Abstract
Synthesis of 1,2,3-triazole-substituted coumarins and also 1,2,3-triazolyl or 1,2,3-triazolylalk-1-inyl-linked coumarin-2,3-furocoumarin hybrids was performed by employing the cross-coupling and copper catalyzed azide-alkyne cycloaddition reaction approaches. The synthesized compounds were evaluated for their in vitro antibacterial activity against Staphylococcus aureus, Bacillius subtilis, Actinomyces viscosus and Escherichia coli bacterial strains. Coumarin-benzoic acid hybrids 4с, 42с and 3-((4-acetylamino-3-(methoxycarbonyl)phenyl)ethynyl)coumarin (29) showed promising activity against S. aureus strains, and the 1,2,3-triazolyloct-1-inyl linked coumarin-2,3-furocoumarin hybrid 37c was endowed with high selectivity against B. subtilis and E. coli species. The in vitro antibacterial activity of 4с, 29, 37c and 42с can potentially be compared with that of a number of modern antibiotic drugs used in the clinic, suggesting promising prospects for further research. A detailed study of the molecular interactions with the targeted protein MurB was performed using docking simulations and the obtained results are quite promising.
1. Introduction
As stated in a WHO report, in the last few decades, the incidence of microbial infections has increased dramatically together with emergence of antimicrobial-resistant strains [1]. Increasing instances of antimicrobial drug resistance requires the design and synthesis of new small molecules with higher affinity and specificity for their potential targets to serve as antibiotics. Coumarins, naturally plant-derived compounds with a benzopyrone moiety, possess a wide variety of biological activities. Series of coumarin derivatives are being extensively studied due to their broad array of biological activities, low toxicity, and lower drug resistance properties [2].
Various naturally-isolated coumarins, as well as their chemically modified analogs, are active against numerous bacterial strains, including those which have developed multidrug resistance [2]. Among these compounds of interest are the coumarin-1,2,3-triazole hybrids (Figure 1) [3,4]. 1,2,3-Triazoles have been nuclei of choice in recent years because of their excellent favorable safety profile, latent ability to form hydrogen bonds, moderate dipole character, rigidity and stability under in vivo conditions and capability of interacting with biomolecular targets [5,6]. A set of coumarin hybrids A (Figure 1) having a 1,2,3-triazole moiety in the C-7 position on the coumarin core was assessed for their in vitro antimicrobial activities against Gram-positive and Gram-negative pathogens [7]. The obtained results showed that all hybrids of type A displayed considerable activity against the tested strains, and SAR studies revealed that the substituent and the length of alkyl spacers in the 1 and 4 position of the triazole ring have profound effects on the antimicrobial potency. Morpholinylmethyl- and piperazinylmethyl (N-R1,R2)-containing hybrids exhibited noticeable activity against various bacterial pathogens and were more potent than the hybrids incorporating a phthalimidomethyl moiety in the 4 position of the triazole ring. The 6,8-disubstituted coumarin-1,2,3-triazole conjugates B (Figure 1) exhibited significant in vitro antibacterial activity [8,9]. Investigation of the antimicrobial activity of this type of hybrid compounds led to the identification of several different structural frameworks. The obtained results suggested that both the length of the spacer (n) and also the nature of the substituent (H, OH, OMe) in the 6 and 8 positions modified the lipophilicity of the hybrids, and this in turn affected the antibacterial activity. In these molecules, the triazole rings are linked with the coumarins by a methylene oxygen. The antibacterial activity was correlated with the length of the spacer (n), and the contribution order of substituents was the following: H > OMe > OH.
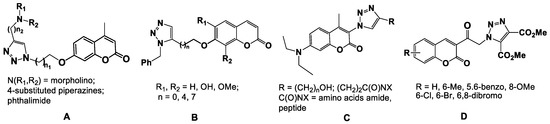
Figure 1.
Chemical structures of coumarin-1,2,3-triazole hybrids of types A–D.
The substituted coumarins with 1,2,3-triazole substituent in the 3 position (compounds C) exhibited activity which was dependent on the nature of the substituent on the triazole ring (alkylamide, alkylpeptide) [10].
All coumarin-1,2,3-triazole hybrids of the type D (Figure 1) displayed promising antibacterial and antifungal properties, which were comparable with the reference drug griseofulvin [11]. The SAR revealed that the substituent at C-5, C-6, and/or C-8 position of coumarin moiety have a great influence on the antimicrobial activity, and 6-Br was favorable for activity against the bacterial strain E. coli and the fungus A. niger, while a 6-Me benefitted the activity against B. subtilis; monohalo hybrids were more potent than their bis-halo counterparts (in the 6 and 8 positions) against all strains.
