Spectroscopic and Structural Study of Some Oligosilanylalkyne Complexes of Cobalt, Molybdenum and Nickel



Abstract
:1. Introduction
2. Results and Discussion
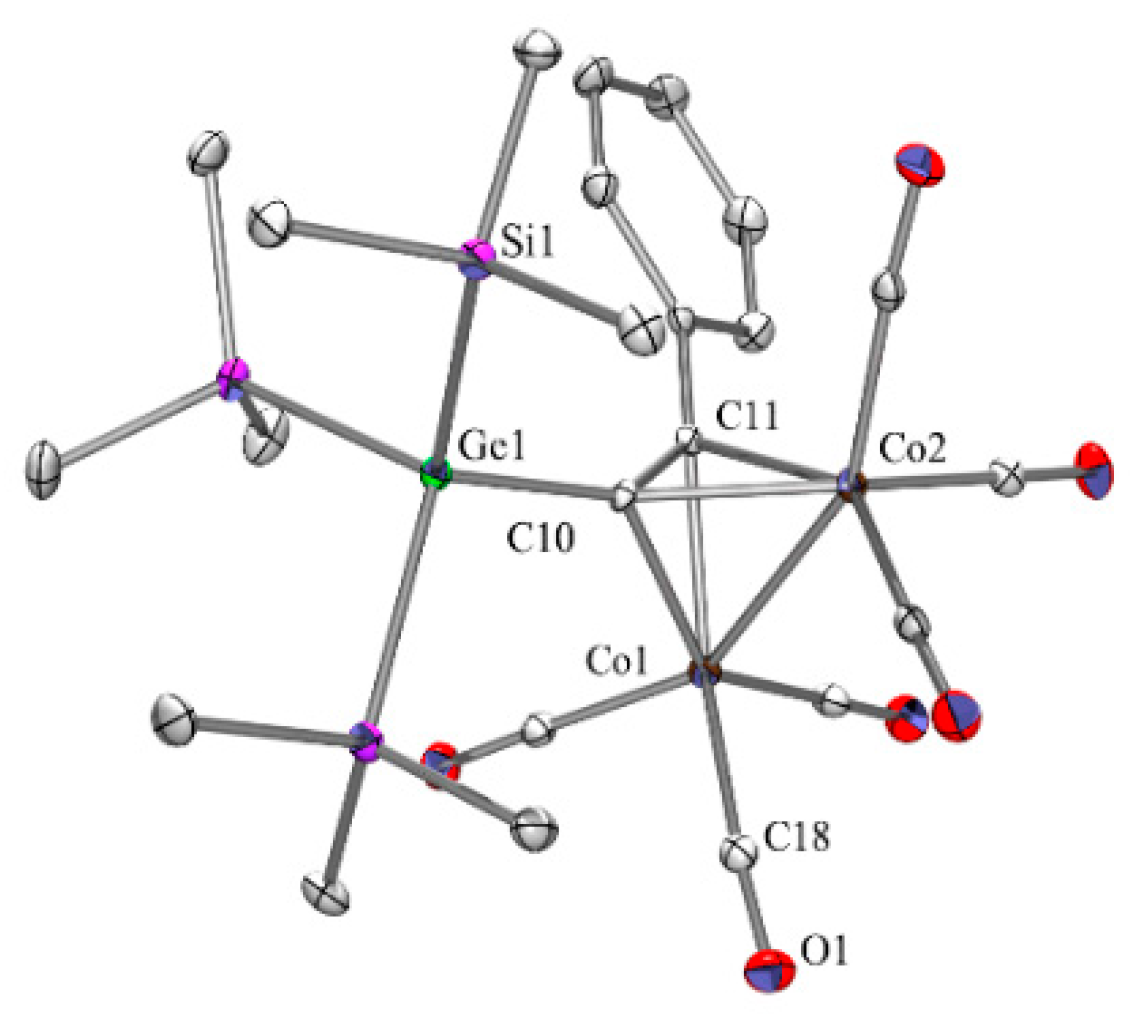
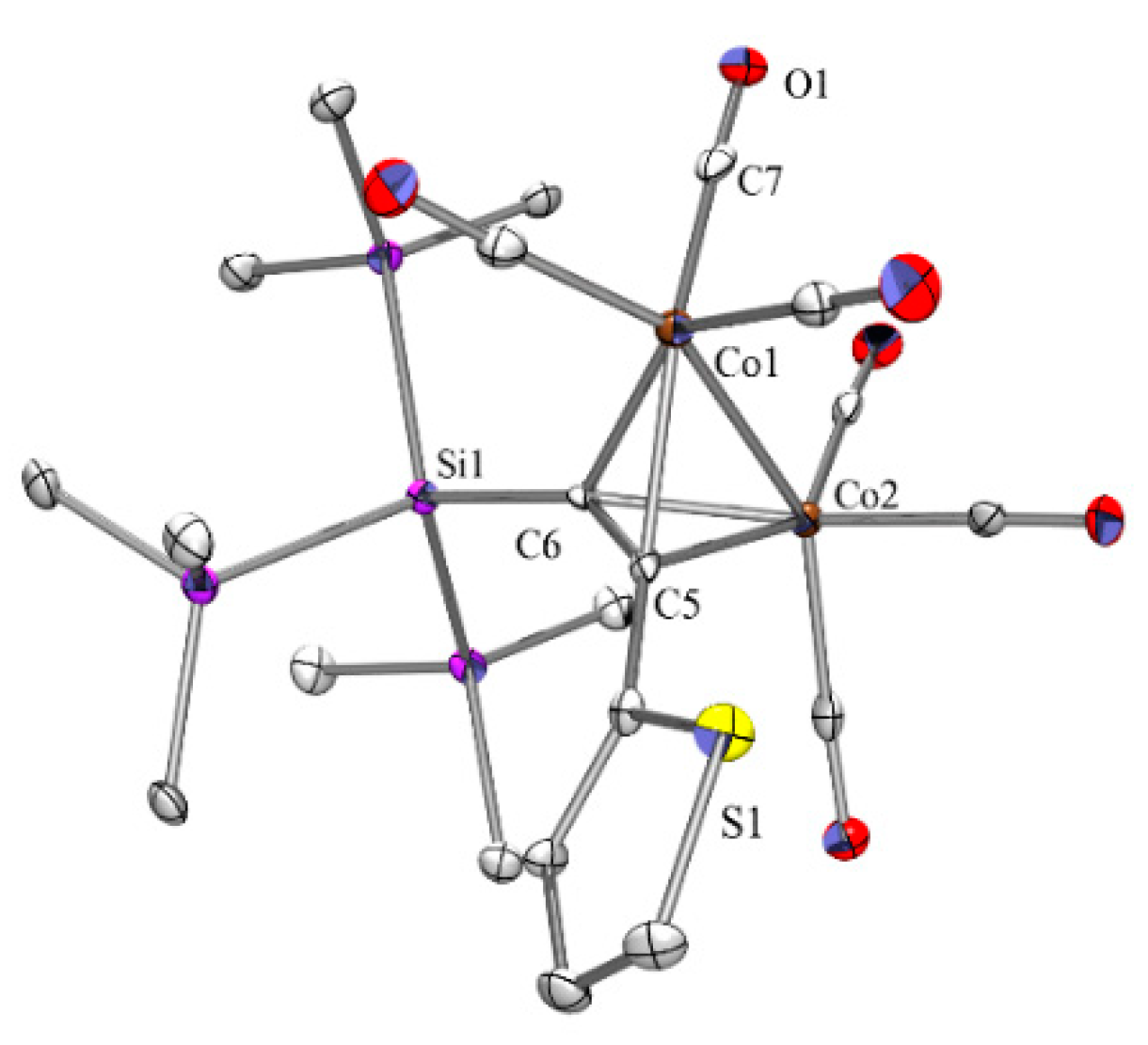
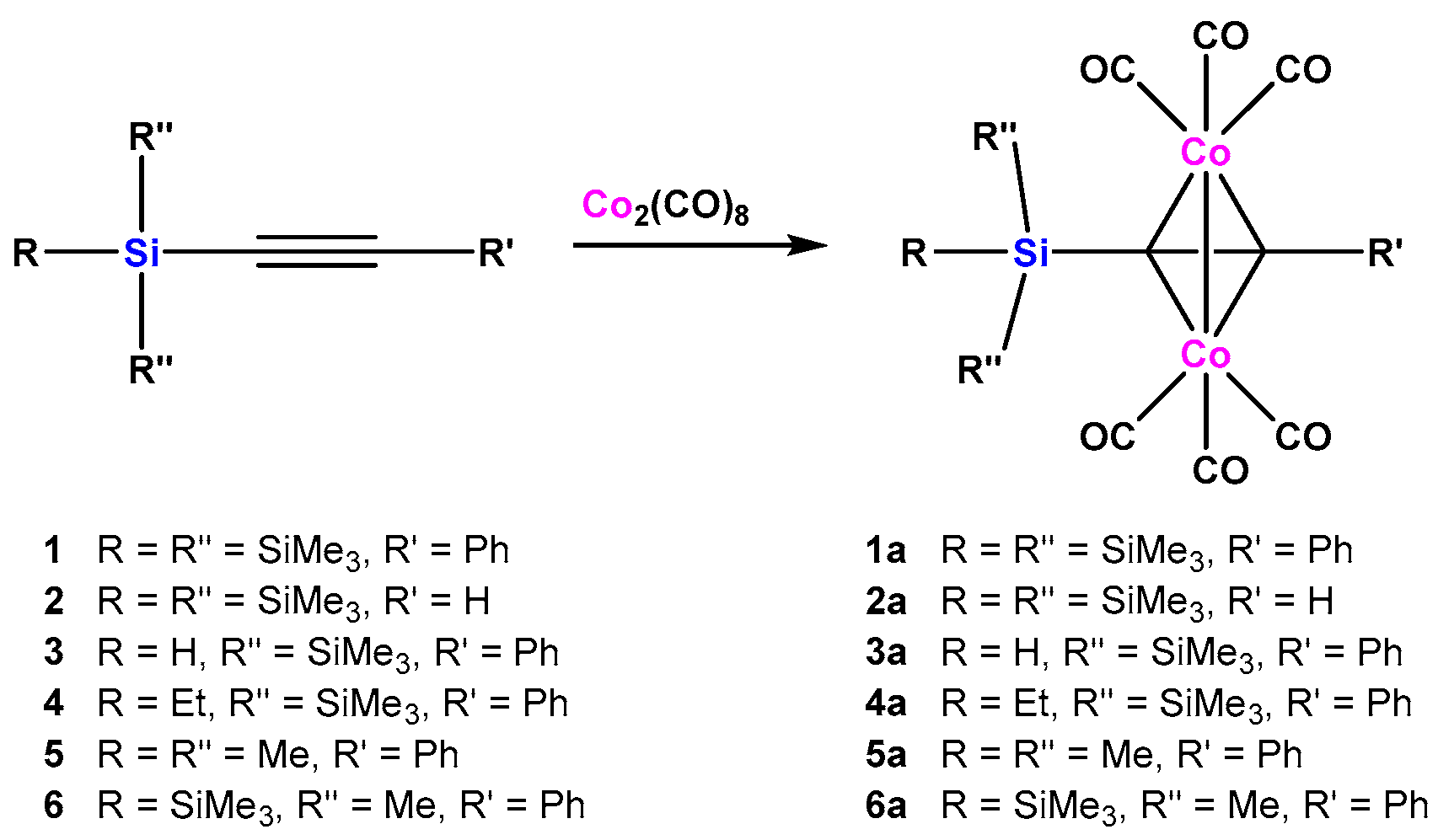
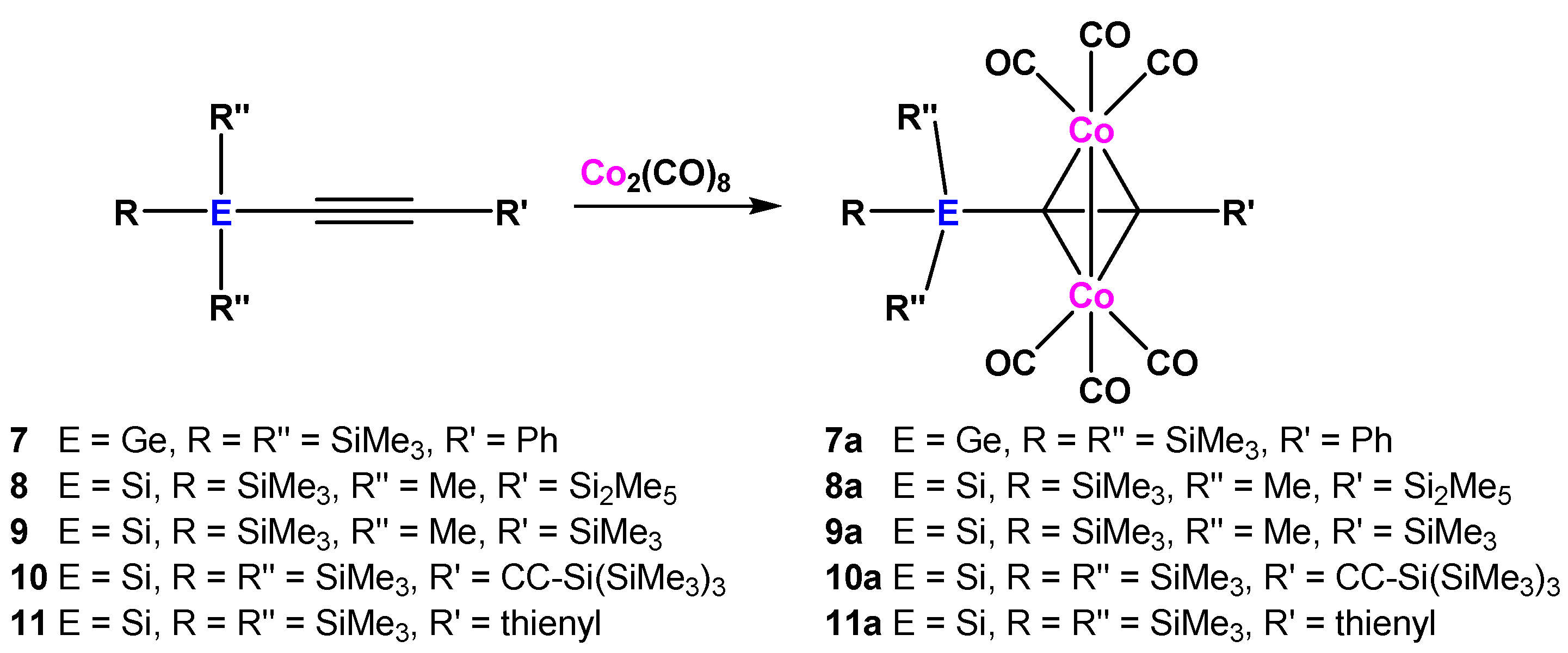
2.1. Cobalt Complexes
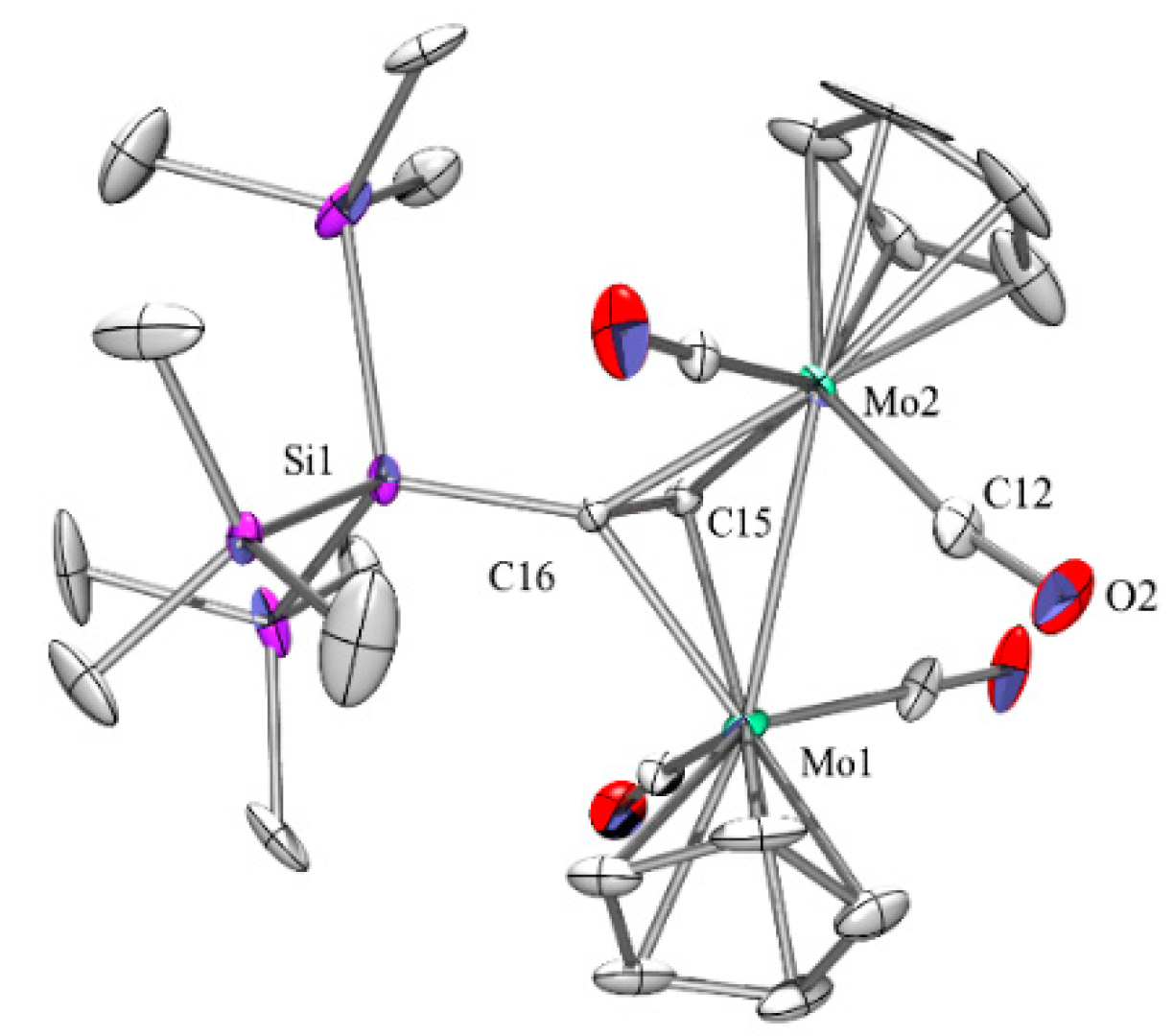
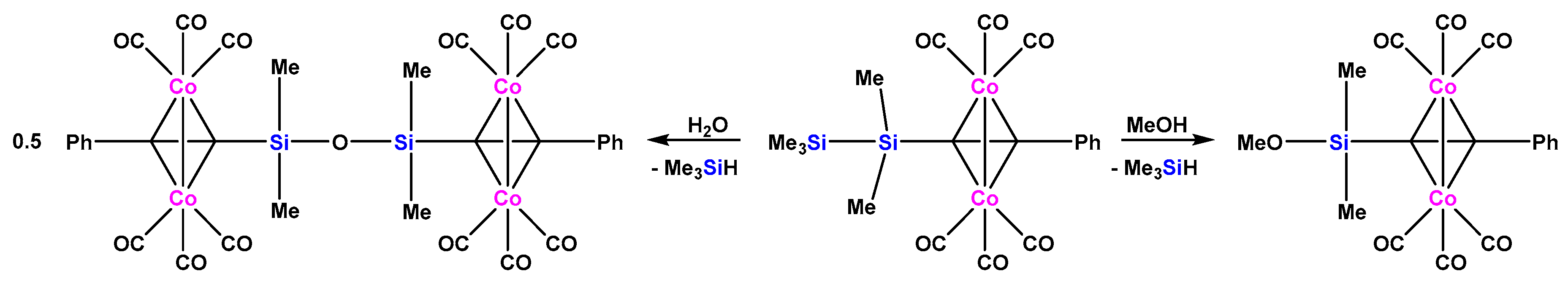
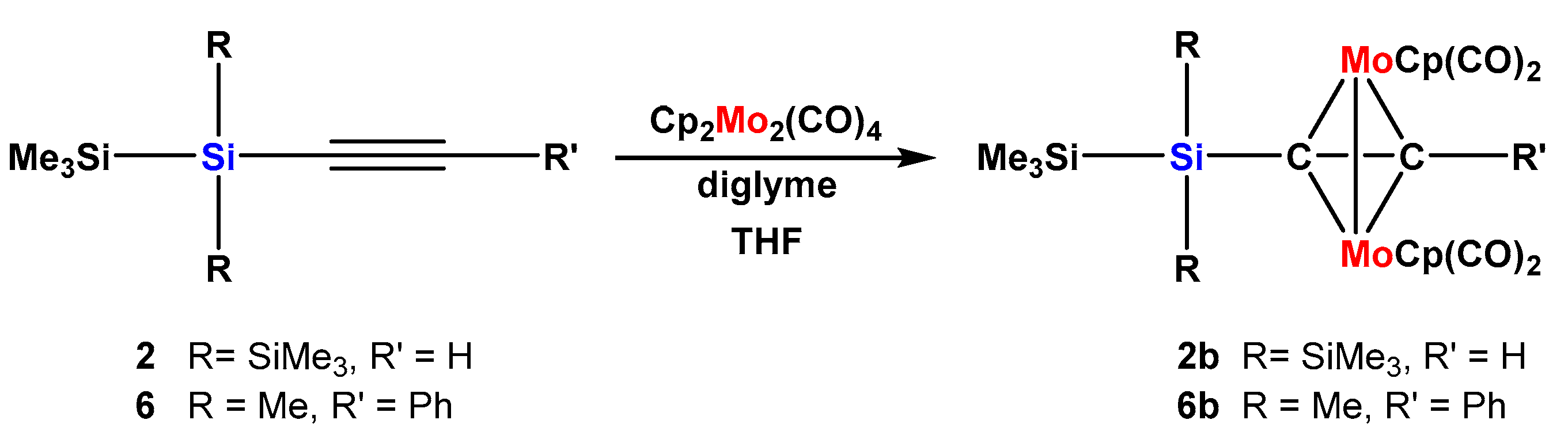
2.2. Molybdenum Complexes
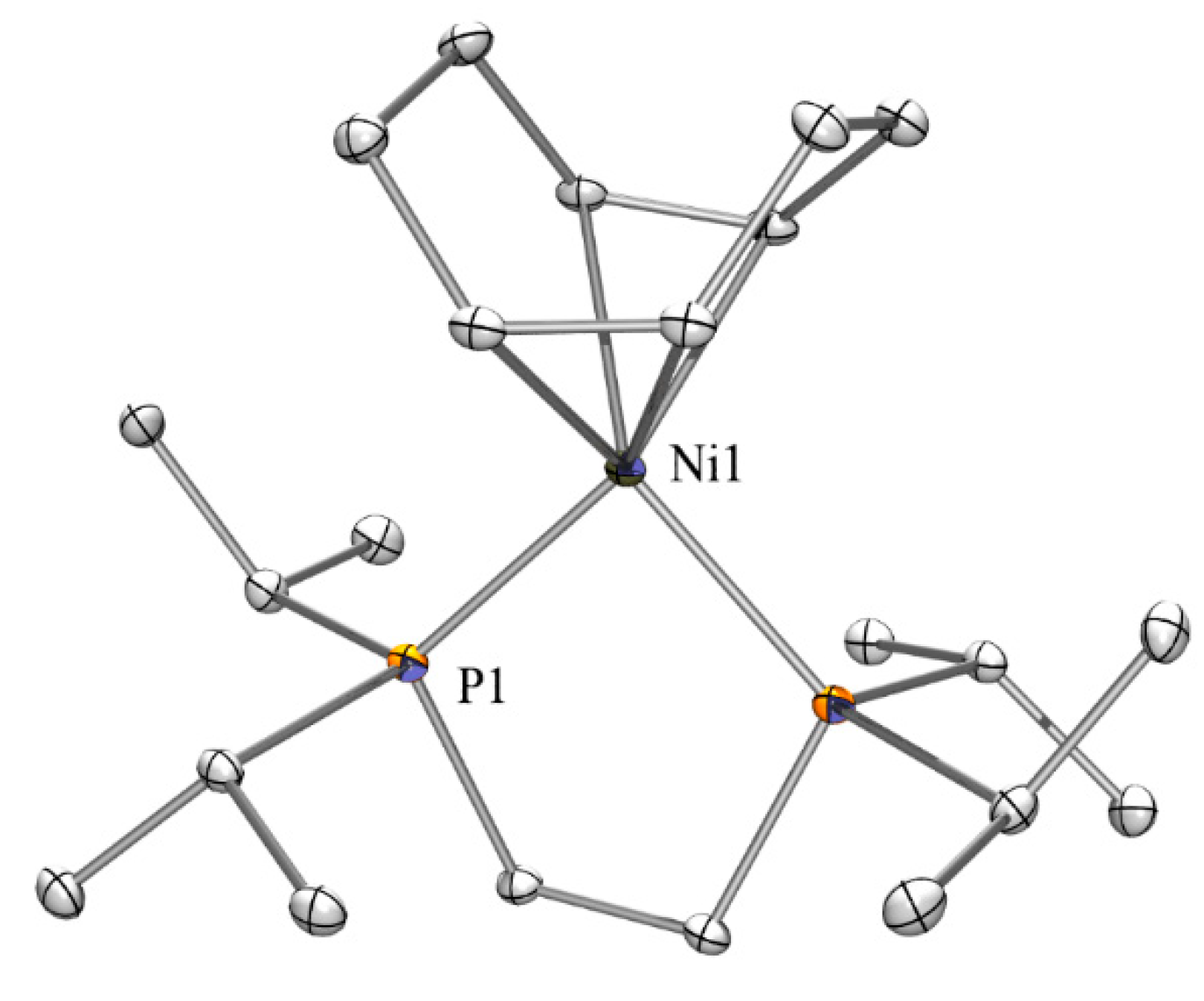
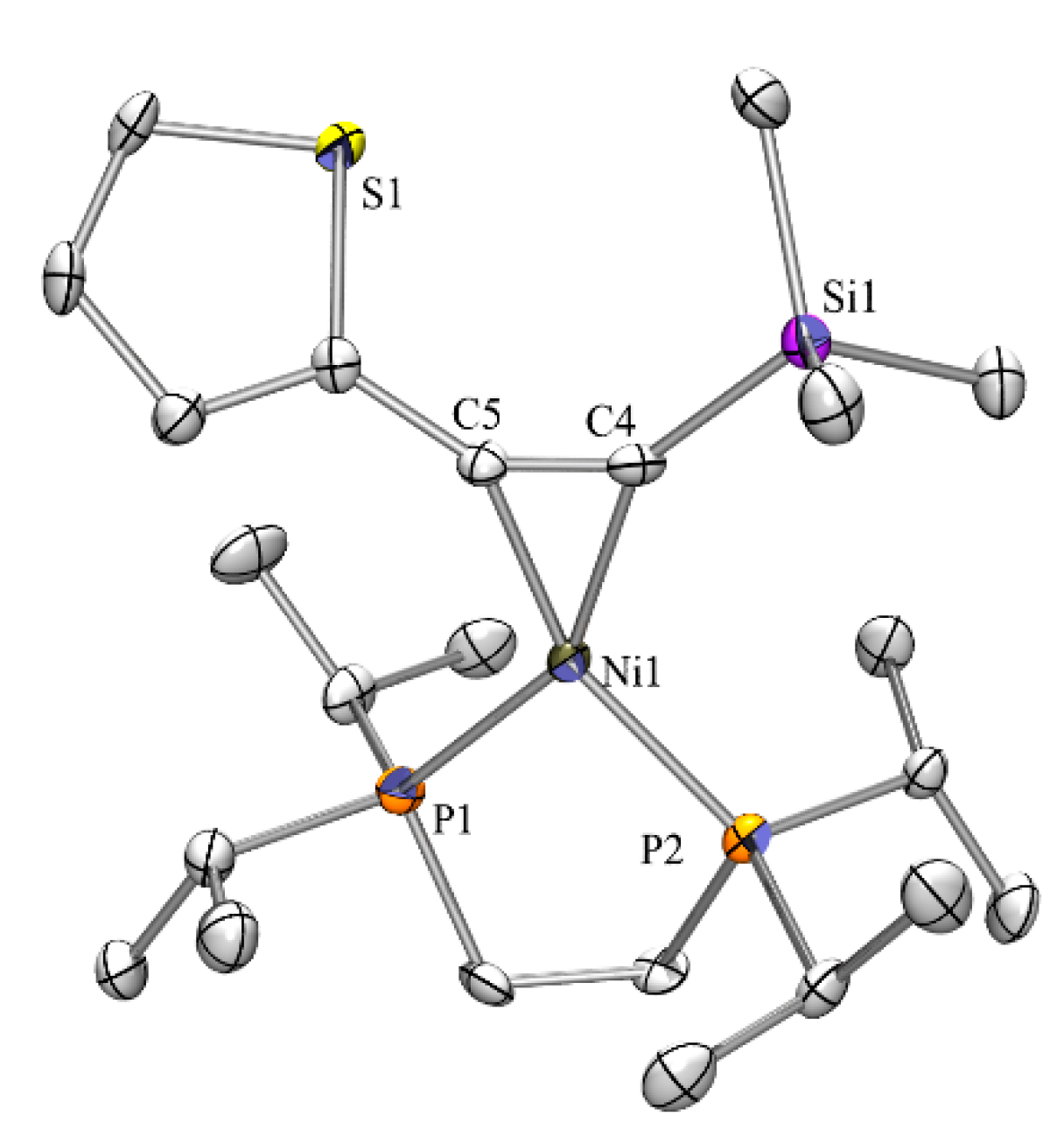
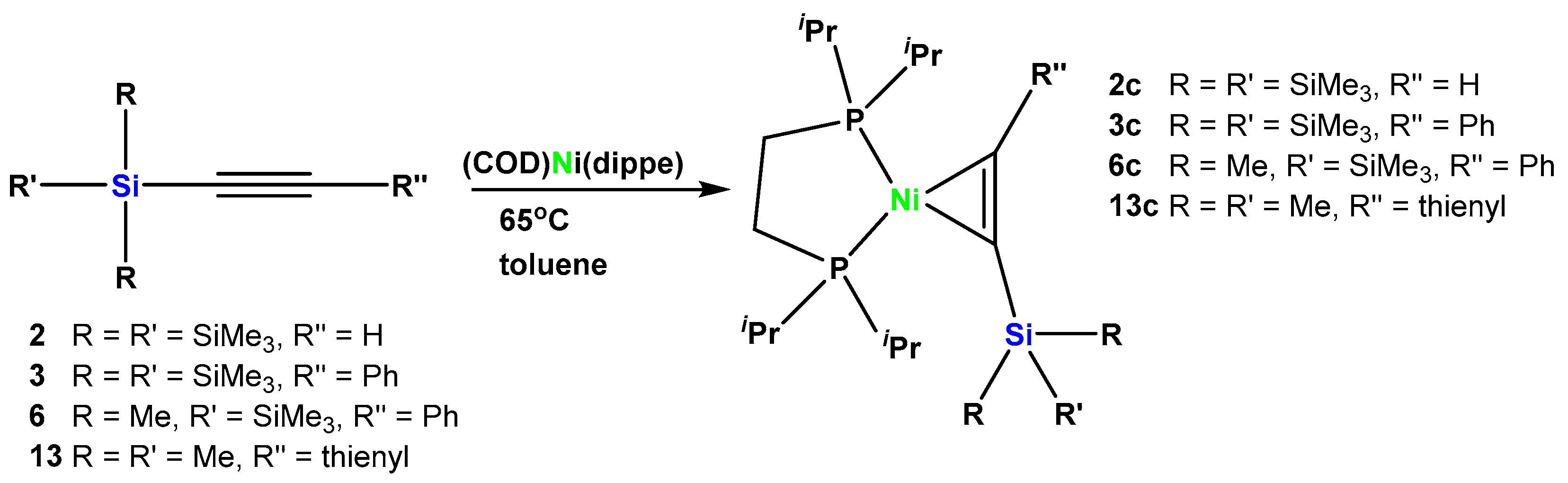
2.3. Nickel Complexes
2.4. NMR Spectroscopic Analysis
2.5. Crystallographic Analysis
3. Experimental Section
3.1. Synthesis of Oligosilanylalkynes
3.2. Oligosilanylalkyne Dicobalthexacarbonyl Complexes
3.3. Oligosilanylalkyne Dimolybdenyum-1,2-dicyclopentadienyltetraacarbonyl Complexes
3.4. Oligosilanylalkyne Nickel-Ethylenebis(diisopropylphosphine) Complexes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hartwig, J. Organotransition Metal Chemistry: From Bonding to Catalysis; University Science Books: Sausalito, CA, USA, 2009; ISBN 1-891389-53-X. [Google Scholar]
- Nicholas, K.M. A Forty Year Odyssey in Metallo–Organic Chemistry. J. Org. Chem. 2015, 80, 6943–6950. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.J.J. Stereoselective Propargylations with Transition-Metal-Stabilized Propargyl Cations. Eur. J. Org. Chem. 2001, 2001, 2021–2033. [Google Scholar] [CrossRef]
- Teobald, B.J. The Nicholas reaction: The use of dicobalt hexacarbonyl-stabilised propargylic cations in synthesis. Tetrahedron 2002, 58, 4133–4170. [Google Scholar] [CrossRef]
