
Development and Optimisation of a Multiresidue Method for the Determination of 40 Anthelmintic Compounds in Environmental Water Samples by Solid Phase Extraction (SPE) with LC-MS/MS Detection

 ,
,  ,
, 

Abstract
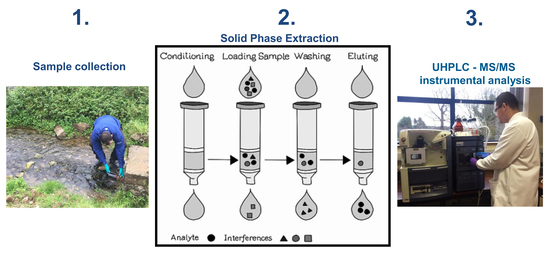
:
1. Introduction
2. Results and Discussion
2.1. Method Development: Sample Preparation—Solid Phase Extraction
2.1.1. Assessment of SPE Sorbents
2.1.2. Elution Solvent Composition and Wash Solvent Assessment
2.1.3. Sample Modification (Organic Modifier and pH)
2.2. Method Validation
2.2.1. Identification
2.2.2. Specificity and Linearity
2.2.3. Trueness and Precision
2.2.4. Recovery, Limits of Detection (LOD) and Limits of Quantification (LOQ)
2.3. Matrix Effects
2.4. Applicability
3. Materials and Methods
3.1. Chemicals, Standards and Consumables
3.2. Preparation of Standard Solutions
3.3. Sample Collection, Control Samples and Quality Control (QC)
3.4. Matrix Matched Calibration
3.5. UHPLC-MS/MS Determination
3.6. Sample Preparation-Solid Phase Extraction
3.6.1. Development and Optimisation
3.6.2. Final Method
3.7. Method Validation Procedure
3.8. Matrix Effects
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tuck, S.; Furey, A.; Danaher, M. Analysis of Anthelmintic and Anticoccidial Drug Residues in Animal-Derived Foods. In Chemical Analysis of Non-antimicrobial Veterinary Drug Residues in Food; Kay, J.F., MacNeil, J.D., Wang, J., Eds.; John Wiley & Sons, Inc.: Whitehouse, NJ, USA, 2016. [Google Scholar]
- Kahn, C.M.; Line, S. The Merck Veterinary Manual, 10th ed.; Kahn, C.M., Line, S., Eds.; Merck & Co. Inc.: Whitehouse, NJ, USA, 2010. [Google Scholar]
- Beynon, S.A. Potential environmental consequences of administration of ectoparasiticides to sheep. Vet. Parasitol. 2012, 189, 125–135. [Google Scholar] [CrossRef]
- Danaher, M.; De Ruyck, H.; Crooks, S.R.; Dowling, G.; O’Keeffe, M. Review of methodology for the determination of benzimidazole residues in biological matrices. J. Chromatogr. B 2007, 845, 1–37. [Google Scholar] [CrossRef]
- Wardhaugh, K.G. Insecticidal activity of synthetic pyrethroids, organophosphates, insect growth regulators and other livestock parasiticides: An australian perspective. Environ. Toxicol. Chem. 2005, 24, 789–796. [Google Scholar] [CrossRef]
- Boxall, A.B.A.; Kolpin, D.W.; Halling-Sorensen, B. Are Veterinary Medicines Causing Environmental Risks? Environ. Sci. Technol. 2003, 37, 286–294. [Google Scholar] [CrossRef]
- Beynon, S.A. Potential environmental consequences of administration of anthelmintics to sheep. Vet. Parasitol. 2012, 189, 113–124. [Google Scholar] [CrossRef]
- Danaher, M.; Howells, L.C.; Crooks, S.R.; Cerkvenik-Flajs, V.; O’Keeffe, M. Review of methodology for the determination of macrocyclic lactone residues in biological matrices. J. Chromatogr. B 2006, 844, 175–203. [Google Scholar] [CrossRef] [PubMed]
- Liebig, M.; Fernandez, A.A.; Blubaum-Gronau, E.; Boxall, A.; Brinke, M.; Carbonell, G.; Egeler, P.; Fenner, K.; Fernandez, C.; Fink, G.; et al. Environmental risk assessment of ivermectin: A case study. Integr. Environ. Asses. Manag. 2010, 6, 567–587. [Google Scholar] [CrossRef] [PubMed]
- McKellar, Q.A. Ecotoxicology and residues of anthelmintic compounds. Vet. Parasitol. 1997, 72, 413–435. [Google Scholar] [CrossRef]
- Charuaud, L.; Jarde, E.; Jaffrezic, A.; Thomas, M.F.; Le Bot, B. Veterinary pharmaceutical residues from natural water to tap water: Sales, occurrence and fate. J. Hazard. Mater. 2019, 361, 169–186. [Google Scholar] [CrossRef] [PubMed]
- Snow, D.D.; Cassada, D.A.; Bartelt-Hunt, S.L.; Li, X.; D’Alessio, M.; Zhang, Y.; Zhang, Y.; Sallach, J.B. Detection, Occurrence and Fate of Emerging Contaminants in Agricultural Environments. Water Environ. Res. 2016, 88, 913–929. [Google Scholar] [CrossRef] [PubMed]