Thus, the design and synthesis of novel coumarinotriazole derivatives offers a prospective route for accessing new molecules with improved antibacterial activity profiles. Our research interests include the design of convenient ways to access polysubstituted natural furocoumarins [12,13,14,15,16] and coumarins [17,18] and the assessment of their potential as bioactive agents. Herein we report the synthesis of a range of coumarins containing a triazole substituent in the 3 or 6 position of the coumarin core. Our attention was concentrated on the synthesis of compounds bearing both a previously identified pharmacophoric moiety and a substituted triazole ring. Thus, we describe the synthesis of a range of mixed compounds having coumarin-2,3-dihydrofurocoumarin hybrid structures linked through an 1H-1,2,3-triazole ring or alkyne-methylene-triazolyl bridge at the C2-C3′ atoms. As starting compounds we used the natural linear furocoumarin peucedanin (1) and the coumarin peuruthenicin (officinalin, 7-hydroxy-6-(methoxycarbonyl)coumarin, 2, Scheme 1). The Pd-catalyzed coupling methods and Cu-catalyzed azide-alkyne cycloaddition (CuAAC reaction) were the main routes of synthesis. The antimicrobial activity of the triazolyl-substituted coumarins against a panel of clinically relevant bacterial strains were also investigated and discussed.
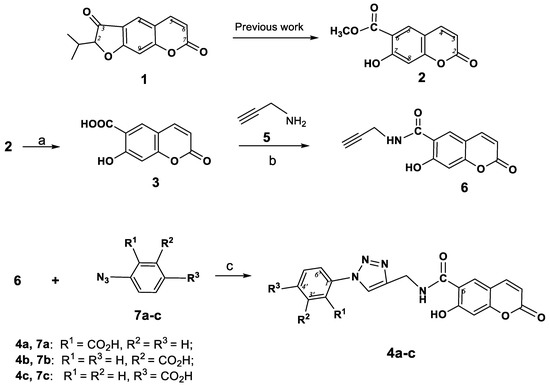
Scheme 1.
Synthesis 6-(1-(carboxyphenyl)-1H-1,2,3-triazol-4-ylcarbamoyl)umbelliferones 4a–c. Reagents and Conditions: (a) 20% eq. NaOH, dioxane, reflux, 20 min; (b) DCC, HOBt, Et3N, DMF, rt, 48 h; (c) CuSO4.5H2O (5 mol%), sodium ascorbate (15 mol%), H2O-CH2Cl2, 20→40 °C.
2. Results and Discussion
2.1. Chemical Synthesis
The umbelliferon carboxylic acid 3, the starting compound for the synthesis of triazolyl substituted coumarins 4a–c (Scheme 1), was conveniently obtained by treatment of peuruthenicin (2) with NaOH in dioxane [19]. The peptide coupling reaction of 3 with propargyl amine 5 using DCC as a coupling reagent gave the corresponding 6-(prop-2-ynylcarbamoyl)umbelliferone (6) in 67% yield. We have chosen the most simple and well-characterized variant, namely, carrying out the reaction in a CH2Cl2-water mixture, with catalysis of the Cu(I) ions generated in situ from CuSO4 and sodium ascorbate [12]. CuAAC-reaction of ethynyl coumarin 6 with three positional azidobenzoic acid isomers 7a–с led to the 6-[1-(carboxyphenyl)-1,2,3-triazol-4-yl]carbamoyl)- umbelliferones 4a–c in 50–62% yields.
The synthetic route followed for the synthesis of the desired novel coumarins substituted with a triazole ring in the C-3 position 8–10 is outlined in Scheme 2. Bromination of peuruthenicin (2) with dioxane dibromide (2.2 eq) in CH2Cl2 proceeds selectively and led to the formation of 3-bromopeuruthenicin (11) as the sole product in 88% isolated yield. 3-Аzidocoumarin 12 was conveniently obtained in 68% yield, by reacting the corresponding 3-bromopeuruthenicin (11) with sodium azide in DMF. The second alkyne building block was obtained from amino acids in two steps. At the first step, 9-aminopelargonic acid, d,l-2-aminobutyric acid or L-phenylalanine hydrochloride were transformed to subsequent methyl ethers of the amino acid hydrochloride in nearly quantitative yield requiring no further purification. Reaction of amino acids methyl esters hydrochloride with propargyl bromide in the presence of K2CO3 in dry DMF gave alkynes 13–15, which were purified through the column chromatography on silica gel using a CHCl3/EtOH mixture as an eluent.
