Tetrahedron Lett. 2004, 45, 7283–7286. [Google Scholar] [CrossRef]
- Roschangar, F.; Sheldon, R.A.; Senanayake, C.H. Overcoming barriers to green chemistry in the pharmaceutical industry-the Green Aspiration LevelTM concept. Green Chem. 2015, 17, 752–768. [Google Scholar] [CrossRef]
- Sun, Y.B.; Cao, C.Y.; Wei, F.; Huang, P.P.; Yang, S.L.; Song, W.G. Nanocarbon-based TEMPO as stable heterogeneous catalysts for partial oxidation of alcohols. Sci. Bull. 2016, 61, 772–777. [Google Scholar] [CrossRef]
- Dornan, L.M.; Cao, Q.; Flanagan, J.C.A.; Crawford, J.J.; Cook, M.J.; Muldoon, M.J. Copper/TEMPO catalysed synthesis of nitriles from aldehydes or alcohols using aqueous ammonia and with air as the oxidant. Chem. Commun. 2013, 49, 6030–6032. [Google Scholar] [CrossRef] [PubMed]
- Sonobe, T.; Oisaki, K.; Kanai, M. Catalytic aerobic production of imines en route to mild, green, and concise derivatizations of amines. Chem. Sci. 2012, 3, 3249–3255. [Google Scholar] [CrossRef]
- Hoover, J.M.; Stahl, S.S. Highly Practical Copper(I)/TEMPO Catalyst System for Chemoselective Aerobic Oxidation of Primary Alcohols. J. Am. Chem. Soc. 2011, 133, 16901–16910. [Google Scholar] [CrossRef]
- Liu, L.; Ji, L.Y.; Wei, Y.Y. Base promoted aerobic oxidation of alcohols to corresponding aldehydes or ketones catalyzed by CuCl/TEMPO. Catal. Commun. 2008, 9, 1379–1382. [Google Scholar]
- Sand, H.; Weberskirch, R. Bipyridine copper functionalized polymer resins as support materials for the aerobic oxidation of alcohols. Polym. Int. 2017, 66, 428–435. [Google Scholar] [CrossRef]
- Kumpulainen, E.T.T.; Koskinen, A.M.P. Catalytic Activity Dependency on Catalyst Components in Aerobic Copper-TEMPO Oxidation. Chem. Eur. J. 2009, 15, 10901–10911. [Google Scholar] [CrossRef]
- Badalyan, A.; Stahl, S.S. Cooperative electrocatalytic alcohol oxidation with electron-proton-transfer mediators. Nature 2016, 535, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Hoover, J.M.; Steves, J.E.; Stahl, S.S. Copper(I)/TEMPO-catalyzed aerobic oxidation of primary alcohols to aldehydes with ambient air. Nat. Protoc. 2012, 7, 1161–1166. [Google Scholar] [CrossRef] [PubMed]
- Hoover, J.M.; Ryland, B.L.; Stahl, S.S. Copper/TEMPO-Catalyzed Aerobic Alcohol Oxidation: Mechanistic Assessment of Different Catalyst Systems. ACS Catal. 2013, 3, 2599–2605. [Google Scholar] [CrossRef] [PubMed]
- Ryland, B.L.; McCann, S.D.; Brunold, T.C.; Stahl, S.S. Mechanism of Alcohol Oxidation Mediated by Copper(II) and Nitroxyl Radicals. J. Am. Chem. Soc. 2014, 136, 12166–12173. [Google Scholar] [CrossRef] [PubMed]
- Lagerblom, K.; Wrigstedt, P.; Keskivali, J.; Parviainen, A.; Repo, T. Iron-Catalysed Selective Aerobic Oxidation of Alcohols to Carbonyl and Carboxylic Compounds. ChemPlusChem 2016, 81, 1160–1165. [Google Scholar] [CrossRef]
