
Aza-Amino Acids Disrupt β-Sheet Secondary Structures


Abstract
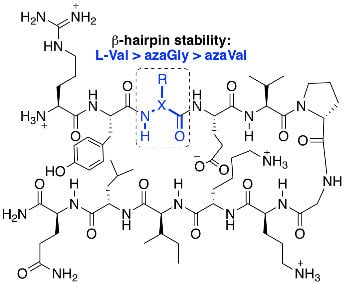
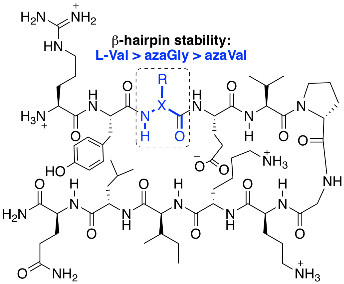
:
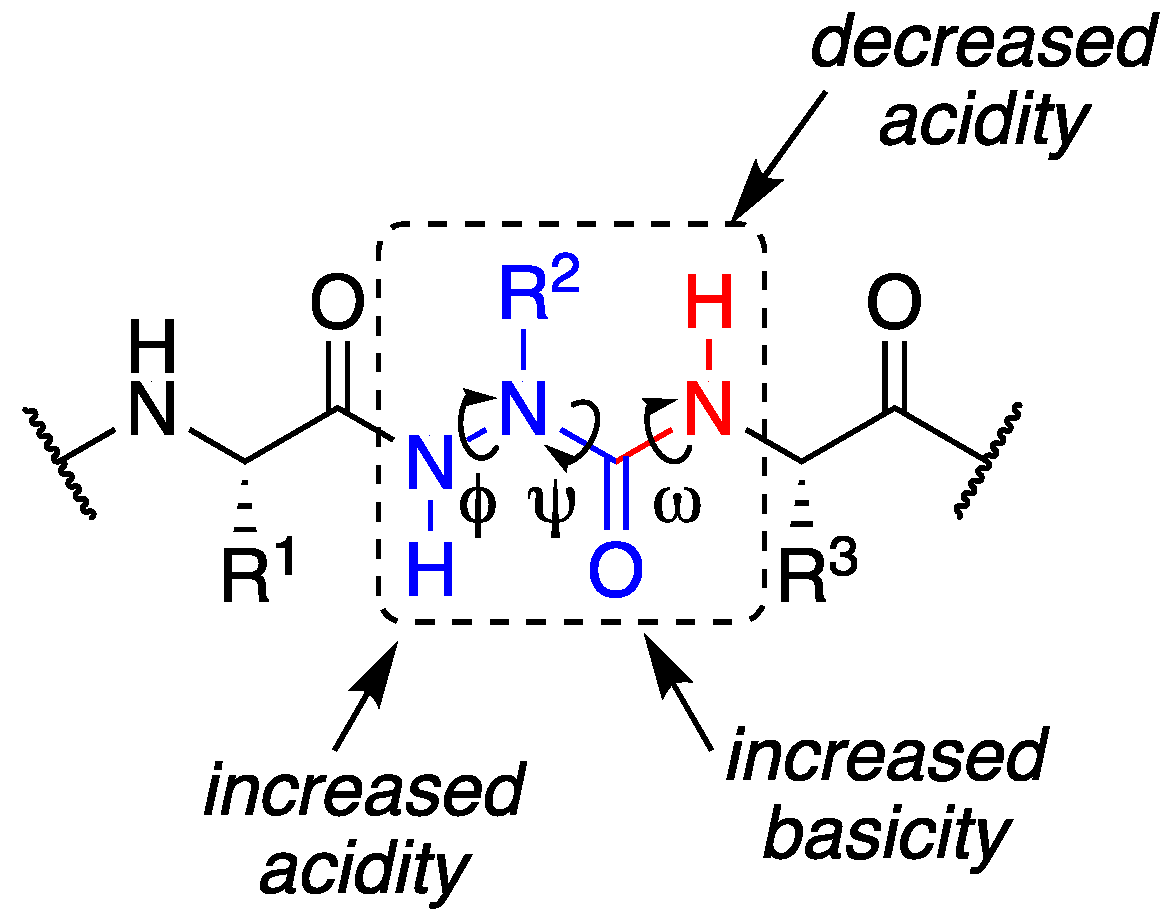
1. Introduction
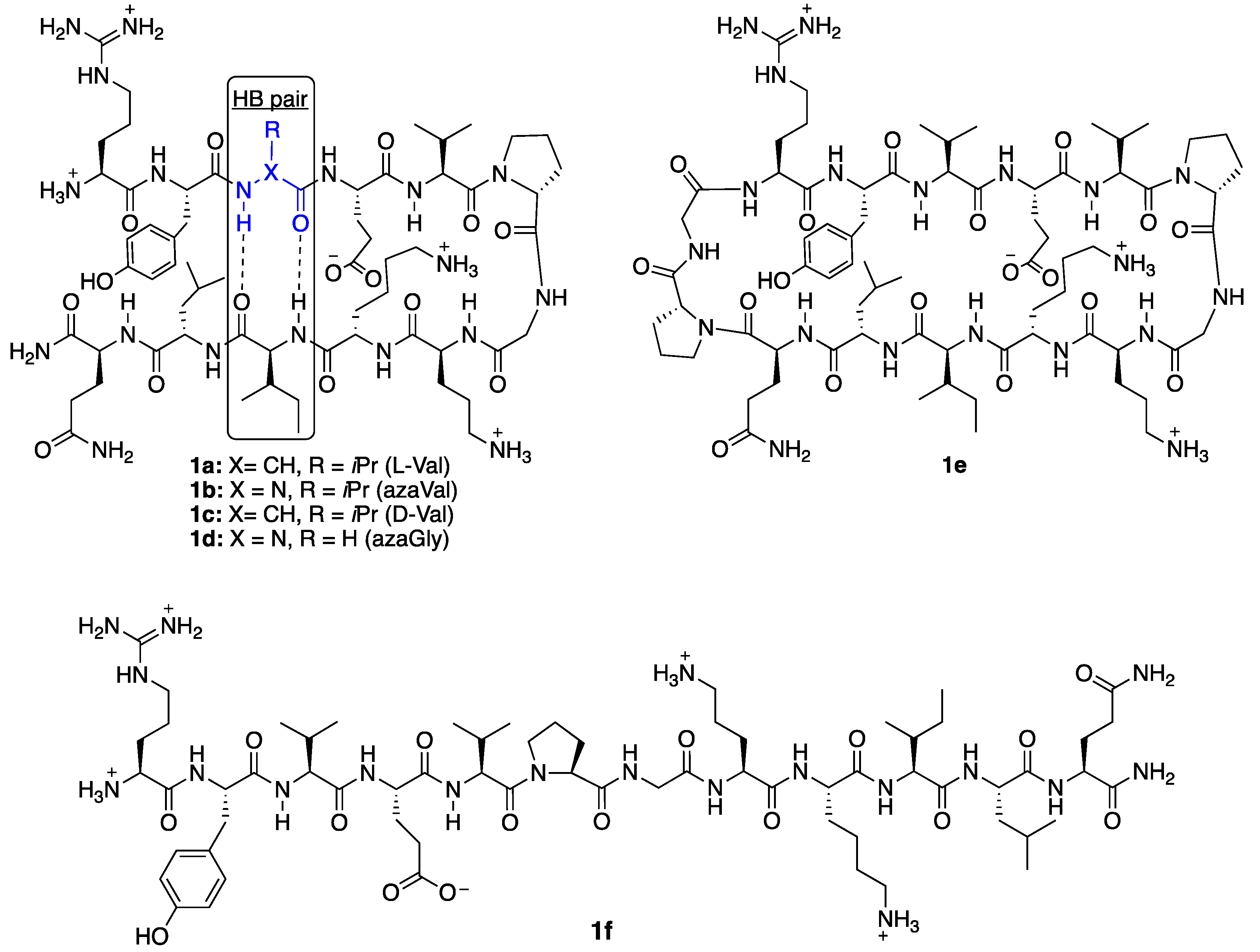
2. Results and Discussion
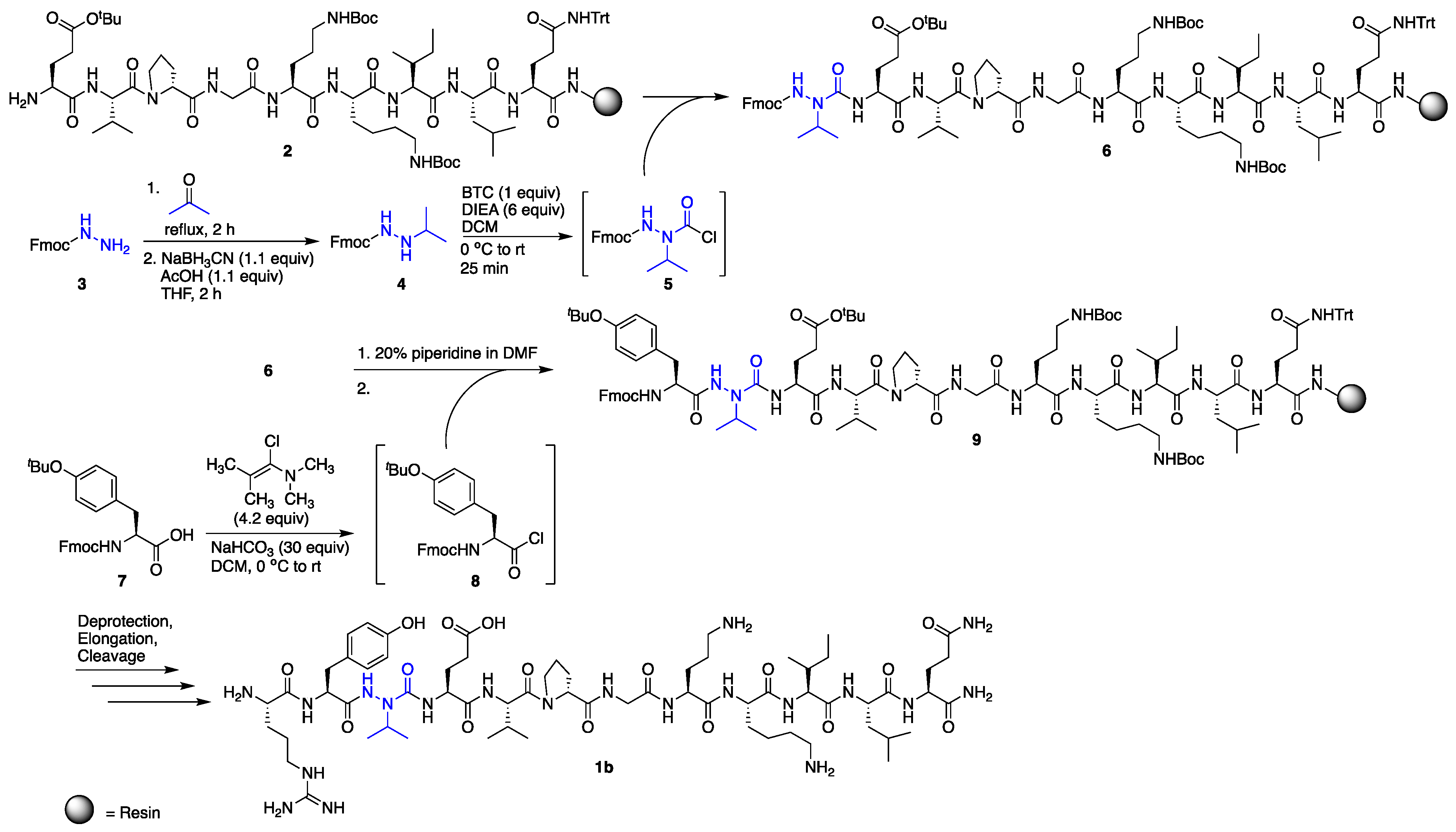
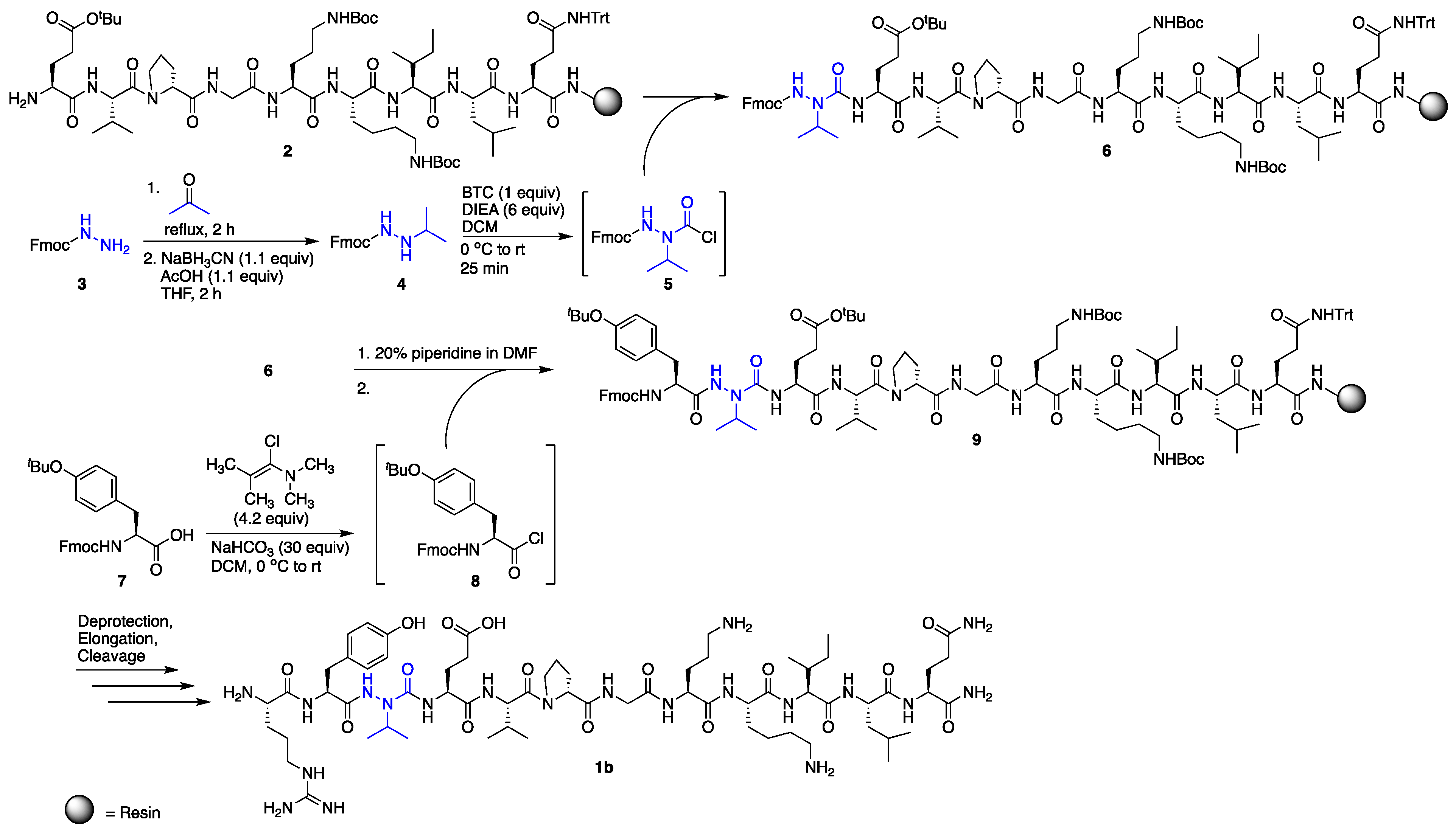
2.1. Synthesis
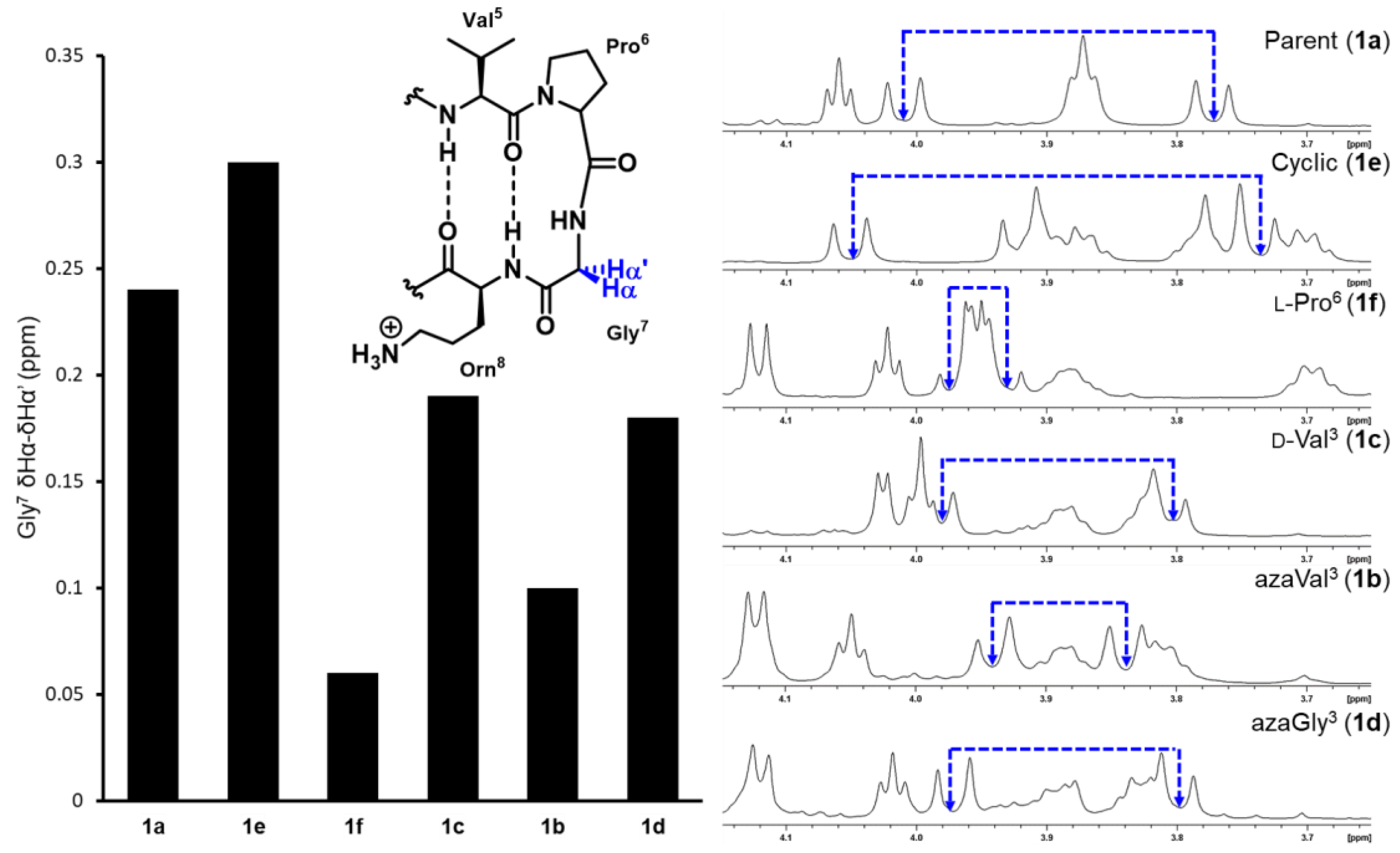
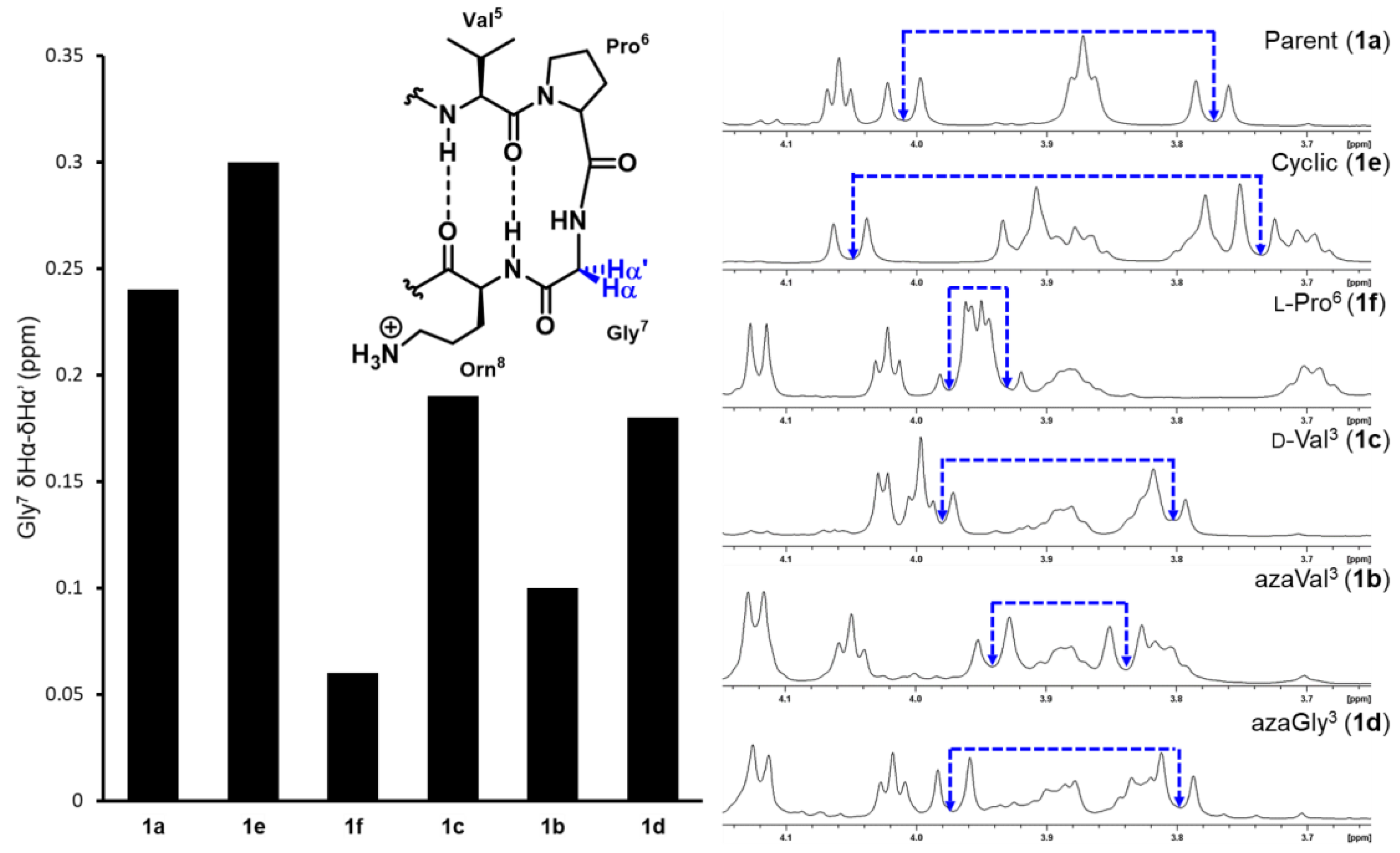
2.2. Conformational Analysis
3. Materials and Methods
3.1. General
3.2. Reagents
3.3. 9-Fluorenylmethoxycarbonyl (Fmoc)-Based Solid Phase Peptide Synthesis (SPPS): Fmoc Deprotection and HBTU Couplings
3.4. Test Cleavages of Resin-Bound Peptides
3.5. N′-Isopropyl-fluoren-9-ylmethyl Carbazate (4)
3.6. Incorporation of Aza-Valine on the Solid Phase
3.7. Coupling to AzaV-EVpGOKILQ
3.7.1. BTC Coupling
3.7.2. PyBOP Coupling
3.7.3. COMU Coupling
3.7.4. NMI/TCFH Coupling
3.7.5. Ghosez Coupling
3.8. Incorporation of Aza-Glycine on the Solid Phase
3.9. Coupling to azaG-EVpGOKILQ
3.10. Synthesis of Cyclic Peptide 1e
3.11. Full Cleavage and Purification of (Aza)Peptides
3.12. NMR Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Notes
- Cheng, P.N.; Pham, J.D.; Nowick, J.S. The Supramolecular Chemistry of beta-Sheets. J. Am. Chem. Soc. 2013, 135, 5477–5492. [Google Scholar] [CrossRef] [PubMed]
