Fungi-Mediated Biotransformation of the Isomeric Forms of the Apocarotenoids Ionone, Damascone and Theaspirane



Abstract
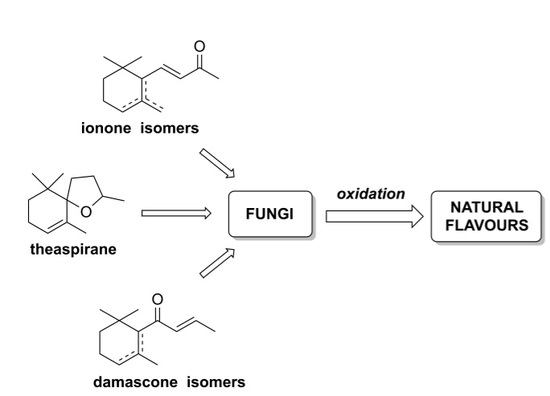
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Materials and General Methods
3.2. Analytical Methods and Characterization of the Products Deriving from the Biotransformation Experiments
3.3. Microorganisms and Biotransformation Experiments
3.3.1. Representative Procedures for Biotransformations
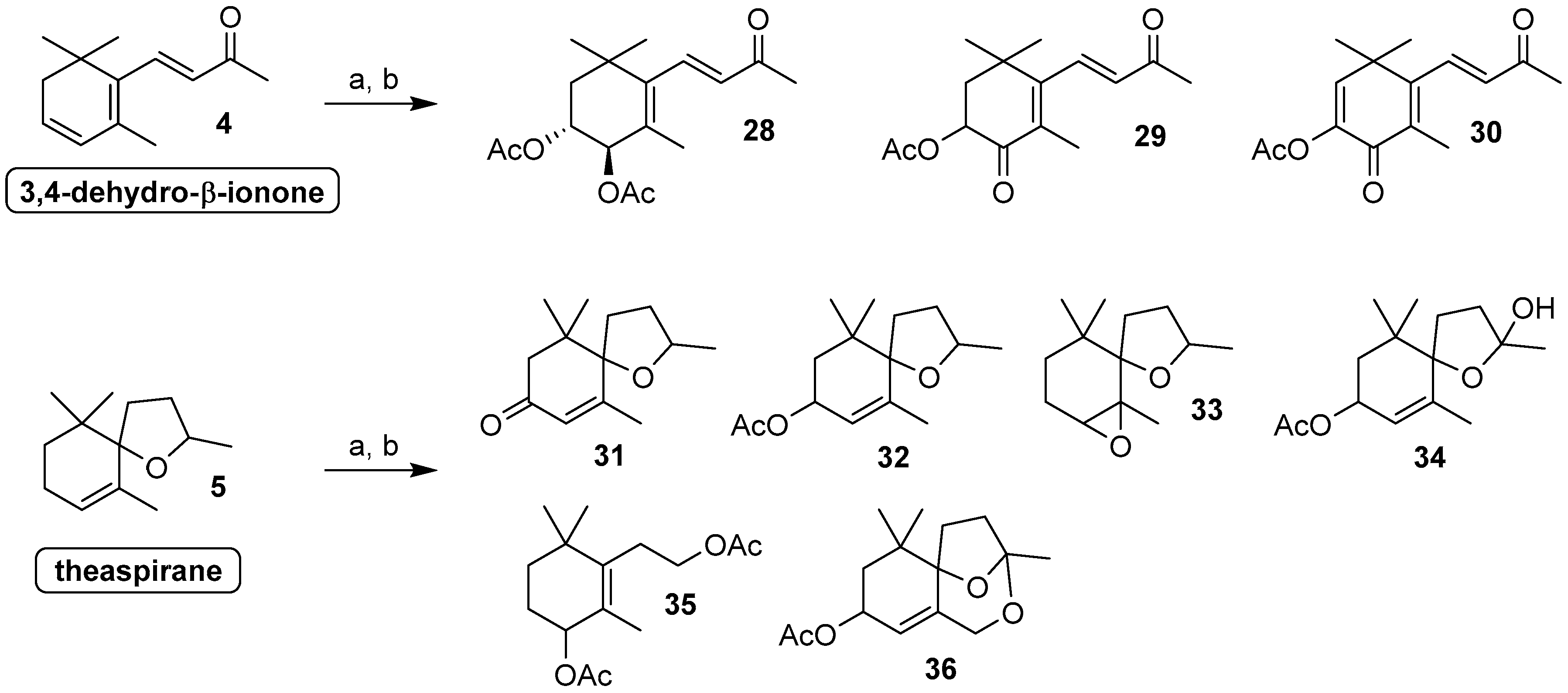
3.3.2. Preparative Biotransformations and Chemical Characterization of Compounds 23a, 25, 28, 35 and 36
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Walter, M.H.; Strack, D. Carotenoids and their cleavage products: Biosynthesis and functions. Nat. Prod. Rep. 2011, 28, 663–692. [Google Scholar] [CrossRef] [PubMed]
- Serra, S. Recent advances in the synthesis of carotenoid-derived flavours and fragrances. Molecules 2015, 20, 12817–12840. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.-G.; Wang, Y.-G.; Liu, W.-M.; Wei, R.-R.; Yang, J.-B.; Wang, A.-G.; Ji, T.-F.; Tian, J.; Su, Y.-L. Hepatoprotective sesquiterpenes and rutinosides from Murraya koenigii (L.) Spreng. J. Agric. Food Chem. 2014, 62, 4145–4151. [Google Scholar] [CrossRef] [PubMed]
- Schievano, E.; Morelato, E.; Facchin, C.; Mammi, S. Characterization of markers of botanical origin and other compounds extracted from unifloral honeys. J. Agric. Food Chem. 2013, 61, 1747–1755. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-Y.; Wu, T.-S. Constituents of the stigmas of Crocus sativus and their tyrosinase inhibitory activity. J. Nat. Prod. 2002, 65, 1452–1456. [Google Scholar] [CrossRef] [PubMed]
- Ina, K.; Etō, H. 3-keto-β-ionone in the essential oil from black tea. Agric. Biol. Chem. 1971, 35, 962–963. [Google Scholar] [CrossRef]
- Fujimori, T.; Kasuga, R.; Matsushita, H.; Kaneko, H.; Noguchi, M. Neutral aroma constituents in burley tobacco. Agric. Biol. Chem. 1976, 40, 303–315. [Google Scholar] [CrossRef]
- Bolt, A.J.N.; Purkis, S.W.; Sadd, J.S. A damascone derivative from Nicotiana tabacum. Phytochemistry 1983, 22, 613–614. [Google Scholar] [CrossRef]
- Sato, S.; Sasakura, S.; Kobayashi, A.; Nakatani, Y.; Yamanishi, T. Flavor of black tea. Part VI. Intermediate and high boiling components of the neutral fraction. Agric. Biol. Chem. 1970, 34, 1355–1367. [Google Scholar] [CrossRef]