- Gamez, P.; Arends, I.W.C.E.; Sheldon, R.A.; Reedijk, J. Room Temperature Aerobic Copper-Catalysed Selective Oxidation of Primary Alcohols to Aldehydes. Adv. Synth. Catal. 2004, 346, 805–811. [Google Scholar] [CrossRef]
- Steves, J.E.; Stahl, S.S. Stable TEMPO and ABNO Catalyst Solutions for User-Friendly (bpy)Cu/Nitroxyl-Catalyzed Aerobic Alcohol Oxidation. J. Org. Chem. 2015, 80, 11184–11188. [Google Scholar] [CrossRef]
- Hoover, J.M.; Ryland, B.L.; Stahl, S.S. Mechanism of Copper(I)/TEMPO-Catalyzed Aerobic Alcohol Oxidation. J. Am. Chem. Soc. 2013, 135, 2357–2367. [Google Scholar] [CrossRef]
- Hossain, M.M.; Shyu, S.G. Efficient and Selective Aerobic Alcohol Oxidation Catalyzed by Copper(II)/2,2,6,6,-Tetramethylpiperidine-1-oxyl at Room Temperature. Adv. Synth. Catal. 2010, 352, 3061–3068. [Google Scholar] [CrossRef]
- Xie, Y.; Mo, W.M.; Xu, D.; Shen, Z.L.; Sun, N.; Hu, B.X.; Hu, X.Q. Efficient NO Equivalent for Activation of Molecular Oxygen and Its Applications in Transition-Metal-Free Catalytic Aerobic Alcohol Oxidation. J. Org. Chem. 2007, 72, 4288–4291. [Google Scholar] [CrossRef]
- He, X.J.; Shen, Z.L.; Mo, W.M.; Sun, N.; Hu, B.X.; Hu, X.Q. TEMPO-tert-Butyl Nitrite: An Efficient Catalytic System for Aerobic Oxidation of Alcohols. Adv. Synth. Catal. 2009, 351, 89–92. [Google Scholar] [CrossRef]
- Shen, Z.L.; Chen, M.; Fang, T.T.; Li, M.C.; Mo, W.M.; Hu, B.X.; Sun, N.; Hu, X.Q. Transformation of ethers into aldehydes or ketones: A catalytic aerobic deprotection/oxidation pathway. Tetrahedron Lett. 2015, 56, 2768–2772. [Google Scholar] [CrossRef]
- Fey, T.; Fischer, H.; Bachmann, S.; Albert, K.; Bolm, C. Silica-Supported TEMPO Catalysts: Synthesis and Application in the Anelli Oxidation of Alcohols. J. Org. Chem. 2001, 66, 8154–8159. [Google Scholar] [CrossRef] [PubMed]
- Shakir, A.J.; Paraschivescu, C.; Matache, M.; Tudose, M.; Mischie, A.; Spafiu, F.; Ionita, P. A convenient alternative for the selective oxidation of alcohols by silica supported TEMPO using dioxygen as the final oxidant. Tetrahedron Lett. 2015, 56, 6878–6881. [Google Scholar] [CrossRef]
- Karimi, B.; Badreh, E. SBA-15-functionalized TEMPO confined ionic liquid: An efficient catalyst system for transition-metal-free aerobic oxidation of alcohols with improved selectivity. Org. Biomol. Chem. 2011, 9, 4194–4198. [Google Scholar] [CrossRef] [PubMed]
- Karimi, B.; Farhangi, E.; Vali, H.; Vahdati, S. SBA-15-Functionalized 3-Oxo-ABNO as Recyclable Catalyst for Aerobic Oxidation of Alcohols under Metal-Free Conditions. ChemSusChem 2014, 7, 2735–2741. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M.; Tomizawa, M.; Suzuki, I.; Iwabuchi, Y. 2-Azaadamantane N-Oxyl (AZADO) and 1-Me-AZADO: Highly Efficient Organocatalysts for Oxidation of Alcohols. J. Am. Chem. Soc. 2006, 128, 8412–8413. [Google Scholar] [CrossRef]