- Nicholas, K.M. Chemistry and synthetic utility of cobalt-complexed propargyl cations. Acc. Chem. Res. 1987, 20, 207–214. [Google Scholar] [CrossRef]
- Krüerke, U.; Hübel, W. Über Organometall-Komplexe, VIII. Reaktionen von Kobaltcarbonyl-Verbindungen mit Alkinen. Chem. Ber. 1961, 94, 2829–2856. [Google Scholar] [CrossRef]
- Seyferth, D.; White, D.L. Silicon-, germanium- and tin-substituted acetylenes and their dicobalt hexacarbonyl complexes. J. Organomet. Chem. 1971, 32, 317–322. [Google Scholar] [CrossRef]
- Pannell, K.H.; Crawford, G.M. Organometalloidal Derivatives of the Transition Metals I. J. Coord. Chem. 1973, 2, 251–256. [Google Scholar] [CrossRef]
- Galow, P.; Sebald, A.; Wrackmeyer, B. Darstellung und NMR-spektroskopie organometallisch substituierter dikobalthexacarbonyl-alkin-komplexe. J. Organomet. Chem. 1983, 259, 253–268. [Google Scholar] [CrossRef]
- Corriu, R.J.P.; Moreau, J.J.E.; Praet, H. Cobalt carbonyl complexes of functional ethynylsilanes. Reactivity at the silicon atom. Organometallics 1989, 8, 2779–2786. [Google Scholar] [CrossRef]
- Corriu, R.J.P.; Moreau, J.J.E.; Praet, H. (Ethynylhydrosilane)cobalt carbonyl complexes. Reactivity of the silicon-hydrogen bond. Organometallics 1990, 9, 2086–2091. [Google Scholar] [CrossRef]
- Connor, R.E.; Nicholas, K.M. Isolation, characterization, and stability of α-[(ethynyl)dicobalt hexacarbonyl] carbonium ions. J. Organomet. Chem. 1977, 125, C45–C48. [Google Scholar] [CrossRef]
- Melikyan, G.G.; Bright, S.; Monroe, T.; Hardcastle, K.I.; Ciurash, J. Overcoming a Longstanding Challenge: X-Ray Structure of a [Co2(CO)6]-Complexed Propargyl Cation. Angew. Chem. Int. Ed. 1998, 37, 161–164. [Google Scholar] [CrossRef]