- Beekwilder, J.; van Rossum, H.M.; Koopman, F.; Sonntag, F.; Buchhaupt, M.; Schrader, J.; Hall, R.D.; Bosch, D.; Pronk, J.T.; van Maris, A.J.A.; et al. Polycistronic expression of a β-carotene biosynthetic pathway in Saccharomyces cerevisiae coupled to β-ionone production. J. Biotechnol. 2014, 192, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Serra, S.; Fuganti, C.; Brenna, E. Biocatalytic preparation of natural flavours and fragrances. Trends Biotechnol. 2005, 23, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Prelog, V.; Meier, H.L. Untersuchungen über organextrakte und harn. 18. Mitteilung. Über die biochemische oxydation von β-jonon im tierkörper. Helv. Chim. Acta 1950, 33, 1276–1284. [Google Scholar] [CrossRef]
- Mikami, Y.; Watanabe, E.; Fukunaga, Y.; Kisaki, T. Formation of 2S-hydroxy-β-ionone and 4-hydroxy-β-ionone by microbial hydroxylation of β-ionone. Agric. Biol. Chem. 1978, 42, 1075–1077. [Google Scholar] [CrossRef]
- Mikami, Y.; Fukunaga, Y.; Arita, M.; Kisaki, T. Microbial transformation of β-ionone and β-methylionone. Appl. Environ. Microbiol. 1981, 41, 610–617. [Google Scholar] [PubMed]
- Krasnobajew, V.; Helmlinger, D. Fermentation of fragrances: Biotransformation of β-ionone by Lasiodiplodia theobromae. Helv. Chim. Acta 1982, 65, 1590–1601. [Google Scholar] [CrossRef]
- Hartman, D.A.; Pontones, M.E.; Kloss, V.F.; Curley, R.W.; Robertson, L.W. Models of retinoid metabolism: Microbial biotransformation of α-ionone and β-ionone. J. Nat. Prod. 1988, 51, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Schoch, E.; Benda, I.; Schreier, P. Bioconversion of α-damascone by Botrytis cinerea. Appl. Environ. Microbiol. 1991, 57, 15–18. [Google Scholar]
- Kakeya, H.; Sugai, T.; Ohta, H. Biochemical preparation of optically active 4-hydroxy-β-ionone and its transformation to (S)-6-hydroxy-α-ionone. Agric. Biol. Chem. 1991, 55, 1873–1876. [Google Scholar] [CrossRef]
- Weidmann, V.; Kliewer, S.; Sick, M.; Bycinskij, S.; Kleczka, M.; Rehbein, J.; Griesbeck, A.G.; Zorn, H.; Maison, W. Studies towards the synthetic applicability of biocatalytic allylic oxidations with the lyophilisate of Pleurotus sapidus. J. Mol. Catal. B Enzym. 2015, 121, 15–21. [Google Scholar] [CrossRef]
- Gliszczyńska, A.; Gładkowski, W.; Dancewicz, K.; Gabryś, B.; Szczepanik, M. Transformation of β-damascone to (+)-(S)-4-hydroxy-β-damascone by fungal strains and its evaluation as a potential insecticide against aphids Myzus persicae and lesser mealworm Alphitobius diaperinus Panzer. Catal. Commun. 2016, 80, 39–43. [Google Scholar] [CrossRef]