- Horvat, A.J.M.; Babić, S.; Pavlović, D.M.; Ašperger, D.; Pelko, S.; Kaštelan-Macan, M.; Petrović, M.; Mance, A.D. Analysis, occurrence and fate of anthelmintics and their transformation products in the environment. TRAC-Trend Anal. Chem. 2012, 31, 61–84. [Google Scholar] [CrossRef]
- Boxall, A.B.A. Veterinary Medicines and the Environment. In Comparative and Veterinary Pharmacology. Handbook of Experimental Pharmacology; Cunningham, F., Elliott, J., Lees, P., Eds.; Springer: Berlin, Germany, 2010. [Google Scholar]
- Kim, K.-R.; Owens, G.; Kwon, S.-I.; So, K.-H.; Lee, D.-B.; Ok, Y.S. Occurrence and Environmental Fate of Veterinary Antibiotics in the Terrestrial Environment. Water Air Soil Poll. 2010, 214, 163–174. [Google Scholar] [CrossRef]
- Boxall, A.B.A.; Fogg, L.; Blackwell, P.A.; Kay, P.; Pemberton, E.J. Review of Veterinary Medicines in the Environment-R&D Technical Report P6-012/8/TR; Environment Agency: Bristol, UK, 2002. [Google Scholar]
- Boxall, A.B.A.; Fogg, L.A.; Blackwell, P.A.; Kay, P.; Pemberton, E.J.; Croxford, A. Prioritisation of veterinary medicines in the UK environment. Toxicol. Lett. 2003, 142, 207–218. [Google Scholar] [CrossRef]
- O’Hea, N.; Kirwan, L.; Giller, P.; Finn, J. Lethal and sub-lethal effects of ivermectin on north temperate dung beetles, Aphodius aterandAphodius rufipes(Coleoptera: Scarabaeidae). Insect. Conserv. Diver. 2010, 3, 24–33. [Google Scholar] [CrossRef]
- European Commission. COUNCIL DIRECTIVE 98/83/EC of 3 November 1998 on the quality of water intended for human consumption. Off. J. Eur. Commun. 1998, L330, 32–54. [Google Scholar]
- European Parliament. DIRECTIVE 2006/118/EC OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 12 December 2006 on the protection of groundwater against pollution and deterioration. Off. J. Eur. Commun. 2006, L372, 19–31. [Google Scholar]
- Le Bizec, B.; Pinel, G.; Antignac, J.P. Options for veterinary drug analysis using mass spectrometry. J. Chromatogr. A 2009, 1216, 8016–8034. [Google Scholar] [CrossRef] [PubMed]
- Sim, W.J.; Kim, H.Y.; Choi, S.D.; Kwon, J.H.; Oh, J.E. Evaluation of pharmaceuticals and personal care products with emphasis on anthelmintics in human sanitary waste, sewage, hospital wastewater, livestock wastewater and receiving water. J. Hazard. Mater. 2013, 248–249, 219–227. [Google Scholar] [CrossRef]
- Van De Steene, J.C.; Lambert, W.E. Validation of a solid-phase extraction and liquid chromatography-electrospray tandem mass spectrometric method for the determination of nine basic pharmaceuticals in wastewater and surface water samples. J. Chromatogr. A 2008, 1182, 153–160. [Google Scholar] [CrossRef]
- Kaufmann, A.; Butcher, P.; Maden, K.; Widmer, M. Quantitative multiresidue method for about 100 veterinary drugs in different meat matrices by sub 2-microm particulate high-performance liquid chromatography coupled to time of flight mass spectrometry. J. Chromatogr. A 2008, 1194, 66–79. [Google Scholar] [CrossRef]
- Geis-Asteggiante, L.; Lehotay, S.J.; Lightfield, A.R.; Dutko, T.; Ng, C.; Bluhm, L. Ruggedness testing and validation of a practical analytical method for >100 veterinary drug residues in bovine muscle by ultrahigh performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2012, 1258, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Zrncic, M.; Gros, M.; Babic, S.; Kastelan-Macan, M.; Barcelo, D.; Petrovic, M. Analysis of anthelmintics in surface water by ultra high performance liquid chromatography coupled to quadrupole linear ion trap tandem mass spectrometry. Chemosphere 2014, 99, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Krogh, K.A.; Bjorklund, E.; Loeffler, D.; Fink, G.; Halling-Sorensen, B.; Ternes, T.A. Development of an analytical method to determine avermectins in water, sediments and soils using liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2008, 1211, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.D.; Haberhauer, G.; Kist, A.; Rathor, M.N.; Gerzabek, M.; Cannavan, A. Multi-class determination of anthelmintics in soil and water by LC-MS/MS. Food Addit. Contam. A 2013, 30, 1128–1137. [Google Scholar] [CrossRef] [PubMed]