- Ruffolo, R.; Decken, A.; Girard, L.; Gupta, H.K.; Brook, M.A.; McGlinchey, M.J. Toward Metal-Stabilized Silylium Cations: An EHMO Study of [(HC≡C-SiH2)Co2(CO)6]+ and X-ray Crystal Structures of (Me3SiC≡CSiPh2H)Mo2(CO)4Cp2 and [(Me3SiC≡CSiMe2)Co2(CO)6]2O. Organometallics 1994, 13, 4328–4335. [Google Scholar] [CrossRef]
- Ruffolo, R.; Kainz, S.; Gupta, H.; Brook, M.; Mcglinchey, M. A synthetic and structural study on metal cluster complexes of allyl—alkynyl—silanes: Does protonation lead to metal-stabilized silyl cations? J. Organomet. Chem. 1997, 547, 217–226. [Google Scholar] [CrossRef]
- Stradiotto, M.; Brook, M.A.; McGlinchey, M.J. Can metal clusters assist silicon migrations? An NMR spectroscopic and X-ray crystallographic study. Inorg. Chem. Commun. 1998, 1, 105–108. [Google Scholar] [CrossRef]
- Zirngast, M.; Marschner, C.; Baumgartner, J. Cobalt-Assisted Silicon−Silicon Bond Activation. Organometallics 2006, 25, 4897–4908. [Google Scholar] [CrossRef]
- Zirngast, M.; Marschner, C.; Baumgartner, J. Group 4 Metallocene Complexes of Tris(trimethylsilyl)silylacetylene and Related Alkynes. Organometallics 2008, 27, 2570–2583. [Google Scholar] [CrossRef]
- West, R.; Quass, L.C. Preparation of ethynylsilanes from polychloroethylenes. J. Organomet. Chem. 1969, 18, 55–67. [Google Scholar] [CrossRef]
- Kerst, C.; Rogers, C.W.; Ruffolo, R.; Leigh, W.J. Direct Detection and Characterization of a Transient 1-Silaallene Derivative in Solution. J. Am. Chem. Soc. 1997, 119, 466–471. [Google Scholar] [CrossRef]