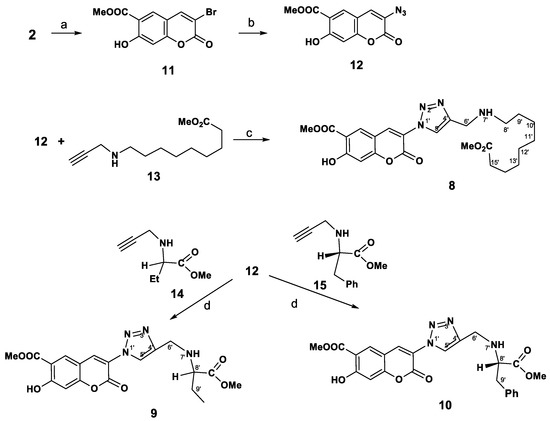
Scheme 2.
Synthesis of 3-(triazolyl)-substituted coumarins 8–10. Reagents and Conditions: (a): dioxane dibromide, CH2Cl2, rt, 16 h; (b): NaN3, DMF, 40 °C, 14 h; (c) CuSO4, sodium ascorbate, eq. CH2Cl2 (1:1), rt, 3 h, then 40 °С, 1 h; (d): CuI, Et3N, MeCN, rt, 10 h.
The interaction of the new azide 12 with methyl 9-(prop-2-ynylamino)nonanoate 13 under above catalytic conditions was carried out at 20 °C for 4 h (monitoring by TLC) and led to the target compound 8 in the isolated yield 67%. The reaction of azide 12 with terminal alkynes 14, 15, proceeds with the formation of substituted coumarins 9 or 10 in high yield by performing this reaction in the presence of CuI and Et3N (method d). By using of CuSO4 and sodium ascorbate as the catalysts in the reaction of 12 with 15, the isolated yield of coumarin 10 was decreased to 34%.
Thus we obtained 3-(1H-1,2,3-triazol-1-yl)peuruthenicin derivatives 8–10. It was also important for us to identify the influence of the position of coumarin as the substituent in the C-4 or C-1 of the triazole ring. To obtain 3-(1H-1,2,3-triazol-4-yl)peuruthenicins we were interested in the synthesis of 3-(ethynyl)peuruthenicin (16, Scheme 3).
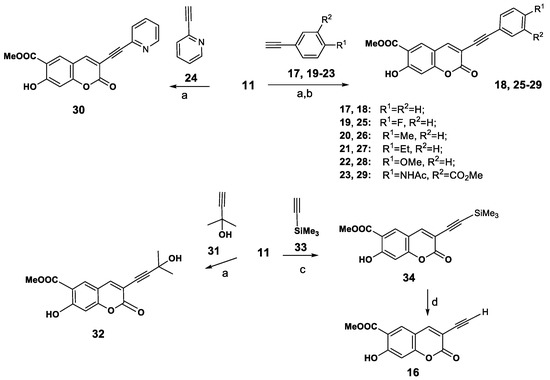
Scheme 3.
Synthesis 3-arylethynylcoumarins 17, 24–29, 3-(alkynyl)coumarins 31, 33, and 3-(ethynyl)peuruthenicin (34). Reagents and Conditions: (a): Pd(PPh3)2Cl2, CuI, Et3N, benzol, 110 °C, 12 h; (b) Pd(PPh3)2Cl2, CuI, Et3N, Bu4NBr, DMF; (c) Pd(PPh3)2Cl2, CuI, Et3N, toluene, MAOS, 100 °C, 2 h; (d) CsF, MeOH, TEBA, rt, 10 h.