- Lutz-Wahl, S.; Fischer, P.; Schmidt-Dannert, C.; Wohlleben, W.; Hauer, B.; Schmid, R.D. Stereo- and regioselective hydroxylation of α-ionone by Streptomyces strains. Appl. Environ. Microbiol. 1998, 64, 3878–3881. [Google Scholar] [PubMed]
- Maurer, S.C.; Schulze, H.; Schmid, R.D.; Urlacher, V. Immobilisation of P450 BM-3 and an NADP+ cofactor recycling system: Towards a technical application of heme-containing monooxygenases in fine chemical synthesis. Adv. Synth. Catal. 2003, 345, 802–810. [Google Scholar] [CrossRef]
- Litzenburger, M.; Bernhardt, R. Selective oxidation of carotenoid-derived aroma compounds by CYP260B1 and CYP267B1 from Sorangium cellulosum So ce56. Appl. Microbiol. Biotechnol. 2016, 100, 4447–4457. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, H.; Beer, S.B.A.d.; Geerke, D.P.; Vermeulen, N.P.E.; Commandeur, J.N.M. Regio- and stereoselective hydroxylation of optically active α-ionone enantiomers by engineered cytochrome P450 BM3 mutants. Adv. Synth. Catal. 2012, 354, 2172–2184. [Google Scholar] [CrossRef]
- Fuganti, C.; Serra, S.; Zenoni, A. Synthesis and olfactory evaluation of (+)- and (-)-gamma-ionone. Helv. Chim. Acta 2000, 83, 2761–2768. [Google Scholar] [CrossRef]
- Serra, S.; Fuganti, C.; Brenna, E. Synthesis, olfactory evaluation, and determination of the absolute configuration of the 3,4-didehydroionone stereoisomers. Helv. Chim. Acta 2006, 89, 1110–1122. [Google Scholar] [CrossRef]
- Serra, S.; Fuganti, C.; Brenna, E. Two easy photochemical methods for the conversion of commercial ionone alpha into regioisomerically enriched gamma-ionone and gamma-dihydroionone. Flavour Fragr. J. 2007, 22, 505–511. [Google Scholar] [CrossRef]
- Serra, S. An expedient preparation of enantio-enriched ambergris odorants starting from commercial ionone alpha. Flavour Fragr. J. 2013, 28, 46–52. [Google Scholar] [CrossRef]
- Brenna, E.; Fuganti, C.; Serra, S.; Kraft, P. Optically active ionones and derivatives: Preparation and olfactory properties. Eur. J. Org. Chem. 2002, 967–978. [Google Scholar] [CrossRef]
- Serra, S.; Fuganti, C. Synthesis of the enantiomeric forms of alpha- and gamma-damascone starting from commercial racemic alpha-ionone. Tetrahedron Asymmetry 2006, 17, 1573–1580. [Google Scholar] [CrossRef]
- Brenna, E.; Fuganti, C.; Serra, S. Synthesis and olfactory evaluation of the enantiomerically enriched forms of 7,11-epoxymegastigma-5(6)-en-9-one and 7,11-epoxymegastigma-5(6)-en-9-ols isomers, identified in passiflora edulis. Tetrahedron Asymmetry 2005, 16, 1699–1704. [Google Scholar] [CrossRef]