- Bruce, M.I.; Low, P.J.; Werth, A.; Skelton, B.W.; White, A.H. Some transition-metal complexes derived from silylated 1,3-diynes. J. Chem. Soc. Dalton Trans. 1996, 1551–1566. [Google Scholar] [CrossRef]
- Ginley, D.S.; Bock, C.R.; Wrighton, M.S. Photogeneration of dinuclear metal carbonyls containing a metal-metal triple bond. Inorg. Chim. Acta 1977, 23, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Curtis, M.D. Reactions of the metal-metal triple bond in Cp2Mo2(CO)4 and related complexes. Polyhedron 1987, 6, 759–782. [Google Scholar] [CrossRef]
- Bonrath, W.; Pörschke, K.R.; Wilke, G.; Angermund, K.; Krüger, C. Ein- und zweikernige Nickel(0)-Komplexe von Butadiin. Angew. Chem. 1988, 100, 853–855. [Google Scholar] [CrossRef]
- Rosenthal, U.; Schulz, W.; Goerls, H. [(C6H5)3P]2Ni(Me3SiC≡CSiMe3). Preparation, properties, and structure of the first stable nickel(0) complex with bis(trimethylsilyl)acetylene. Z. Anorg. Chem. 1987, 550, 169–176. [Google Scholar] [CrossRef]
- Bartik, T.; Happ, B.; Iglewsky, M.; Bandmann, H.; Boese, R.; Heimbach, P.; Hoffmann, T.; Wenschuh, E. Synthesis and characterization of bis(phosphine)nickel(0) complexes containing nonsymmetrically substituted acetylenes. Organometallics 1992, 11, 1235–1241. [Google Scholar] [CrossRef]
- Edelbach, B.L.; Lachicotte, R.J.; Jones, W.D. Catalytic Carbon-Carbon and Carbon-Silicon Bond Activation and Functionalization by Nickel Complexes. Organometallics 1999, 18, 4660–4668. [Google Scholar] [CrossRef]
- Edelbach, B.L.; Lachicotte, R.J.; Jones, W.D. Catalytic Carbon−Carbon Bond Activation and Functionalization by Nickel Complexes. Organometallics 1999, 18, 4040–4049. [Google Scholar] [CrossRef]