- Watkins, A.M.; Arora, P.S. Anatomy of beta-Strands at Protein-Protein Interfaces. ACS Chem. Biol. 2014, 9, 1747–1754. [Google Scholar] [CrossRef]
- Hughes, R.M.; Waters, M.L. Model systems for beta-hairpins and beta-sheets. Curr. Opin. Struct. Biol. 2006, 16, 514–524. [Google Scholar] [CrossRef]
- Loughlin, W.A.; Tyndall, J.D.A.; Glenn, M.P.; Fairlie, D.P. Beta-strand mimetics. Chem. Rev. 2004, 104, 6085–6117. [Google Scholar] [CrossRef]
- Doig, A.J. A three stranded beta-sheet peptide in aqueous solution containing N-methyl amino acids to prevent aggregation. Chem. Commun. 1997, 22, 2153–2154. [Google Scholar] [CrossRef]
- Nowick, J.S.; Chung, D.M.; Maitra, K.; Maitra, S.; Stigers, K.D.; Sun, Y. An unnatural amino acid that mimics a tripeptide beta-strand and forms beta-sheet like hydrogen-bonded dimers. J. Am. Chem. Soc. 2000, 122, 7654–7661. [Google Scholar] [CrossRef]
- Sarnowski, M.P.; Kang, C.W.; Elbatrawi, Y.M.; Wojtas, L.; Del Valle, J.R. Peptide N-Amination Supports beta-Sheet Conformations. Angew. Chem. Int. Ed. 2017, 56, 2083–2086. [Google Scholar] [CrossRef]
- Proulx, C.; Sabatino, D.; Hopewell, R.; Spiegel, J.; Ramos, Y.G.; Lubell, W.D. Azapeptides and their therapeutic potential. Future Med. Chem. 2011, 3, 1139–1164. [Google Scholar] [CrossRef]