- Santaladchaiyakit, Y.; Srijaranai, S. A simplified ultrasound-assisted cloud-point extraction method coupled with high performance liquid chromatography for residue analysis of benzimidazole anthelmintics in water and milk samples. Anal. Method 2012, 4, 3864. [Google Scholar] [CrossRef]
- Whelan, M.; Kinsella, B.; Furey, A.; Moloney, M.; Cantwell, H.; Lehotay, S.J.; Danaher, M. Determination of anthelmintic drug residues in milk using ultra high performance liquid chromatography-tandem mass spectrometry with rapid polarity switching. J. Chromatogr. A 2010, 1217, 4612–4622. [Google Scholar] [CrossRef]
- Peters, R.J.B.; Bolck, Y.J.C.; Rutgers, P.; Stolker, A.A.M.; Nielen, M.W.F. Multiresidue screening of veterinary drugs in egg, fish and meat using high-resolution liquid chromatography accurate mass time-of-flight mass spectrometry. J. Chromatogr. A 2009, 1216, 8206–8216. [Google Scholar] [CrossRef]
- Babic, S.; Mutavdzic Pavlovic, D.; Asperger, D.; Perisa, M.; Zrncic, M.; Horvat, A.J.; Kastelan-Macan, M. Determination of multi-class pharmaceuticals in wastewater by liquid chromatography-tandem mass spectrometry (LC-MS-MS). Anal. Bioanal. Chem. 2010, 398, 1185–1194. [Google Scholar] [CrossRef]
- Sebestyén, I.; Monostory, K.; Hirka, G. Environmental risk assessment of human and veterinary medicinal products - Challenges and ways of improvement. Microchem. J. 2018, 136, 67–70. [Google Scholar] [CrossRef] [Green Version]
- Thompson, T.S.; Noot, D.K.; Forrest, F.; van den Heever, J.P.; Kendall, J.; Keenliside, J. Large volume injection for the direct analysis of ionophores and avermectins in surface water by liquid chromatography-electrospray ionization tandem mass spectrometry. Anal. Chim. Acta 2009, 633, 127–135. [Google Scholar] [CrossRef]
- Berendsen, B.J.; Stolker, L.A.; Nielen, M.W. The (un)certainty of selectivity in liquid chromatography tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2013, 24, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Whelan, M.; O’Mahony, J.; Moloney, M.; Cooper, K.M.; Furey, A.; Kennedy, D.G.; Danaher, M. Maximum residue level validation of triclabendazole marker residues in bovine liver, muscle and milk matrices by ultra high pressure liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2013, 1275, 41–47. [Google Scholar] [CrossRef] [PubMed]
- European Commission. 2002/657/EC. COMMISSION DECISION of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off. J. Eur. Commun. 2002, L221, 8–36. [Google Scholar]
- European Commission. SANTE/11813/2017. Guidance Document on Analytical Quality Control and Method Validation Procedures for Pesticide Residues and Analysis in Food and Feed. European Commission Directorate-General for Health and Food Safety 2017. Available online: https://ec.europa.eu/food/sites/food/files/plant/docs/pesticides_mrl_guidelines_wrkdoc_2017-11813.pdf (accessed on 20 May 2019).
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the Assessment of Matrix Effect in Quantitative Bioanalytical Methods Based on HPLC-MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef]
- O’Neill, M.J. The Merck Index: An. Encyclopedia of Chemicals, Drugs, and Biologicals, 13th ed.; O’Neil, M.J., Ed.; Merck & Co., Inc.: Whitehouse, NJ, USA, 2001. [Google Scholar]
- Van der Velde-Koerts, T. Emamectin Benzoate (247) {Monograph}; Centre for Substances and Integrated Risk Assessment, National Institute of Public Health and the Environment: Bilthoven, The Netherlands, 2014; Available online: http://fao.org/fileadmin/templates/agphome/documents/Pests_Pesticides/JMPR/Evaluation11/Emamectin.pdf (accessed on 20 May 2019).