- Tillack, A.; Pulst, S.; Baumann, W.; Baudisch, H.; Kortus, K.; Rosenthal, U. Hydrosilylation of symmetric disubstituted alkynes and butadiynes with L2Ni(0)-butadiyne complexes [L = Ph3P, (o-Tol-O)3P] as catalysts. J. Organomet. Chem. 1997, 532, 117–123. [Google Scholar] [CrossRef]
- Rosenthal, U.; Pulst, S.; Arndt, P.; Baumann, W.; Tillack, A.; Kempe, R. Influence of substituents in new Ni(0) butadiyne complexes. Z. Naturforsch. B 1995, 50, 377–384. [Google Scholar] [CrossRef]
- Rosenthal, U.; Pulst, S.; Arndt, P.; Baumann, W.; Tillack, A.; Kempe, R. Influence of ligands in new Ni(0) complexes of disubstituted butadiynes. Z. Naturforsch. B 1995, 50, 368–376. [Google Scholar] [CrossRef]
- Rosenthal, U.; Pulst, S.; Arndt, P.; Ohff, A.; Tillack, A.; Baumann, W.; Kempe, R.; Burlakov, V.V. Heterobimetallic σ,π-Acetylide-Bridged Complexes from Disubstituted 1,3-Butadiynes. Organometallics 1995, 14, 2961–2968. [Google Scholar] [CrossRef]
- Rosenthal, U.; Oehme, G.; Burlakov, V.V.; Petrovskii, P.V.; Shur, V.B.; Vol’pin, M.E. Carbon-13 and proton NMR studies of selected transition metal alkyne complexes. J. Organomet. Chem. 1990, 391, 119–122. [Google Scholar] [CrossRef]
- Chan, W.Y.; Berenbaum, A.; Clendenning, S.B.; Lough, A.J.; Manners, I. Toward Highly Metalized Polymers: Synthesis and Characterization of Silicon-Bridged [1]Ferrocenophanes with Pendent Cluster Substituents. Organometallics 2003, 22, 3796–3808. [Google Scholar] [CrossRef]
- Aime, S.; Milone, L.; Rossetti, R.; Stanghellini, P.L. 1H and 13C NMR studies of acetylenic complexes of Co2(CO)8. Inorg. Chim. Acta 1977, 22, 135–139. [Google Scholar] [CrossRef]