For this purpose we studied the activity of 3-bromopeuruthenicin (11) in the Sonogashira cross-coupling reaction. Besides providing data about the reactivity of 3-bromopeuruthenicin (11) in the cross-coupling reaction with various terminal alkynes, this study served to make available a larger panel of peuruthenicin derivatives for testing their antimicrobial activity. Therefore, the reaction between 3-bromopeurutenicine (11) with phenyl acetylene (17) was optimized first, attempting to obtain the cross-coupling derivative 18 under different conditions. After considerable experimentation, we found that the cross-coupling proceeds in a benzene solution in the presence of a catalytic amounts of trans-dichlorobis(triphenylphosphine)palladium(II), copper(I) iodide, and Et3N as a base (conditions a, Scheme 3) and led to the corresponding 3-(phenylethynyl)coumarin (18) in 65%yield after column chromatography on silica gel. Using Bu4NBr as an additive, DMF as a solvent and Et3N as a base did not improve the yield of compound 18 (conditions b). On the other hand, using K2CO3 as a base in benzene or DMF at 80–100 °C also met with failure, and the reaction afforded yields of 45% or 42%, respectively, and a yield of 20% was recorded when the transformation was performed using (iPr)2NH as a base. With the set of optimum conditions in hands, we found that the reaction of coumarin 11 with aryl(hetaryl)acetylenes 19–24 proceeds with the formation of compounds 25–30 (52–68% yields. A perspective route for synthesis of terminal alkynes (for example, 3-ethynylpeuruthenicin (16) was the Sonogashira coupling of 11 with accessible 2-methylbut-3-yn-2-ol (31) and subsequent elimination of acetone from the 3-alkynylcoumarin 32. We found that the reaction of 11 with alkyne 31 (conditions a) led to the formation of 3-(3-hydroxy-3-methylbut-1-ynyl)coumarin (32, (66% yield). However, the transformation of the alcohol 32 into the terminal alkyne using the known conditions [20] was unsuccessful. The copper and palladium-catalyzed Sonogashira coupling of 3-bromopeuruthenicin 11 with trimethylsilylacetylene (33) (using conditions b) rqwuired a long reaction time and proceeds with the formation of traces of 3-(trimethylsilyl- ethynyl)peuruthenicin (34). Those results forced us improve this reaction. Currently, microwave-assisted organic synthesis (MAOS) is receiving increasing attention as a valuable alternative to the conventional heating to speed up chemical reactions, and microwave irradiation has been applied to Sonogashira coupling reactions [21,22]. It should be noted that MAOS has significant advantages that include simplicity in operation, increased reaction rates, and improved reaction yields. Performing the microwave-assisted reaction of 3-bromopeuruthenicin (11) and trimethylsilylacetylene (33) using Pd(PPh3)2Cl2, CuI as catalysts and Et3N as a base in toluene solution under microwave irradiation at 100 °C for 2 h give the cross-coupling product 34 in 64% isolated yield. Thus, the use of microwaves was found to significantly improve the reaction yield and shorten the reaction time. Desilylation of compound 34 using CsF in MeOH in the presence of benzyltriethylammonium chloride (TEBA) afforded 3-ethynylpeuruthenicin (16, 85% yield) which was purified by column chromatography on silica gel using CHCl3 as an eluent.
At the next step of our study, the acetylene building block 16 was reacted with 2-azidooreozelone (35) [14]. Carrying out the reaction in a СH2Сl2-water mixture, with catalysis of the Cu(I) ions generated in situ from CuSO4 and sodium ascorbate we obtained the coumarin-2,3-dihydrofurocoumarin hybrid linked through an 1H-1,2,3-triazole ring 36 isolated in 70% yield (Scheme 4).
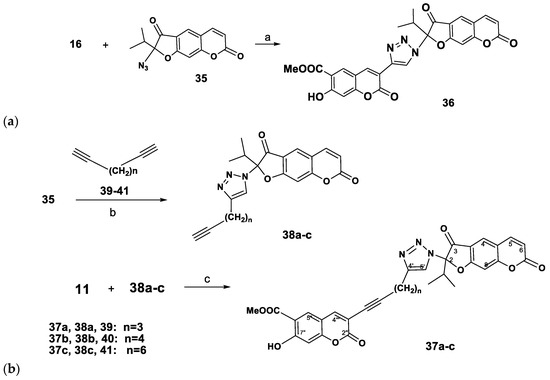
Scheme 4.
Synthesis of coumarin-2,3-dihydrofurocoumarin hybrids linked with 1H-1,2,3-triazole rings. Reagents and Conditions: (a): CuSO4.5H2O (5 mol%), sodium ascorbate (15 mol%), H2O-CH2Cl2, 20→40 °C; (b): CuSO4.5H2O, sodium ascorbate, H2O-CH2Cl2, 40 °C, 5 h; (c) Pd(PPh3)2Cl2, CuI, Et3N, benzene, 80 °C, 14 h.
We also performed the synthesis of coumarin-2,3-dihydrofurocoumarin hybrids 37a–c, containing a triazolylmethylene-1-inyl linker group (Scheme 4). The synthetic route involved the Sonogashira’s reaction of new 2-ethynylalkyltriazolyl substituted furocoumarins 38a–c with 3-bromopeuruthenicin (11, Scheme 4). Compounds 38a–c (yield 62–74%) were prepared by the CuAAC reaction of 2-azidooreoselone (35), with 1,6-heptadiyne (39), 1,7-octadiyne (40) or 1,9-decadiyne (41). Reaction of 3-bromopeuruthenicin (16) with alkynes 38а–с in the Sonogashira reaction conditions afforded the coumarin-2,3-dihydrofurocoumarin hybrids linked through 1H-1,2,3-triazolylalk-1-inyl groups 37a–c (52–57% yields).