Sample Availability: Not available. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Performance Criteria | Guideline a |
|---|---|---|
| Identification | ||
| Points | Minimum 3 | 2002/657 |
| Relative retention (RRT) | ≤2.5% | 2002/657 |
| Ion ratio tolerance (ΔR) | 20–50% 30% | 2002/657 SANTE |
| Selectivity | Interferences: ≤10% lowest calibrant Interferences: ≤30% lowest calibrant | 2002/657 SANTE |
| Linearity | Regression coefficient R2 ≥ 0.98 Residuals ± 20% | 2002/657 SANTE |
| Trueness (WLR and WLr) | 70–120% | SANTE |
| Precision (RSDwR and RSDr) | ≤20% | SANTE |
| Recovery | 70–120% | SANTE |
| Analyte | Abbreviation | P/TP | Labelled IS Used | Calibration Range (ng L−1) | Linearity R2 | Mean ME (%) (n = 30) | ME RANGE (%) | RSD No IS (n = 30) (%) | RSD with IS (n = 30) (%) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Min | Max | |||||||||
| Benzimidazoles | ||||||||||
| Albendazole | ABZ | P | ABZ-d3 | 1–1000 | 0.997 | 27.1 | 8.2 | 47.3 | 9.3 | 3.0 |
| Albendazole sulphoxide | ABZ-SO | TP | ABZ-SO-d3 | 1–1000 | 0.994 | 93.4 | 13.8 | 212 | 31.6 | 7.1 |
| Albendazole sulphone | ABZ-SO2 | TP | ABZ-SO2-d3 | 1–1000 | 0.996 | 60.8 | 29 | 120 | 18.2 | 6.5 |
| Albendazole-amino-sulphone | ABZ-NH2-SO2 | TP | ABZ-NH2-SO2-d3 | 0.5–1000 | 0.998 | 16.9 | −1.4 | 28.0 | 6.9 | 4.0 |
| Cambendazole | CAM | P | FBZ-d3 | 0.5–1000 | 0.997 | 9.7 | −5.1 | 24.2 | 6.6 | 7.2 |
| Fenbendazole | FBZ | P | FBZ-d3 | 0.5–1000 | 0.995 | 23.1 | 1.0 | 44.9 | 9.3 | 2.3 |
| Oxfendazole | OXF | TP | FBZ-SO-d3 | 1–1000 | 0.993 | 42.0 | 11.6 | 106.2 | 18.5 | 6.4 |
| Fenbendazole sulphone | FBZ-SO2 | TP | FBZ-SO2-d3 | 1–1000 | 0.998 | 47.5 | 8.1 | 165.7 | 25.5 | 3.3 |
| Flubendazole | FLU | P | FLU-d3 | 1–1000 | 0.996 | 33.3 | 7.4 | 108.2 | 14.1 | 3.7 |
| Amino-flubendazole | FLU-NH2 | TP | TCB-NH2 (pos) | 1–1000 | 0.995 | 11.5 | −3.7 | 29.8 | 8.0 | 8.8 |
| Hydroxy-flubendazole | FLU-OH | TP | MBZ-OH-d3 | 1–1000 | 0.997 | 3.7 | −12.9 | 27.4 | 12.1 | 7.6 |
| Mebendazole | MBZ | P | MBZ-d3 | 1–1000 | 0.994 | 45.0 | 11.4 | 104.2 | 18.1 | 3.6 |
| Amino-mebendazole | MBZ-NH2 | TP | TCB-NH2 (pos) | 1–1000 | 0.995 | 15.1 | 0 | 36.3 | 7.2 | 8.5 |
| Hydroxy-mebendazole | MBZ-OH | TP | MBZ-OH-d3 | 1–1000 | 0.998 | 27.4 | 3.8 | 64.2 | 13.1 | 4.6 |
| Oxibendazole | OXI | P | OXI-d7 | 0.5–1000 | 0.994 | 9.3 | −2.5 | 21.6 | 5.8 | 4.5 |
| Triclabendazole | TCB | P | TCB-d3 | 0.5–1000 | 0.997 | 3.6 | −14.2 | 27.4 | 8.0 | 3.3 |
| Triclabendazole-sulphoxide | TCB-SO | TP | TCB-NH2(neg) | 4–20 | 0.967 | −3.0 | −45.0 | 47.8 | 25.2 | 24.7 |