- Serra, S.; Barakat, A.; Fuganti, C. Chemoenzymatic resolution of cis- and trans-3,6-dihydroxy-alpha-ionone. Synthesis of the enantiomeric forms of dehydrovomifoliol and 8,9-dehydrotheaspirone. Tetrahedron Asymmetry 2007, 18, 2573–2580. [Google Scholar] [CrossRef]
- Mazur, M.; Grudniewska, A.; Wawrzeńczyk, C. Microbial transformations of halolactones with p-menthane system. J. Biosci. Bioeng. 2015, 119, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Simeo, Y.; Sinisterra, J.V. Biotransformation of terpenoids: A green alternative for producing molecules with pharmacological activity. Mini-Rev. Org. Chem. 2009, 6, 128–134. [Google Scholar] [CrossRef]
- Bhatti, H.N.; Khera, R.A. Biological transformations of steroidal compounds: A review. Steroids 2012, 77, 1267–1290. [Google Scholar] [CrossRef]
- Carballeira, J.D.; Álvarez, E.; Sinisterra, J.V. Biotransformation of cyclohexanone using immobilized Geotrichum candidum NCYC49: Factors affecting the selectivity of the process. J. Mol. Catal. B Enzym. 2004, 28, 25–32. [Google Scholar] [CrossRef]
- Bankar, A.V.; Kumar, A.R.; Zinjarde, S.S. Environmental and industrial applications of Yarrowia lipolytica. Appl. Microbiol. Biotechnol. 2009, 84, 847. [Google Scholar] [CrossRef]
- Johnson, E.A. Phaffia rhodozyma: Colorful odyssey. Int. Microbiol. 2003, 6, 169–174. [Google Scholar] [CrossRef]
- Knapp, H.; Weigand, C.; Gloser, J.; Winterhalter, P. 2-hydroxy-2,6,10,10-tetramethyl-1-oxaspiro[4.5]dec-6-en-8-one: Precursor of 8,9-dehydrotheaspirone in white-fleshed nectarines. J. Agric. Food Chem. 1997, 45, 1309–1313. [Google Scholar] [CrossRef]
- Näf, R.; Velluz, A.; Decorzant, R.; Näf, F. Structure and synthesis of two novel ionone-type compounds identified in quince brandy (Cydonia oblonga Mil.). Tetrahedron Lett. 1991, 32, 753–756. [Google Scholar] [CrossRef]
- Serra, S. MnO2/TBHP: A versatile and user-friendly combination of reagents for the oxidation of allylic and benzylic methylene functional groups. Eur. J. Org. Chem. 2015, 2015, 6472–6478. [Google Scholar] [CrossRef]
- Serra, S.; Lissoni, V. First enantioselective synthesis of marine diterpene ambliol-A. Eur. J. Org. Chem. 2015, 2015, 2226–2234. [Google Scholar] [CrossRef]
- Dess, D.B.; Martin, J.C. Readily accessible 12-I-5 oxidant for the conversion of primary and secondary alcohols to aldehydes and ketones. J. Org. Chem. 1983, 48, 4155–4156. [Google Scholar] [CrossRef]
- Audran, G.; Galano, J.M.; Monti, H. Enantioselective synthesis and determination of the absolute configuration of natural (−)-elegansidiol. Eur. J. Org. Chem. 2001, 2293–2296. [Google Scholar] [CrossRef]