- Iwabuchi, Y. Discovery and Exploitation of AZADO: The Highly Active Catalyst for Alcohol Oxidation. Chem. Pharm. Bull. 2013, 61, 1197–1213. [Google Scholar] [CrossRef]
- Shibuya, M.; Osada, Y.; Sasano, Y.; Tomizawa, M.; Iwabuchi, Y. Highly Efficient, Organocatalytic Aerobic Alcohol Oxidation. J. Am. Chem. Soc. 2011, 133, 6497–6500. [Google Scholar] [CrossRef]
- Hamada, S.; Furuta, T.; Wada, Y.; Kawabata, T. Chemoselective Oxidation by Electronically Tuned Nitroxyl Radical Catalysts. Angew. Chem. Int. Ed. 2013, 52, 8093–8097. [Google Scholar] [CrossRef]
- Lauber, M.B.; Stahl, S.S. Efficient Aerobic Oxidation of Secondary Alcohols at Ambient Temperature with an ABNO/NOx Catalyst System. ACS Catal. 2013, 3, 2612–2616. [Google Scholar] [CrossRef]
- Ma, J.Q.; Hong, C.; Wan, Y.; Li, M.C.; Hu, X.Q.; Mo, W.M.; Hu, B.X.; Sun, N.; Jin, L.Q.; Shen, Z.L. Aerobic oxidation of secondary alcohols in water with ABNO/tert-butyl nitrite/KPF6 catalytic system. Tetrahedron Lett. 2017, 58, 652–657. [Google Scholar] [CrossRef]
- Yoshida, J.I.; Kataoka, K.; Horcajada, R.; Nagaki, A. Modern Strategies in Electroorganic Synthesis. Chem. Rev. 2008, 108, 2265–2299. [Google Scholar] [CrossRef] [PubMed]
- Francke, R.; Little, R.D. Redox catalysis in organic electrosynthesis: Basic principles and recent developments. Chem. Soc. Rev. 2014, 45, 2492–2521. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.H.; Gao, X.F.; Gao, J.; Yuan, G.Q. A highly efficient electrochemical route for the conversion of aldehydes to nitriles. Sci. China Chem. 2015, 58, 747–750. [Google Scholar] [CrossRef]
- Chen, C.; Niu, P.F.; Shen, Z.L.; Li, M.C. Electrochemical Sulfenylation of Indoles with Disulfides Mediated by Potassium Iodide. J. Electrochem. Soc. 2018, 165, 67–74. [Google Scholar] [CrossRef]
- Christopher, C.; Lawrence, S.; Kulandainathan, M.A.; Kulangiappar, K.; Raja, M.E.; Xavier, N.; Raja, S. Electrochemical selective oxidation of aromatic alcohols with sodium nitrate mediator in biphasic medium at ambient temperature. Tetrahedron Lett. 2012, 53, 2802–2804. [Google Scholar] [CrossRef]
- Lu, J.J.; Ma, J.Q.; Yi, J.M.; Shen, Z.L.; Zhong, Y.J.; Ma, C.A.; Li, M.C. Electrochemical Polymerization of Pyrrole Containing TEMPO Side Chain on Pt Electrode and Its Electrochemical Activity. Electrochim. Acta 2014, 130, 412–417. [Google Scholar] [CrossRef]
- Wang, X.; Ma, J.Q.; Song, D.D.; Shen, Z.L.; Li, M.C.; Ma, C.A. Characterization and Electrocatalytic Activity of Poly(4-thienylacetyl-oxy-2,2,6,6-tetramethylpiperidin-1-yloxy) Prepared by Electrochemical Polymerization. ECS Electrochem. Lett. 2014, 3, 12–15. [Google Scholar] [CrossRef]
- Tang, D.Y.; Yang, X.J.; Chen, Q.G.; Shen, Z.L.; Li, M.C. Efficient Electrooxidation of Alcohols Using TEMPO-Modified Polyaniline Electrode Prepared by Electrochemical Polymerization. J. Electrochem. Soc. 2016, 163, 321–326. [Google Scholar] [CrossRef]