| Triclabendazole-sulphone | TCB-SO2 | TP | TCB-NH2(neg) | 4–20 | 0.891 | 5.2 | −25.4 | 57.8 | 18.2 | 19.8 |
| Thiabendazole | TBZ | P | TBZ-13C6 | 0.5–1000 | 0.999 | 9.1 | −6.8 | 26.6 | 6.7 | 2.7 |
| 5-Hydroxy-Thiabendazole | TBZ-OH | TP | ABZ-NH2-SO2-d3 | 0.5–200 | 0.991 | −6.4 | −23.8 | 12.7 | 9.6 | 7.2 |
| Macrocyclic lactones (Avermectins & Milbemycins) | ||||||||||
| Abamectin | ABA | P | SEL | 10–2000 | 0.996 | 20.4 | −4.1 | 45.7 | 9.7 | 7.5 |
| Doramectin | DORA | P | SEL | 10–1000 | 0.993 | 77.8 | 13.8 | 130.9 | 16.0 | 15.2 |
| Emamectin | EMA | P | SEL | 0.5–200 | 0.996 | 24.8 | 3.4 | 37.8 | 7.7 | 8.2 |
| Eprinomectin | EPRINO | P | SEL | 20–2000 | 0.997 | 6.8 | −17.9 | 25.7 | 9.9 | 8.3 |
| Ivermectin | IVER | P | SEL | 10–2000 | 0.996 | 5.2 | −22.5 | 27.2 | 9.6 | 7.9 |
| Moxidectin | MOXI | P | SEL | 10–2000 | 0.996 | 34.9 | −9.1 | 76.3 | 16.3 | 13.7 |
| Salicylanilides and substituted phenols | ||||||||||
| Bithionol | BITH | P | RAFOX-13C6 | 5–1000 | 0.995 | 32.0 | −1.4 | 50 | 10.6 | 5.4 |
| Closantel | CLOS | P | CLOS-13C6 | 2–1000 | 0.997 | −3.9 | −12.1 | 5.2 | 5.4 | 2.8 |
| Niclosamide | NICLOS | P | SAL | 1–200 | 0.991 | 13.0 | −5 | 33.3 | 8.3 | 5.1 |
| Nitroxynil | NITROX | P | NITROX-13C6 | 10–1000 | 0.993 | 28.6 | −5.7 | 73.2 | 14.9 | 14.2 |
| Oxyclozanide | OXY | P | OXY-13C6 | 5–1000 | 0.996 | 42.8 | 18.2 | 70.9 | 9.5 | 10.4 |
| Rafoxanide | RAFOX | P | RAFOX-13C6 | 2–1000 | 0.994 | 23.0 | 2 | 41.2 | 10.5 | 3.4 |
| Tetrahydropyrimidines | ||||||||||
| Morantel | MOR | P | TBZ-13C6 | 1–1000 | 0.997 | 13.3 | −2.5 | 34.1 | 7.4 | 1.6 |
| Imidazothiazoles | ||||||||||
| Levamisole | LEV | P | LEVA-d5 | 0.5–1000 | 0.999 | 12.4 | −2.5 | 33.7 | 7.4 | 2.0 |
| Organophosphates | ||||||||||
| Coumaphos | COUMA | P | ABZ-d3 | 5–200 | 0.986 | 47.0 | 10.1 | 87.7 | 12.9 | 8.4 |
| Coumaphos-Oxon | COUMA-O | P | FBZ-d3 | 1–1000 | 0.992 | 16.2 | 3.9 | 31.9 | 6.0 | 7.6 |
| Haloxon | HALOX | P | ABZ-d3 | 5–500 | 0.989 | 25.5 | −73.8 | 55 | 12.0 | 7.2 |
| Amino-acetonitrile derivatives | ||||||||||
| Monepantel | MONE | P | CLOS-13C6 | 5–400 | 0.991 | 16.7 | −6.1 | 31.8 | 7.2 | 8.5 |
| Monepantel-sulphone | MONE-SO2 | TP | CLOS-13C6 | 1–400 | 0.993 | 14.0 | −4.4 | 28.8 | 7.0 | 7.4 |
| Miscellaneous | ||||||||||
| Clorsulon | CLOR | P | SAL | 40–2000 | 0.991 | −15.1 | −48.8 | 9.2 | 18.7 | 15.6 |
| Analyte | Validated Levels L1, L2, L3 (ng L−1) | WLr Trueness (RSDr) (%) a | WLR Trueness (RSDWR) (%) b | LOD c (ng L−1) | LOQ d (ng L−1) | Recovery % (RSD%, n = 3) at | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| L1 | L2 | L3 | L1 | L2 | L3 | 20/40 ng L−1 | 200/400 ng L−1 | ||||