- Serra, S.; Gatti, F.G.; Fuganti, C. Lipase-mediated resolution of the hydroxy-cyclogeraniol isomers: Application to the synthesis of the enantiomers of karahana lactone, karahana ether, crocusatin C and gamma-cyclogeraniol. Tetrahedron Asymmetry 2009, 20, 1319–1329. [Google Scholar] [CrossRef]
- Kaiser, R.; Lamparsky, D. Inhaltsstoffe des Osmanthus-absolues. 1. Mitteilung: 2,5-epoxy-megastigma-6,8-dien. Helv. Chim. Acta 1978, 61, 373–382. [Google Scholar] [CrossRef]
- Tu, V.A.; Kaga, A.; Gericke, K.-H.; Watanabe, N.; Narumi, T.; Toda, M.; Brueckner, B.; Baldermann, S.; Mase, N. Synthesis and characterization of quantum dot nanoparticles bound to the plant volatile precursor of hydroxy-apo-10′-carotenal. J. Org. Chem. 2014, 79, 6808–6815. [Google Scholar] [CrossRef]
- Khachik, F.; Chang, A.-N. Synthesis of (3S)- and (3R)-3-hydroxy-β-ionone and their transformation into (3S)- and (3R)-β-cryptoxanthin. Synthesis 2011, 2011, 509–516. [Google Scholar] [CrossRef]
- Takei, Y.; Mori, K.; Matsui, M. Synthesis of a stereoisomeric mixture of 3-hydroxy-α-damascone. Agric. Biol. Chem. 1973, 37, 2927–2928. [Google Scholar] [CrossRef]
- Pascual, A.; Bischofberger, N.; Frei, B.; Jeger, O. Photochemical reactions. 149th communication. Photochemistry of 7,8-dihydro-4-hydroxy-β-ionone and derivatives. Helv. Chim. Acta 1988, 71, 374–388. [Google Scholar] [CrossRef]
- Buschor, D.J.; Eugster, C.H. Synthese der (3S,4R,3′S,4′R)- und (3S,4R,3′S,4′S)crustaxanthine sowie weiterer verbindungen mit 3,4-dihydroxy-β-endgruppen. Helv. Chim. Acta 1990, 73, 1002–1021. [Google Scholar] [CrossRef]
- Irie, H.; Matsumoto, R.; Nishimura, M.; Zhang, Y. Synthesis of (±)-heritol, a sesquiterpene lactone belonging to the aromatic cadinane group. Chem. Pharm. Bull. 1990, 38, 1852–1856. [Google Scholar] [CrossRef]
- Cooper, R.D.G.; Davis, J.B.; Leftwick, A.P.; Price, C.; Weedon, B.C.L. Carotenoids and related compounds. Part XXXII. Synthesis of astaxanthin, phoenicoxanthin, hydroxyechinenone, and the corresponding diosphenols. J. Chem. Soc. Perkin Trans. 1 1975, 2195–2204. [Google Scholar] [CrossRef]
- Serra, S.; Piccioni, O. A new chemo-enzymatic approach to the stereoselective synthesis of the flavors tetrahydroactinidiolide and dihydroactinidiolide. Tetrahedron Asymmetry 2015, 26, 584–592. [Google Scholar] [CrossRef]