2.2. Biological Screening
The new coumarins 4a–c, 11, 29, 30, coumarin-dihydrofurocoumarin hybrids 36, 37a–c, and the 7-triazolylsubstituted coumarins 42a–c (Figure 2), the synthesis of which was described in our previous paper [23] were screened for their in vitro antibacterial activity against Gram positive bacterial strain Staphylococcus aureus 209 ATCC 6538P. The antibacterial activity was studied by serial dilution in a liquid nutrient medium [24]. The minimum inhibition concentrations (MIC) of the test cultures were determined. The most promising substances were those with indicators of MIC = 100 µg/mL and less. Table 1 reveals that coumarino-1,2,3-triazole derivatives 4c and 42c and 3-arylethynylpeuruthenicin 29 showed excellent antibacterial activity, with MIC values ranging from 0.16–0.41 μg/mL against the tested microorganism and they prove to be better than that of the reference drugs ceftriaxone (0.97 µg/mL) and streptomycin (1.89 µg/mL).
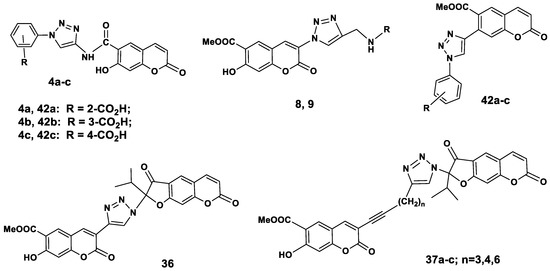
Figure 2.
Chemical structures of antibacterial coumarin-benzoic acid hybrids and coumarin-furocoumarin hybrids with a 1,2,3-triazolyl linker.

Table 1.
The in vitro antibacterial activity of compounds 4a–c, 11, 26, 29, 30, 37a–c, 42a–c and starting compounds 1–3 against bacterial strain S. aureus 209p ATCC6538P.
In the series of coumarin-benzoic acid hybrids 4a–c, 42a–c in view of the position of the carboxylic group in the benzoic acid moiety, we considered that compounds containing this substituent in the 4 position 4c, 42c were more active than compounds 4a,b and 42a,b, respectively, so the carboxylic acid group is an excellent microbial ligand which can bind more effectively at the active site of receptor. The difference in antibacterial activity profile of 3-bromocoumarin 11 and 3-(arylethynyl) coumarins 26, 29, 30 indicates the importance of the nature of the functional substituent on the triple bond in the C-3 position of coumarin. Thus, compound 29 containing an anthranilic acid methyl ester fragment on the 3-ethynylcoumarin nucleus showed greater activity than compounds with 4-tolyl- or pyridine substituents at the triple bond in 3-ethynylcoumarin. It should be noted that the biological effect of anthranilic acid derivatives is based on their ability to act as modulators of the nuclear peroxisome proliferator activated receptors (PRAR) and the farnesoid X (FXR) receptors, which fulfill crucial roles in metabolic balance [25,26]. In this direction the antibacterial activity of compounds 37a–c, having a methylene-triazolyl-furocoumarin substituent on the triple bond of 3-ethynylcoumarin was dependent on the length of the C-methylene linker. The activity of the most active hybrid compound 37c is due the presence of a hexamethylene-1-inyl linker group. Coumarin-2,3-dihydrofurocoumarin hybrid 37c was found to exhibit good potency at 51.25 mg/mL of MIC, while the parent compounds 1 and 2 exhibited a lack of activity (Table 1).
The results of study of promising substances 4с, 29, 37c and 42с on other strains of Staphylococcus aureus and Actinomyces viscosus U-18 are presented in Table 2. Further study on the S. aureus strain confirmed the high activity of compounds 4с, 29 and 42c. Compound 4c (carboxamidotriazolyl- benzoic acid substitution at the C-6 position of the coumarin core) showed good activity against A. viscosus compared with compound 42c which showed moderate activity against A. viscosus. Characteristically, that compound 42c with a triazolylbenzoic acid substituent in the C-7 position possessed the highest activity against S. aureus “Viotko” bacterial strains. Of interest was also the high antibacterial activity of 3-ethynylcoumarin with methylanthranilate substituent 29 on the all tested S. aureus strains. This compound will be further used as the scaffold for structural optimization to develop more potent and selective antibacterial agents.