- Rafiee, M.; Miles, K.C.; Stahl, S.S. Electrocatalytic Alcohol Oxidation with TEMPO and Bicyclic Nitroxyl Derivatives: Driving Force Trumps Steric Effects. J. Am. Chem. Soc. 2015, 137, 14751–14757. [Google Scholar] [CrossRef] [PubMed]
- Herath, A.C.; Becker, J.Y. 2,2,6,6-Tetramethyl piperidine-1-oxyl (TEMPO)-mediated catalytic oxidation of benzyl alcohol in acetonitrile and ionic liquid 1-butyl-3-methyl-imidazolium hexafluorophosphate [BMIm][PF6]: Kinetic analysis. Electrochim. Acta 2008, 53, 4324–4330. [Google Scholar] [CrossRef]
- Fan, Z.Q.; Yang, X.J.; Chen, C.; Shen, Z.L.; Li, M.C. One-Pot Electrochemical Oxidation of Alcohols to Nitriles Mediated by TEMPO. J. Electrochem. Soc. 2017, 164, 54–58. [Google Scholar] [CrossRef]
- Das, A.; Stahl, S.S. Noncovalent Immobilization of Molecular Electrocatalysts for Chemical Synthesis: Efficient Electrochemical Alcohol Oxidation with a Pyrene-TEMPO Conjugate. Angew. Chem. Int. Ed. 2017, 56, 8892–8897. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.N.; Yoo, S.J.; Li, L.J.; Zeng, C.C.; Little, R.D. A comparative study of organic electron transfer redox mediators: Electron transfer kinetics for triarylimidazole and triarylamine mediators in the oxidation of 4-methoxybenzyl alcohol. Electrochim. Acta 2014, 142, 254–260. [Google Scholar] [CrossRef]
- Zhang, K.Y.; Lu, N.N.; Yoo, S.J.; Hu, L.M.; Little, R.D.; Zeng, C.C. Electrochemical analysis of the triarylimidazole-type organic redox catalysts: Chemical stability and homogeneous electron transfer kinetics for the oxidation of 4-methoxybenzyl alcohol. Electrochim. Acta 2016, 199, 357–365. [Google Scholar] [CrossRef]
- Yi, J.M.; Tang, D.Y.; Song, D.D.; Wu, X.H.; Shen, Z.L.; Li, M.C. Selective oxidation of benzyl alcohol on poly(4-(3-(pyrrol-1-yl)propionamido)-2,2,6,6-tetramethylpiperidin-1-yloxy) electrode. J. Solid State Electrochem. 2015, 19, 2291–2297. [Google Scholar] [CrossRef]
- Timmiati, S.N.; Jalil, A.A.; Triwahyono, S.; Setiabudi, H.D.; Annuar, N.H.R. Formation of acidic Brönsted (MoOx)-(Hy)+ evidenced by XRD and 2,6-lutidine FTIR spectroscopy for cumene cracking. Appl. Catal. A Gen. 2013, 459, 8–16. [Google Scholar] [CrossRef]
- Wang, M.L.; Zhang, Y.Y.; Xie, Q.J.; Yao, S.Z. In situ FT-IR spectroelectrochemical study of electrooxidation of pyridoxol on a gold electrode. Electrochim. Acta 2005, 51, 1059–1068. [Google Scholar] [CrossRef]
- Song, D.D.; Chen, Q.G.; Tang, D.Y.; Shen, Z.L.; Li, M.C.; Ma, C.A. Electropolymerization and Electrocatalytic Activity of Poly(4-thienylacetyl-amino-2,2,6,6-tetramethylpiperidinyl-1-yloxy)/(2,2-bithiophene) Copolymer. J. Electrochem. Soc. 2015, 162, 251–255. [Google Scholar] [CrossRef]