- Zhang, Y.T.; Malamakal, R.M.; Chenoweth, D.M. Aza-Glycine Induces Collagen Hyperstability. J. Am. Chem. Soc. 2015, 137, 12422–12425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.T.; Herling, M.; Chenoweth, D.M. General Solution for Stabilizing Triple Helical Collagen. J. Am. Chem. Soc. 2016, 138, 9751–9754. [Google Scholar] [CrossRef]
- Thormann, M.; Hofmann, H.J. Conformational properties of azapeptides. J. Mol. Struct. THEOCHEM 1999, 469, 63–76. [Google Scholar] [CrossRef]
- Reynolds, C.H.; Hormann, R.E. Theoretical study of the structure and rotational flexibility of diacylhydrazines: Implications for the structure of nonsteroidal ecdysone agonists and azapeptides. J. Am. Chem. Soc. 1996, 118, 9395–9401. [Google Scholar] [CrossRef]
- Stanger, H.E.; Gellman, S.H. Rules for antiparallel beta-sheet design: D-Pro-Gly is superior to L-Asn-Gly for beta-hairpin nucleation. J. Am. Chem. Soc. 1998, 120, 4236–4237. [Google Scholar] [CrossRef]
- Syud, F.A.; Espinosa, J.F.; Gellman, S.H. NMR-based quantification of beta-sheet populations in aqueous solution through use of reference peptides for the folded and unfolded states. J. Am. Chem. Soc. 1999, 121, 11577–11578. [Google Scholar] [CrossRef]