Table 2.
The in vitro antibacterial activity results of compounds 4c, 29, 37c and 42c against S. aureus and A. viscosus U-18 strains.
Next set of experiments was dedicated to the analysis of the antibacterial potential of coumarin-2,3-dihydrofurocoumarin hybrids 36, 37a–c (Table 3). The in vitro antibacterial activity of those compounds was additionally tested against Bacillius subtilis and Escherichia coli (JM 109) bacterial strains. The obtained results were compared with the known tumorogenic compound 4-nitroquinolin-1-oxide (NQO) and presented as an average concentration of inhibitory 50% bacterial proliferation (incubation time 20 h). It can be observed that from the series of coumarin-2,3-dihydrofurocoumarin hybrids only dimeric compound 37c with the 1,2,3-triazolyloct-1-inyl linker group displayed promising activity against B. subtilis and also E. coli (JM 109) bacterial strains. On the contrary to this, variously 1,2,3-triazolyl or 1,2,3-triazolylhex-1-inyl or 1,2,3-triazolylpent-1-inyl linked hybrids 36, 37a,b were deprived of anti-bacterial activities (MICs were >1000 mg/mL).
Table 3.
Antimicrobial activities of coumarin-furocoumarin hybrids 36, 37a–c and parent compounds 1, 2 against B. subtilis and E. coli strains (* P < 0.05 relative to the control).
Molecular Docking
Molecular docking protocol is an essential tool to mimic the natural course of interaction of the ligand with active sites of receptors through lowest binding energies in which two molecules fit together in 3D space. Here, we discuss the mechanism of interaction between coumarinotriazole derivatives 3, 4 and 5 with the MurB protein (PDB ID: 1HSK). MurB is an attractive target protein in bacterial infection diseases and it is an essential enzyme which takes part in amino sugar metabolism which reduces the enolpyruvyluridine diphosphate N-acetyl glucosamine (EP-UNAG) as an intermediate in the assembly of the UNAM-pentapeptide (m-A2pm) protein to uridine diphosphate N-acetylMuamic acid (UNAM) of cell wall precursor [27]. For the calculations, the XRD model of S. aureus N-acetylenolpyruvylglucosamine reductase (MurB) with PDB ID 1HSK was chosen (resolution 2.3 Å) [28]. To model a possible mechanism of MurB inhibition, molecular docking of new coumarins was performed at the binding site of flavin adenine dinucleotide (FAD) in the Glide application [29]. We have screened the coumarinotriazoles 4a–c, 8, 9, and 42a–c and also coumarin-furocoumarin hybrids 36, 37b, and 37c. The molecules 4a–c, 37c and 42a,c strongly approach the MurB protein receptor as shown by their minimum binding energies −8.416–−8.983 Kcal/mol (Table 4). The docking results were found to be in good agreement with in vitro antibacterial experimental MIC values (Table 1, Table 2 and Table 3). The FAD binding site is saturated with polar amino acids. This facilitates formation of a large number of hydrogen bonds due to the large number of polar groups in the FAD molecule. Additional stabilization in the binding site provided stacking interactions of aromatic cycles of adenine. Inspection of the binding mode demonstrated, that compounds 4c, 37c and 42c successfully combine in their structure a large number of polar groups and π-systems (Figure 3). The presence of the carboxy function in compounds 4c, 42c allowed the formation of hydrogen bond with amino acid ARG225 residue (Figure 3A–C). As shown in Figure 3C, the inhibitor 37c formed addition hydrogen bonding interactions with the active site residues: the furan ring C=O with SER 238 (2.92 Å) and the coumarin ring C=O with TYR 155. All those interactions contribute to a spatial orientation close to FAD and the formation of hydrogen bonds with the same amino acid residues as FAD (Figure 3D). We found that the MurB protein receptor amino acids ASN80, SER82, SER238, GLY81 are the most active sites responsible for interactions with the ligand. Important interaction centers at the binding site are arginines 225 and 310, protein chain section 79–83, glycines 145, 146 and 153, isoleucine 140 and proline 141, valine 199. New coumarins interact with almost all these centers, except for 140–141, and also form bonds characteristic only for these molecules. The formation of interactions with aromatic amino acid residues increases the stability of the conformations of new coumarins at the binding site.
Table 4.
Values of the minimum binding energy for new compounds to MurB protein.