- Happ, B.; Bartik, T.; Zucchi, C.; Rossi, M.C.; Ghelfi, F.; Palyi, G.; Varadi, G.; Szalontai, G.; Horvath, I.T.; Guastini, C. On the Reactivity of Acetylenes Coordinated to Cobalt. 9. Effects of Substitution and Coordination on the 13C-NMR Chemical Shifts of the sp Carbons of (μ2-R1C2R2)Co2(CO)6 Complexes. Molecular Structure of μ2-2-PhC2SiPh3)Co2(CO)6. Organometallics 1995, 14, 809–819. [Google Scholar] [CrossRef]
- Mechtler, C.; Zirngast, M.; Baumgartner, J.; Marschner, C. Synthesis and Reactions of Alkynyl Oligosilanes. Eur. J. Inorg. Chem. 2004, 2004, 3254–3261. [Google Scholar] [CrossRef]
- Markov, J.; Baumgartner, J.; Marschner, C.; Oehme, H.; Gross, T. Vinyloligosilyl anions—A new class of compounds. In Organosilicon Chemistry VI; Auner, N., Weis, J., Eds.; Wiley-VCH: Hoboken, NJ, USA, 2005; pp. 309–313. [Google Scholar]
- Kempe, R.; Sieler, J.; Walther, D. Crystal structure of bis(ethyldiphenylphosphane)cycloocta-1,5-dienenickel(0), C36H42NiP2. Z. Kristallogr. 2010, 211, 565–566. [Google Scholar] [CrossRef]
- Yin, G.; Kalvet, I.; Englert, U.; Schoenebeck, F. Fundamental Studies and Development of Nickel-Catalyzed Trifluoromethylthiolation of Aryl Chlorides: Active Catalytic Species and Key Roles of Ligand and Traceless MeCN Additive Revealed. J. Am. Chem. Soc. 2015, 137, 4164–4172. [Google Scholar] [CrossRef]
- Maciejewski, H.; Sydor, A.; Marciniec, B.; Kubicki, M.; Hitchcock, P.B. Intermediates in nickel(0)–phosphine complex catalyzed dehydrogenative silylation of olefins. Inorg. Chim. Acta 2006, 359, 2989–2997. [Google Scholar] [CrossRef]