- Hess, H.J.; Moreland, W.T.; Laubach, G.D. N- [2-Isopropyl-3-(L-Aspartyl-L-Arginyl)-Carbazoyl]-L-Tyrosyl-L-Valyl-L-Histidyl-L-Prolyl-L-Phenylalanine, an Isostere of Bovine Angiotensin Ii. J. Am. Chem. Soc. 1963, 85, 4040–4041. [Google Scholar] [CrossRef]
- Meienhofer, J.; Waki, M.; Heimer, E.P.; Lambros, T.J.; Makofske, R.C.; Chang, C.D. Solid-Phase Synthesis without Repetitive Acidolysis. Int. J. Pept. Prot. Res. 1979, 13, 35–42. [Google Scholar] [CrossRef]
- Boeglin, D.; Lubell, W.D. Aza-amino acid scanning of secondary structure suited for solid-phase peptide synthesis with Fmoc chemistry and aza-amino acids with heteroatomic side chains. J. Comb. Chem. 2005, 7, 864–878. [Google Scholar] [CrossRef]
- Chingle, R.; Proulx, C.; Lubell, W.D. Azapeptide Synthesis Methods for Expanding Side-Chain Diversity for Biomedical Applications. Acc. Chem. Res. 2017, 50, 1541–1556. [Google Scholar] [CrossRef]
- Sabatino, D.; Proulx, C.; Klocek, S.; Bourguet, C.B.; Boeglin, D.; Ong, H.; Lubell, W.D. Exploring Side-Chain Diversity by Submonomer Solid-Phase Aza-Peptide Synthesis. Org. Lett. 2009, 11, 3650–3653. [Google Scholar] [CrossRef]
- Kurian, L.A.; Silva, T.A.; Sabatino, D. Submonomer synthesis of azapeptide ligands of the Insulin Receptor Tyrosine Kinase domain. Bioorg. Med. Chem. Lett. 2014, 24, 4176–4180. [Google Scholar] [CrossRef]