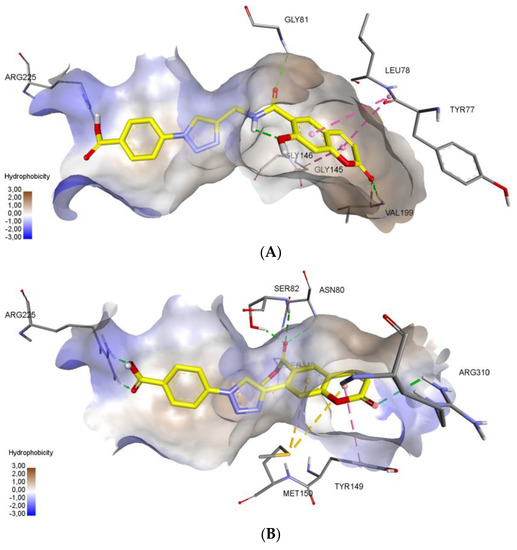
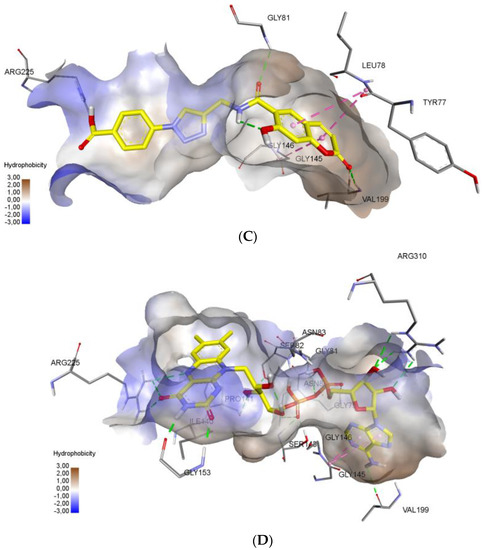
Figure 3.
Noncovalent interactions of compounds (3A—4c, 3B—42c, 3C—37c, 3D—FAD) are shown by dotted lines: green—hydrogen bonds, purple—stacking interactions, orange—electrostatic interactions. Hydrophobic interactions and nonpolar hydrogens are omitted. (A) 3D interactions of ligand 4c at the FAD binding site of MurB Protein. (B) 3D interactions of ligand 42c at the FAD binding site of MurB Protein. (C) 3D interactions of ligand 37c at the FAD binding site of MurB Protein. (D) 3D interactions of FAD molecule at the binding site of MurB Protein.
3. Conclusions
In summary, we have synthesized new series of coumarinotriazole compounds using [2 + 3]-cycloaddition reactions or Sonogashira cross-coupling reactions as the main approaches. The synthesized coumarino-triazole type derivatives were screened for their in vitro antimicrobial activity. Compounds 4c and 42c, having the 4-(carboxyphenyl)triazolyl substituent in the 6 or 7 position of the coumarin ring showed excellent antibacterial activity against S. aureus strains, with MIC values of 0.16–3.75 μg/mL and 0.21–6.28 μg/mL respectively. Most of the compounds of series 11, 29 and 30 with bromine, and aryl(hetaryl)substituents in the 3 position revealed significant MIC values (between 0.41 to 2.0 μg/mL for compound 29), indicating that 3-substituted coumarin analogues show promising antibacterial activity and are key compounds for further development as antibacterial agents. Coumarin-2,3-dihydrofurocoumarin hybrid compound 37c was found to be selective against Bacillius subtilis and E. coli, with MIC values of 0.02–0.15 μg/mL. A molecular docking study was performed for the most active compounds against the MurB protein. Molecular docking results were well corroborated with the in vitro antibacterial activity findings. Finally, the successful synthesis and antimicrobial evaluation along with docking study of new coumarino-triazole scaffolds obtained on the base of accessible plant coumarins peucedanin and peuruthenicin will provide further advantages to design and develop triazole derivatives with selective antibacterial activity.