| Benzimidazoles | |||||||||||
| ABZ | 5, 50, 200 | 100 (5.6) | 100 (3.0) | 97 (1.5) | 102 (3.6) | 100 (3.2) | 98 (2.8) | 0.125 | 1.0 | 94 (4.7) | 94 (0.5) |
| ABZ-SO | 5, 50, 200 | 113 (10.8) | 97 (7.3) | 101 (4.7) | 107 (13.5) | 99 (9.9) | 99 (5.2) | 0.2 | 1.0 | 95 (1.3) | 114 (5.5) |
| ABZ-SO2 | 5, 50, 200 | 95 (7.3) | 96 (4.5) | 99 (2.5) | 105 (6.4) | 99 (3.2) | 99 (3.2) | 0.165 | 1.0 | 92 (2.1) | 105 (5.6) |
| ABZ-NH2-SO2 | 5, 50, 200 | 103 (3.1) | 101 (1.4) | 101 (1.1) | 101 (3.7) | 99 (2.3) | 100 (3.9) | 0.165 | 0.5 | 93 (4.0) | 91 (7.6) |
| CAM | 5, 50, 200 | 103 (4.0) | 96 (1.4) | 97 (1.1) | 102 (4.3) | 101 (3.9) | 100 (3.1) | 0.165 | 0.5 | 94 (3.2) | 92 (6.0) |
| FBZ | 5, 50, 200 | 103 (4.5) | 97 (2.0) | 100 (1.3) | 105 (6.7) | 100 (3.8) | 99 (2.3) | 0.1 | 0.5 | 89 (4.6) | 109 (1.9) |
| OXF | 5, 50, 200 | 87 (11.4) | 100 (5.4) | 101 (3.3) | 101 (15.1) | 98 (6.3) | 98 (6.4) | 0.25 | 1.0 | 94 (6.5) | 103 (4.8) |
| FBZ-SO2 | 5, 50, 200 | 99 (2.7) | 96 (1.6) | 97 (0.8) | 101 (5.1) | 99 (3.0) | 99 (1.7) | 0.20 | 1.0 | 97 (3.2) | 102 (5.5) |
| FLU | 5, 50, 200 | 107 (7.2) | 95 (5.5) | 95 (2.1) | 102 (7.1) | 97 (4.3) | 100 (3.3) | 0.1 | 1.0 | 97 (4.9) | 97 (2.5) |
| FLU-NH2 | 5, 50, 200 | 107 (3.6) | 104 (3.4) | 97 (2.4) | 105 (4.8) | 103 (2.9) | 98 (3.4) | 0.05 | 1.0 | 94 (5.1) | 102 (1.8) |
| FLU-OH | 5, 50, 200 | 97 (6.8) | 109 (4.4) | 103 (2.3) | 99 (5.6) | 102 (4.3) | 101 (3.1) | 0.3 | 1.0 | 95 (4.3) | 99 (3.7) |
| MBZ | 5, 50, 200 | 105 (5.3) | 99 (3.6) | 97 (2.0) | 102 (6.1) | 97 (3.9) | 98 (2.6) | 0.125 | 1.0 | 97 (4.0) | 102 (0.9) |
| MBZ-NH2 | 5, 50, 200 | 104 (3.4) | 104 (3.1) | 96 (3.8) | 105 (4.8) | 104 (3.5) | 100 (4.1) | 0.3 | 1.0 | 92 (2.0) | 101 (2.4) |
| MBZ-OH | 5, 50, 200 | 102 (2.6) | 107 (1.0) | 100 (1.0) | 103 (4.3) | 101 (4.2) | 99 (2.5) | 0.2 | 1.0 | 96 (3.6) | 104 (5.2) |
| OXI | 5, 50, 200 | 102 (2.7) | 99 (2.7) | 97 (1.0) | 106 (5.2) | 101 (3.3) | 98 (3.2) | 0.125 | 0.5 | 103 (3.3) | 98 (2.4) |
| TCB | 5, 50, 200 | 96 (6.9) | 105 (4.5) | 102 (3.5) | 100 (7.6) | 102 (3.5) | 100 (3.4) | 0.125 | 0.5 | 91 (2.0) | 100 (4.0) |
| TCB-SO | 6, 14, 20 | - | - | - | - | - | - | 1.0 | 4.0 | 80 (4.8) | 92 (6.6) |
| TCB-SO2 | 6, 14, 20 | - | - | - | - | - | - | 1.0 | 4.0 | 97 (7.5) | 103 (4.8) |
| TBZ | 5, 50, 200 | 102 (3.8) | 99 (1.0) | 98 (0.6) | 103 (3.2) | 99 (2.4) | 100 (2.0) | 0.1 | 0.5 | 99 (3.1) | 98 (3.2) |
| TBZ-OH | 5, 50, 150 | 110 (1.5) | 101 (1.2) | 93 (0.7) | 109 (3.3) | 100 (2.1) | 92 (4.1) | 0.1 | 0.5 | 104 (2.0) | 80 (5.1) |