- Spiegel, J.; Mas-Moruno, C.; Kessler, H.; Lubell, W.D. Cyclic Aza-peptide Integrin Ligand Synthesis and Biological Activity. J. Org. Chem. 2012, 77, 5271–5278. [Google Scholar] [CrossRef] [PubMed]
- Isocyanate and/or hydantoin byproducts likely arise from activation of the resin-bound semicarbazide with the carbonyl donor, followed by intramolecular attack by the nitrogen of the preceding amino acid residue. See: Quibell, M.; Turnell, W.G.; Johnson, T., Synthesis of Azapeptides by the Fmoc Tert-Butyl Polyamide Technique. J. Chem. Soc. Perk. Trans 1 1993, 22, 2843–2849. [CrossRef]
- Paradis-Bas, M.; Tulla-Puche, J.; Albericio, F. The road to the synthesis of “difficult peptides”. Chem. Soc. Rev. 2016, 45, 631–654. [Google Scholar] [CrossRef]
- Murray, J.K.; Farooqi, B.; Sadowsky, J.D.; Scalf, M.; Freund, W.A.; Smith, L.M.; Chen, J.D.; Gellman, S.H. Efficient synthesis of a beta-peptide combinatorial library with microwave irradiation. J. Am. Chem. Soc. 2005, 127, 13271–13280. [Google Scholar] [CrossRef]
- Chingle, R.; Ratni, S.; Claing, A.; Lubell, W.D. Application of Constrained aza-Valine Analogs for Smac Mimicry. Biopolymers 2016, 106, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Arujoe, M.; Ploom, A.; Mastitski, A.; Jarv, J. Comparison of various coupling reagents in solid-phase aza-peptide synthesis. Tetrahedron Lett. 2017, 58, 3421–3425. [Google Scholar] [CrossRef]