- Comminges, C.; Barhdadi, R.; Doherty, A.P.; O’Toole, S.; Troupel, M. Mechanism of 2,2′6,6′-Tetramethylpiperidin-N-oxyl-Mediated Oxidation of Alcohols in Ionic Liquids. J. Phys. Chem. A 2008, 112, 7848–7855. [Google Scholar] [CrossRef] [PubMed]
- Malyala, R.V.; Rode, C.V.; Arai, M.; Hegde, S.G.; Chaudhari, R.V. Activity, selectivity and stability of Ni and bimetallic Ni-Pt supported on zeolite Y catalysts for hydrogenation of acetophenone and its substituted derivatives. Appl. Catal. A Gen. 2000, 193, 71–86. [Google Scholar] [CrossRef]
- Yang, X.J.; Fan, Z.Q.; Shen, Z.L.; Li, M.C. Electrocatalytic synthesis of nitriles from aldehydes with ammonium acetate as the nitrogen source. Electrochim. Acta 2017, 226, 53–59. [Google Scholar] [CrossRef]
- Souto, R.M.; Rodriguez, J.L.; Pastor, E. Spectroscopic Investigation of the Adsorbates of Benzyl Alcohol on Palladium. Langmuir 2000, 16, 8456–8462. [Google Scholar] [CrossRef]
- Kim, M.J.; Jung, Y.E.; Lee, C.Y.; Kim, J. HKUST-1/ABNO-catalyzed aerobic oxidation of secondary benzyl alcohols at room temperature. Tetrahedron Lett. 2018, 59, 2722–2725. [Google Scholar] [CrossRef]
- Steves, J.E.; Stahl, S.S. Copper(I)/ABNO-Catalyzed Aerobic Alcohol Oxidation: Alleviating Steric and Electronic Constraints of Cu/TEMPO Catalyst Systems. J. Am. Chem. Soc. 2013, 135, 15742–15745. [Google Scholar] [CrossRef] [PubMed]
- Gerken, J.B.; Pang, Y.Q.; Lauber, M.B.; Stahl, S.S. Structural Effects on the pH-Dependent Redox Properties of Organic Nitroxyls: Pourbaix Diagrams for TEMPO, ABNO, and Three TEMPO Analogs. J. Org. Chem. 2018, 83, 7323–7330. [Google Scholar] [CrossRef]
- Walroth, R.C.; Miles, K.C.; Lukens, J.T.; MacMillan, S.N.; Stahl, S.S.; Lancaster, K.M. Electronic Structural Analysis of Copper(II)-TEMPO/ABNO Complexes Provides Evidence for Copper(I)-Oxoammonium Character. J. Am. Chem. Soc. 2017, 139, 13507–13517. [Google Scholar] [CrossRef]
- Kadoh, Y.; Tashiro, M.; Oisaki, K.; Kanai, M. Organocatalytic Aerobic Oxidation of α-Fluoroalkyl Alcohols to Fluoroalkyl Ketones at Room Temperature. Adv. Synth. Catal. 2015, 357, 2193–2198. [Google Scholar] [CrossRef]
- Zhou, Z.Y.; Wang, Q.; Lin, J.L.; Tian, N.; Sun, S.G. In situ FTIR spectroscopic studies of electrooxidation of ethanol on Pd electrode in alkaline media. Electrochim. Acta 2010, 55, 7995–7999. [Google Scholar] [CrossRef]
- Sun, S.G.; Lin, Y. Kinetics of isopropanol oxidation on Pt(111), Pt(110), Pt(100), Pt(610) and Pt(211) single crystal electrodes-Studies of in situ time-resolved FTIR spectroscopy. Electrochim. Acta 1998, 44, 1153–1162. [Google Scholar] [CrossRef]
- Chen, Q.G.; Fang, C.J.; Shen, Z.L.; Li, M.C. Electrochemical synthesis of nitriles from aldehydes using TEMPO as a mediator. Electrochem. Commun. 2016, 64, 51–55. [Google Scholar] [CrossRef]
Sample Availability: Not available. |
















































© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).