| Macrocyclic lactones (Avermectins & Milbemycins) | |||||||||||
| ABA | 40,150,500 | 104 (5.4) | 99 (5.0) | 98 (7.3) | 98 (8.5) | 100 (5.6) | 99 (3.2) | 1.0 | 10.0 | 110 (9.0) | 90 (6.0) |
| DORA | 20, 80, 200 | 103 (4.7) | 97 (5.3) | 103 (4.3) | 98 (7.9) | 97 (7.3) | 99 (4.5) | 0.5 | 10.0 | 105 (6.8) | 87 (1.5) |
| EMA | 5, 50, 150 | 107 (4.5) | 96 (9.6) | 104 (8.7) | 108 (5.6) | 104 (6.5) | 102 (5.5) | 0.05 | 0.5 | 102 (5.0) | 87 (4.5) |
| EPRINO | 40, 150, 500 | 96 (3.4) | 99 (4.9) | 104 (2.6) | 100 (8.9) | 101 (3.1) | 102 (2.4) | 5 | 20.0 | 109 (0.8) | 91 (5.6) |
| IVER | 40, 150, 500 | 104 (4.1) | 100 (2.7) | 107 (5.4) | 98 (7.5) | 100 (2.9) | 103 (4.6) | 2.5 | 10.0 | 113 (10.9) | 72 (8.7) |
| MOXI | 40, 150, 500 | 96 (6.4) | 92 (8.7) | 91 (6.5) | 101 (7.8) | 100 (8.0) | 98 (6.5) | 2.0 | 10.0 | 95 (10.8) | 59 (5.0) |
| Salicylanilides and substituted phenols | |||||||||||
| BITH | 20, 80, 200 | 112 (5.6) | 112 (4.7) | 104 (2.7) | 114 (7.2) | 106 (4.8) | 101 (3.8) | 1.0 | 5.0 | 98 (10.8) | 84 (3.7) |
| CLOS | 5, 50, 200 | 105 (4.8) | 104 (2.0) | 101 (1.0) | 105 (7.1) | 101 (3.7) | 99 (3.2) | 0.5 | 2.0 | 103 (3.6) | 76 (3.5) |
| NICLOS | 5, 50, 150 | 107 (10.3) | 106 (3.7) | 96 (2.0) | 114 (9.5) | 105 (7.2) | 96 (6.9) | 0.125 | 1.0 | 94 (7.0) | 100 (5.4) |
| NITROX | 20, 80, 200 | 107 (19.5) | 107 (13.2) | 91 (4.6) | 96 (19.4) | 104 (12.4) | 96 (8.7) | 2.5 | 10.0 | 105 (4.6) | 56 (4.7) |
| OXY | 20, 80, 200 | 113 (6.7) | 108 (7.4) | 101 (2.4) | 109 (9.6) | 103 (8.6) | 101 (4.1) | 1.5 | 5.0 | 93 (7.7) | 104 (5.6) |
| RAFOX | 5, 50, 200 | 105 (8.7) | 101 (3.0) | 99 (1.8) | 102 (10.3) | 102 (4.3) | 101 (2.5) | 0.3 | 2.0 | 97 (5.8) | 86 (4.8) |
| Tetrahydropyrimidines | |||||||||||
| MOR | 5, 50, 200 | 101 (1.8) | 98 (1.4) | 95 (1.8) | 100 (2.3) | 97 (1.9) | 98 (2.8) | 0.3 | 1.0 | 100 (4.0) | 100 (2.5) |
| Imidazothiazoles | |||||||||||
| LEV | 5, 50, 200 | 102 (1.5) | 100 (1.4) | 100 (0.7) | 102 (2.1) | 100 (1.1) | 101 (1.7) | 0.125 | 0.5 | 89 (5.7) | 96 (1.9) |
| Organophosphates | |||||||||||
| COUMA | 10, 50, 150 | 83 (9.3) | 93 (2,9) | 104 (3.8) | 88 (8.3) | 95 (5.8) | 106 (4.7) | 1.0 | 5.0 | 84 (6.0) | 99 (3.6) |
| COUMA-O | 5, 50, 200 | 95 (3.7) | 89 (3.9) | 98 (1.6) | 96 (6.6) | 92 (3.4) | 99 (3.2) | 0.25 | 1.0 | 93 (5.6) | 102 (2.5) |
| HALOX | 20, 80, 200 | 94 (11.7) | 94 (3.6) | 100 (2.0) | 90 (11.8) | 94 (5.3) | 102 (3.1) | 1.0 | 5.0 | 83 (0.8) | 99 (0.6) |
| Amino-acetonitrile derivatives | |||||||||||
| MONE | 10, 50, 150 | 103 (5.1) | 96 (4,3) | 93 (3.2) | 104 (12.1) | 97 (6.0) | 94 (5.2) | 0.5 | 5.0 | 90 (6.9) | 96 (3.0) |