- Arujoe, M.; Ploom, A.; Mastitski, A.; Jarv, J. Influence of steric effects in solid-phase aza-peptide synthesis. Tetrahedron Lett. 2018, 59, 2010–2013. [Google Scholar] [CrossRef]
- Beutner, G.L.; Young, I.S.; Davies, M.L.; Hickey, M.R.; Park, H.; Stevens, J.M.; Ye, Q.M. TCFH-NMI: Direct Access to N-Acyl Imidazoliums for Challenging Amide Bond Formations. Org. Lett. 2018, 20, 4218–4222. [Google Scholar] [CrossRef] [PubMed]
- Devos, A.; Remion, J.; Frisquehesbain, A.M.; Colens, A.; Ghosez, L. Synthesis of Acyl Halides under Very Mild Conditions. J. Chem. Soc. Chem. Comm. 1979, 24, 1180–1181. [Google Scholar] [CrossRef]
- Gibson, C.; Goodman, S.L.; Hahn, D.; Ho¨lzemann, G.; Kessler, H. Novel Solid-Phase Synthesis of Azapeptides and Azapeptoides via Fmoc-Strategy and Its Application in the Synthesis of RGD-Mimetics. J. Org. Chem. 1999, 64, 7388–7394. [Google Scholar] [CrossRef]
- Bourguet, C.B.; Sabatino, D.; Lubell, W.D. Benzophenone Semicarbazone Protection Strategy for Synthesis of Aza-Glycine Containing Aza-Peptides. Biopolymers 2008, 90, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Chingle, R.; Mulumba, M.; Chung, N.N.; Nguyen, T.M.; Ong, H.; Ballet, S.; Schiller, P.W.; Lubell, W.D. Solid-Phase