- Oritani, T.; Yamamoto, H.; Yamashita, K. Synthesis of (±)-4′-hydroxy-γ-ionylideneacetic acids, fungal biosynthetic intermediates of abscisic acid. Agric. Biol. Chem. 1990, 54, 125–130. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 23a, 25, 28, 35 and 36 are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Biotransformation Products | Fungal Strains and Distribution of the Biotransformation Products 1 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A. niger | N. oryzae | G. candidum | Y. lipolytica | P. roqueforti | R. stolonifer | P. corylophilum | M. isabellina | C. lunata | X. dendrorhous | F. culmorum | ||
| α-ionone (1) | 1 | 21 | 2 | 100 | 87 | 82 | 85 | 84 | 62 | - | 88 | 24 |
| 8 | 13 | 27 | - | - | 4 | 2 | 4 | 1 | 2 | - | 10 | |
| 9a | 33 | 7 | - | 1 | 3 | 3 | 4 | 9 | 7 | 2 | 21 | |
| 9b | 28 | 25 | - | 1 | 10 | 1 | 6 | 12 | - | 1 | 25 | |
| 10 | - | - | - | - | - | - | - | - | 10 | - | 3 | |
| 11 | - | - | - | 8 | - | - | - | 3 | - | 2 | - | |
| 12 | - | 10 | - | - | - | - | - | - | 7 | - | 2 | |
| N.D. 2 | 5 | 31 | - | 3 | 1 | 9 | 2 | 13 | 74 | 7 | 15 | |
| β-ionone (2) | 2 | 45 | 15 | 76 | 82 | 87 | 66 | 61 | 84 | 16 | 90 | 40 |
| 13 | 1 | - | 5 | 4 | 1 | 3 | 3 | 1 | - | - | 4 | |
| 14 | 39 | 26 | 8 | 1 | 8 | 14 | 16 | 7 | 15 | 3 | 11 | |
| 15 | - | - | - | - | - | - | 1 | - | - | - | - | |
| 16 | - | - | - | - | - | - | - | - | 23 | - | 3 | |
| 17 | - | 26 | - | - | - | - | - | - | - | - | - | |
| 18 | 11 | - | - | - | - | - | 8 | - | - | - | - | |
| 19 | - | - | - | - | 2 | - | - | - | - | - | - | |
| 20 | - | - | 1 | - | - | - | - | - | - | - | - | |
| 21 | - | 2 | - | 11 | - | - | 3 | 1 | 12 | - | 3 | |
| 22 | - | 5 | - | 1 | - | - | - | - | - | - | - | |
| N.D. 2 | 4 | 26 | 10 | 1 | 2 | 17 | 8 | 7 | 34 | 7 | 39 | |
| γ-ionone (3) | 3 | - | 6 | 100 | 43 | 83 | 100 | 44 | 85 | 26 | 78 | 21 |
| 23a | 35 | - | - | - | 5 | - | 14 | - | - | - | - | |
| 23b | - | - | - | - | - | - | - | - | - | - | - | |
| 24 | 2 | - | - | - | - | - | - | 2 | 8 | - | 13 | |
| 25 | 49 | 4 | - | - | - | - | - | - | - | - | 10 | |
| 26 | - | 2 | - | 20 | - | - | 19 | - | - | 8 | 7 | |
| 27 | - | 2 | - | 16 | 1 | - | 6 | 2 | 20 | 5 | - | |
| N.D. 2 | 14 | 86 | - | 21 | 11 | - | 17 | 11 | 46 | 9 | 49 | |
| Substrate | Biotransformation Products | Fungal Strains and Distribution of the Biotransformation Products 1 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A. niger | N. oryzae | G. candidum | Y. lipolytica | P. roqueforti | R. stolonifer | P. corylophilum | M. isabellina | C. lunata | X. dendrorhous | F. culmorum | ||
| 3,4-dehydro-β-ionone (4) | 4 | 48 | - | 45 | 87 | 45 | 80 | 76 | 71 | 52 | 6 | - |
| 28 | 25 | 69 | 18 | - | 32 | 7 | 22 | 8 | 26 | 26 | 35 | |