| MONE-SO2 | 5, 50, 150 | 94 (8.1) | 91 (6.2) | 93 (3.2) | 98 (8.9) | 94 (4.6) | 98 (5.3) | 0.2 | 1.0 | 92 (2.6) | 102 (1.7) |
| Miscellaneous | |||||||||||
| CLOR | 80, 300, 800 | 95 (12.8) | 97 (5.8) | 95 (4.9) | 96 (14.9) | 95 (10.0) | 94 (8.4) | 10 | 40.0 | 101 (11.6) | 110 (3.6) |
| Spiking Vol. (µL) | Calibration Level | Concentration Ranges (ng L−1) for Analyte Group a: | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| A | B | C | D | E | F | G | H | I | J | ||
| 100 | 0.5 × L1 | 20 | 10 | 5 | 0.5 | 1 | 0.5 | 2 | 2 | 2.5 | 2.5 |
| 200 | L1 | 40 | 20 | 10 | 1 | 2 | 1 | 4 | 4 | 5 | 5 |
| 200 | L2 | 80 | 40 | 20 | 5 | 5 | 5 | 6 | 6 | 20 | 10 |
| 200 | L3 | 200 | 100 | 40 | 20 | 20 | 20 | 10 | 10 | 40 | 20 |
| 200 | L4 | 300 | 150 | 80 | 50 | 50 | 50 | 14 | 14 | 80 | 50 |
| 200 | L5 | 400 | 200 | 100 | 100 | 100 | 100 | 18 | 18 | 100 | 100 |
| 200 | L6 | 800 | 500 | 200 | 150 | 200 | 200 | 20 | 20 | 200 | 150 |
| 200 | L7 | 1000 | 1000 | 500 | 200 | 500 | 500 | 22 | 22 | 500 | 200 |
| 400 | L8 (2 × L7) | 2000 | 2000 | 1000 | 400 | 1000 | 1000 | 25 | 25 | 1000 | 400 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mooney, D.; Coxon, C.; Richards, K.G.; Gill, L.; Mellander, P.-E.; Danaher, M. Development and Optimisation of a Multiresidue Method for the Determination of 40 Anthelmintic Compounds in Environmental Water Samples by Solid Phase Extraction (SPE) with LC-MS/MS Detection. Molecules 2019, 24, 1978. https://doi.org/10.3390/molecules24101978
Mooney D, Coxon C, Richards KG, Gill L, Mellander P-E, Danaher M. Development and Optimisation of a Multiresidue Method for the Determination of 40 Anthelmintic Compounds in Environmental Water Samples by Solid Phase Extraction (SPE) with LC-MS/MS Detection. Molecules. 2019; 24(10):1978. https://doi.org/10.3390/molecules24101978
Chicago/Turabian StyleMooney, Damien, Catherine Coxon, Karl G Richards, Laurence Gill, Per-Erik Mellander, and Martin Danaher. 2019. "Development and Optimisation of a Multiresidue Method for the Determination of 40 Anthelmintic Compounds in Environmental Water Samples by Solid Phase Extraction (SPE) with LC-MS/MS Detection" Molecules 24, no. 10: 1978. https://doi.org/10.3390/molecules24101978
APA StyleMooney, D., Coxon, C., Richards, K. G., Gill, L., Mellander, P. -E., & Danaher, M. (2019). Development and Optimisation of a Multiresidue Method for the Determination of 40 Anthelmintic Compounds in Environmental Water Samples by Solid Phase Extraction (SPE) with LC-MS/MS Detection. Molecules, 24(10), 1978. https://doi.org/10.3390/molecules24101978