Azopeptide Diels-Alder Chemistry for Aza-Pipecolyl Residue Synthesis to Study Peptide Conformation. J. Org. Chem. 2019, 84, 6006–6016. [Google Scholar] [CrossRef] [PubMed]
- Searle, M.S.; Griffiths-Jones, S.R.; Skinner-Smith, H. Energetics of weak interactions in a beta-hairpin peptide: Electrostatic and hydrophobic contributions to stability from lysine salt bridges. J. Am. Chem. Soc. 1999, 121, 11615–11620. [Google Scholar] [CrossRef]
- Bouayad-Gervais, S.H.; Lubell, W.D. Examination of the potential for adaptive chirality of the nitrogen chiral center in aza-aspartame. Molecules 2013, 18, 14739. [Google Scholar] [CrossRef]
- It should be noted that replacement of valine for an aza-glycine residue results in an overall decrease in chiral environment, which may affect diastereotopic glycine splitting.
- Kiehna, S.E.; Waters, M.L. Sequence dependence of beta-hairpin structure: Comparison of a salt bridge and an aromatic interaction. Protein Sci. 2003, 12, 2657–2667. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Coupling Reagent | Equiv. | Base | Equiv. | Solvent | % Coupled | % Uncoupled | % Isocyanate/Hydantoin Byproduct [22] |
|---|---|---|---|---|---|---|---|---|
| 1 | BTC | 1 | 2,4,6-collidine | 10 | THF | 33 | 21 | 28 |