| 29 | 3 | 2 | 17 | 1 | 9 | 3 | - | 7 | 8 | 22 | 20 | |
| 30 | 2 | 3 | 12 | 1 | 6 | 2 | - | 5 | 6 | 21 | 17 | |
| N.D. 2 | 22 | 26 | 8 | 3 | 8 | 8 | 2 | 9 | 8 | 25 | 28 | |
| theaspirane (5) | 5 | 20 | - | 56 | 82 | 31 | - | 22 | - | 8 | 46 | - |
| 31 | 9 | 12 | 11 | 4 | 19 | 5 | 16 | - | - | 3 | 28 | |
| 32 | 51 | 25 | 15 | 1 | 28 | 62 | 34 | - | 3 | 28 | 34 | |
| 33 | - | - | - | - | 2 | - | - | - | - | 2 | 2 | |
| 34 | - | 28 | - | - | - | 6 | - | 50 | 8 | - | 9 | |
| 35 | - | - | - | - | - | - | - | - | 12 | - | - | |
| 36 | - | - | - | - | - | - | - | - | 9 | - | - | |
| N.D. 2 | 20 | 35 | 18 | 1 | 20 | 27 | 28 | 50 | 60 | 21 | 27 | |
| Substrate | Biotransformation Products | Fungal Strains and Distribution of the Biotransformation Products 1 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A. niger | N. oryzae | G. candidum | Y. lipolytica | P. roqueforti | R. stolonifer | P. corylophilum | M. isabellina | C. lunata | X. dendrorhous | F. culmorum | ||
| α-damascone (6) | 6 | 56 | 89 | 55 | 93 | 100 | 100 | 24 | 30 | 96 | 39 | 56 |
| 37 | 20 | 5 | 10 | - | - | - | - | 22 | - | 10 | 5 | |
| 38 | 5 | 2 | 10 | - | - | - | 14 | 17 | 3 | 21 | 30 | |
| 39 | - | - | - | - | - | - | 2 | - | 3 | - | ||
| 40 | - | - | - | - | - | 13 | - | - | - | - | ||
| N.D. 2 | 19 | 4 | 25 | 7 | - | - | 49 | 29 | 1 | 27 | 9 | |
| β-damascone (7) | 7 | 98 | 70 | 95 | 95 | 100 | 88 | 95 | 95 | 92 | 95 | 57 |
| 41 | - | 2 | - | - | - | 1 | - | - | - | - | 4 | |
| 42 | 1 | 23 | 1 | 1 | - | 2 | - | 2 | 7 | 1 | 22 | |
| N.D. 2 | 1 | 5 | 4 | 4 | - | 9 | 5 | 3 | 1 | 4 | 17 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serra, S.; De Simeis, D. Fungi-Mediated Biotransformation of the Isomeric Forms of the Apocarotenoids Ionone, Damascone and Theaspirane. Molecules 2019, 24, 19. https://doi.org/10.3390/molecules24010019
Serra S, De Simeis D. Fungi-Mediated Biotransformation of the Isomeric Forms of the Apocarotenoids Ionone, Damascone and Theaspirane. Molecules. 2019; 24(1):19. https://doi.org/10.3390/molecules24010019
Chicago/Turabian StyleSerra, Stefano, and Davide De Simeis. 2019. "Fungi-Mediated Biotransformation of the Isomeric Forms of the Apocarotenoids Ionone, Damascone and Theaspirane" Molecules 24, no. 1: 19. https://doi.org/10.3390/molecules24010019
APA StyleSerra, S., & De Simeis, D. (2019). Fungi-Mediated Biotransformation of the Isomeric Forms of the Apocarotenoids Ionone, Damascone and Theaspirane. Molecules, 24(1), 19. https://doi.org/10.3390/molecules24010019