- Liu, N.; Li, X.; Xu, X.; Wang, Z.; Sun, H. Synthesis, structure and DFT study of dinuclear iron, cobalt and nickel complexes with cyclopentadienyl-metal moieties. Dalton Trans. 2011, 40, 6886–6892. [Google Scholar] [CrossRef]
- Freund, R.R.A.; Görls, H.; Langer, J. Nickelalactones with an allyl subunit—The effect of penta-coordination on structures and stability. Dalton Trans. 2014, 43, 13988–14000. [Google Scholar] [CrossRef] [PubMed]
- Skelton, B.W.; Clavell, K.J.; Nielsen, D.J. (Cycloocta-1,5-diene)-((ethane-1,2-diyl)-bis(diphenylphosphane))-nickel. CSD Commun. 2017. Refcode: RECHIT. [Google Scholar]
- Pangborn, A.B.; Giardello, M.A.; Grubbs, R.H.; Rosen, R.K.; Timmers, F.J. Safe and Convenient Procedure for Solvent Purification. Organometallics 1996, 15, 1518–1520. [Google Scholar] [CrossRef]
- Morris, G.A.; Freeman, R. Enhancement of Nuclear Magnetic Resonance Signals by Polarization Transfer. J. Am. Chem. Soc. 1979, 101, 760–762. [Google Scholar] [CrossRef]
- Helmer, B.J.; West, R. Enhancement of 29Si NMR Signals by Proton Polarization Transfer. Organometallics 1982, 1, 877–879. [Google Scholar] [CrossRef]
- Bruker. SAINTPLUS: Software Reference Manual, Version 6.45; Bruker-AXS: Madison, WI, USA, 1997–2003.
- Blessing, R.H. An empirical correction for absorption anisotropy. Acta Cryst. A 1995, 51, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SADABS. Version 2.10; Bruker AXS Inc.: Madison, WI, USA, 2003. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An Update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- POVRAY 3.6; Persistence of Vision Pty. Ltd.: Williamstown, Australia, 2004; Available online: http://www.povray.org/download/ (accessed on 9 July 2008).