| 2 | PyBOP | 3 | DIEA | 6 | DMF | 6 | 35 | 41 |
| 3 | COMU | 5 | DIEA | 10 | DMF | 13 | 17 | 53 |
| 4 | TCFHa | 3 | NMI | 9 | CH2Cl2 | 89a | 0 | 10 |
| 5 | Ghosez | 4.2 | NaHCO3 | 30 | CH2Cl2 | 67 | 0 | 22 |
| Peptide | % Folding | |||
|---|---|---|---|---|
| Gly7 δHα–δHα’ | Orn8 | Ile10 | Average from Orn8/Ile10 | |
| 1a | 75 | 72 | 64 | 68 |
| 1b | 17 | 36 | 0 | 18 |
| 1c | 54 | 56 | 15 | 35.5 |
| 1d | 50 | 50 | 49 | 49.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McMechen, M.A.; Willis, E.L.; Gourville, P.C.; Proulx, C. Aza-Amino Acids Disrupt β-Sheet Secondary Structures. Molecules 2019, 24, 1919. https://doi.org/10.3390/molecules24101919
McMechen MA, Willis EL, Gourville PC, Proulx C. Aza-Amino Acids Disrupt β-Sheet Secondary Structures. Molecules. 2019; 24(10):1919. https://doi.org/10.3390/molecules24101919
Chicago/Turabian StyleMcMechen, Michael A., Evan L. Willis, Preston C. Gourville, and Caroline Proulx. 2019. "Aza-Amino Acids Disrupt β-Sheet Secondary Structures" Molecules 24, no. 10: 1919. https://doi.org/10.3390/molecules24101919
APA StyleMcMechen, M. A., Willis, E. L., Gourville, P. C., & Proulx, C. (2019). Aza-Amino Acids Disrupt β-Sheet Secondary Structures. Molecules, 24(10), 1919. https://doi.org/10.3390/molecules24101919