- Fischer, R.; Frank, D.; Gaderbauer, W.; Kayser, C.; Mechtler, C.; Baumgartner, J.; Marschner, C. α,ω-Oligosilyl Dianions and Their Application in the Synthesis of Homo- and Heterocyclosilanes. Organometallics 2003, 22, 3723–3731. [Google Scholar] [CrossRef]
- Benkeser, R.A.; Hickner, R.A. The Stereochemistry of the Addition of Silicochloroform to Acetylenes. J. Am. Chem. Soc. 1958, 80, 5298–5300. [Google Scholar] [CrossRef]
- Fischer, J.; Baumgartner, J.; Marschner, C. Silylgermylpotassium Compounds. Organometallics 2005, 24, 1263–1268. [Google Scholar] [CrossRef]
- Ishikawa, M.; Kumada, M.; Sakurai, H. Preparation of some polysilicon halides by aluminum halide catalyzed interchange of methyl and halogen on silicon. J. Organomet. Chem. 1970, 23, 63–69. [Google Scholar] [CrossRef]
- Marschner, C.; Baumgartner, J. 4.4.5 Product Subclass 5: Disilanes and Oligosilanes. In Science of Synthesis: Houben-Weyl Methods of Molecular Transformations; Oestreich, M., Ed.; Thieme: Stuttgart, Germany, 2013. [Google Scholar]
Sample Availability: Not available. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
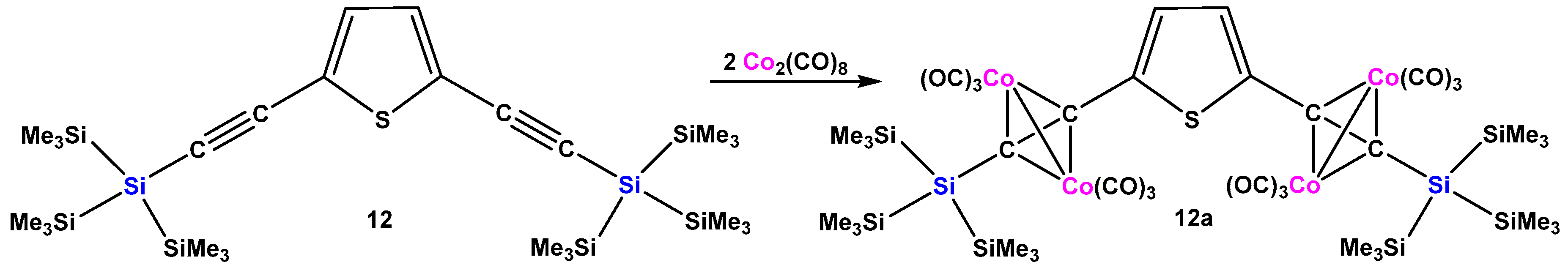{kind=link}
{kind=link}
{kind=link}
| Entry | Alkyne | 29Si | 13C a | Metal | 29Si | 13C a |
|---|---|---|---|---|---|---|
| 1 | (Me3Si)3SiCA≡CBPh (1) b | −100.5 | 88.4/108.6 | Co (1a) b Ni (3c) | −67.1 −90.0 | 75.8/111.4 118.6/157.9 |
| 1a | (Me3Si)3GeCA≡CBPh (7) c | n.a. | 89.6/106.8 | Co (7a) | n.a. | 80.9/109.7 |
| 2 | (Me3Si)3SiCA≡CBH (2) b | −100.7 | 83.4/96.8 | Co (2a) b Mo (2b) Ni (2c) | −67.7 −58.8 −89.4 | 82.9/90.5 96.6/101.9 124.3/145.3 |
| 3 | H(Me3Si)2SiCA≡CBPh (3) b | −90.2 | 86.2/110.2 | Co (3a) b | −53.3 | 71.5/107.9 |
| 4 | Et(Me3Si)2SiCA≡CBPh (4) b | −57.7 | 90.2/109.9 | Co (4a) b | −33.8 | 77.9/109.1 |
| 5 | Me3SiCA≡CBPh (5) b | −18.2 | 94.1/105.8 | Co (5a) b | 0.7 | 79.8/106.0 |
| 6 | Me5Si2CA≡CBPh (6) b | −36.9 | 93.1/108.1 | Co (6a) b Mo (6b) Ni (6c) | −18.0 −16.8 −31.4 | 80.1/107.0 92.1/116.0 132.2/159.6 |
| 7 | (Me3Si)3SiCA≡CBthio (11) | −102.2 | 93.5/100.4 | Co (11c) | −67.1 | 76.6/96.6 |
| 8 | [(Me3Si)3SiC≡C]2 (10) | −100.5 | 79.2/93.4 | Co (10a) | −65.9 | 72.6/88.6 |
| 9 | Me5Si2CA≡CBSiMe3 (9) | −37.6/−18.6 | 112.9/116.5 | Co (9a) | −18.6/0.3 | 93.9/92.4 |
| 10 | Me5Si2C≡CSi2Me5 (8) | −38.2 | 115.3 | Co (8a) | −18.3 | 93.1 |
| Entry | Alkyne | M | M-M Distance (Å) | C-C Distance (Å) | C-Si Distance (Å) | CA-M Distance (Å) | CB-M Distance (Å) |
|---|---|---|---|---|---|---|---|
| 1 | (Me3Si)3SiCA≡CBPh (1) a | Co (1a) | 2.4573(7) | 1.349(5) | 1.873(4) | 2.032(4)/2.022(4) | 1.965(4)/1.987(3) |
| 2 | (Me3Si)3SiCA≡CBH (2) | Co (2a) Mo (2b) | 2.469(2) 2.966(1) | 1.310(15) 1.337(10) | 1.878(10) 1.875(9) | 2.021(9)/2.012(10) 2.278(7)/2.223(10) | 1.938(10)/1.958(10) 2.139(7)/2.170(10) |
| 5 | Me3SiCA≡CBPh (5) a | Co (5a) | 2.4717(13) | 1.330(10) | 1.858(7) | 1.998(7)/1.991(7) | 1.987(7)/1.966(7) |
| 6 | Me5Si2CA≡CBPh (6) a | Co (6a) | 2.4798(9) | 1.345(4) | 1.855(3) | 1.996(3)/1.993(3) | 1.984(3)/1.970(3) |
| 7 | (Me3Si)3SiCA≡CBthio (11) | Co (11a) | 2.456(1)/ 2.473(1) | 1.343(5)/ 1.331(6) | 1.884(4)/ 1.883(4) | 2.023(5)/2.013(4)/ 2.022(4)/2.004(5) | 1.947(6)/1.983(5)/ 1.947(6)/1.983(5) |
| 8 | (Me3Si)3GeCA≡CBPh (7) | Co (7a) | 2.4582(5) | 1.338(3) | 1.966(2) C-Ge | 2.020(2)/2.009(2) | 1.974(2)/1.982(2) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zirngast, M.; Marschner, C.; Baumgartner, J. Spectroscopic and Structural Study of Some Oligosilanylalkyne Complexes of Cobalt, Molybdenum and Nickel. Molecules 2019, 24, 205. https://doi.org/10.3390/molecules24010205
Zirngast M, Marschner C, Baumgartner J. Spectroscopic and Structural Study of Some Oligosilanylalkyne Complexes of Cobalt, Molybdenum and Nickel. Molecules. 2019; 24(1):205. https://doi.org/10.3390/molecules24010205
Chicago/Turabian StyleZirngast, Michaela, Christoph Marschner, and Judith Baumgartner. 2019. "Spectroscopic and Structural Study of Some Oligosilanylalkyne Complexes of Cobalt, Molybdenum and Nickel" Molecules 24, no. 1: 205. https://doi.org/10.3390/molecules24010205
APA StyleZirngast, M., Marschner, C., & Baumgartner, J. (2019). Spectroscopic and Structural Study of Some Oligosilanylalkyne Complexes of Cobalt, Molybdenum and Nickel. Molecules, 24(1), 205. https://doi.org/10.3390/molecules24010205