4. Experimental Section
4.1. General Information
NMR spectra were acquired on Bruker AV-400 (1H: 400.13 MHz, 13C: 100.78 MHz) or Bruker AV-600 (1H: 600.30 MHz, 13C: 150.95 MHz) instruments (Bruker BioSpin GmbH, Rheinstetten, Germany), using tetramethylsilane (TMS) as an internal standart. In the description of the 1H- and 13C-NMR spectra, the furocoumarin and coumarin skeleton atoms numeration system given in structures 1 and 2 was used. Chemical shifts are reported in parts per million (ppm). The IR spectra were recorded by means of the KBr pellet technique on a Bruker Vector-22 spectrometer. The UV spectrum were obtained on an HP 8453 UV Vis spectrometer (Hewlett-Packard, Waldbronn, Germany). HRMS spectra were recorded on a DFS mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA), evaporator temperature 180–220 °C, EI ionization at 70 eV). The specific rotation values [α]D were determined on a PolAAr 3005 polarimeter (Rudolph Research Analytical, Hackettstown, NJ, USA), and expressed in (deg·mL)/(g·dm), while concentration was expressed in g per 100 mL of solution. Melting points were determined using Stuart SMP30 melting point apparatus (Bibby Scientific, Staffordshire, UK). The microwave irradiation reaction was performed in a Microwave 50 reactor (Anton Paar, Graz, Austria). Elemental analysis was carried out on an 1106 Elemental analysis instrument (Carlo-Erba, Milan, Italy). The molecular weights of compounds 37a–c, 38c were determined using a Knauer vapor pressure osmometer (Knauer, Berlin, Germany). Spectral and analytical investigations were carried out at Collective Chemical Service center of Siberian Branch of the Russian Academy of Sciences.
Reaction products were isolated by column chromatography on silica gel 60 (0.063–0.200 mm, Merck KGaA, Darmstadt, Germany) eluting with chloroform and chloroform-ethanol (100:1; to 25:l). The reaction progress and the purity of the obtained compounds were monitored by TLC on Silufol UV-254 plates (CHCl3-EtOH, 9:1; detection under UV light or by treatment with iodine vapor).
The chemicals used: dioxane dibromide, trifluoromethanesulfonic anhydride, NaN3, CuSO4, sodium ascorbate, CuSO4, CuI, N,N-dicyclohexylcarbodiimide (DCC), propargyl amine hydrochloride (5), propargyl bromide (80% in toluene solution), aryl acetylenes 17, 19–2, 24, 2-methylbut-3-yn-2-ol (31), trimethylsilylacetylene (33), 1,6-heptadiyne (39), 1,7-octadiyne (40) and 1,9-decadiyne (41) were purchased from Aldrich (St. Louis, MO, USA) or Alfa Aesar (GmbH, Karlsruhe, Germany). Dichlorobis(triphenylphosphine)palladium(II) was obtained as described in [30]. Peucedanin (1) was isolated from Peucedanum morisonii according to [31]. Peuruthenicin (2) [32], umbelliferone carboxylic acid 3 [19], azidobenzoic acids 7а–с [33], methyl ether of 9-aminopelargonic acid hydrochloride [34], D,L-2-aminobutyric acid and L-phenylalanine hydrochlorides [35], methyl 2-(prop-2-ynylamino)butanoate (14), and (S)-methyl 3-phenyl-2-(prop-2-ynylamino)propanoate (15) [36], methyl ethynylantranilate (23) [37] and 2-azidooreoselone (35) [14] were synthesized according to published procedures. Solvents (CH2Cl2, CHCl3, MeCN, DMF, THF, benzene, dioxane) and Et3N were purified by standard methods and distilled under a stream of argon just before use. Copies of NMR spectra (1H & 13C) are given in Supplementary Materials.
4.2. Syntheses and Spectral Data
7-Hydroxy-2-oxo-N-(prop-2-ynyl)-2H-chromene-6-carboxamide (6) To a stirred solution of umbelliferone-6-carboxylic acid (3, 500 mg, 2 mmol) and hydroxybenzotriazole (135 mg, 1 mmol) in DMF (5 mL) was added propargylamine hydrochloride (5, 120 mg, 2.1 mmol) and Et3N (0.27 mL, 2 mmol) and the reaction mixture was left for 5 min. Then diisopropylcarbodiimide (300 mg, 2.4 mmol) was added and the mixture was stirred for 2 days. The reaction mixture was poured into a Petri dish to evaporate in air, the residue was dissolved in CH2Cl2 (10 mL) and acetone was added (5 mL). The precipitate was filtered, the solution was evaporated and residue was dried. The product was isolated by fractional precipitation from acetone. Compound 6 was obtained in 67% yield (320 mg). Colorless powder. М.р. 188 °С (dec.). IR (KBr, ν, cm−1): 3388, 3286, 3243, 3059, 2921, 2852, 2100, 1737, 1708, 1649, 1620, 1598, 1570, 1500, 1392, 1344, 1310, 1234, 1213, 1144, 911, 854, 837, 735, 688, 639, 628. UV (EtOH) λmax, (lgε): 222(3.22), 240 (3.35), 329 (2.89), 372 (2.88) nm. 1H-NMR (CDCl3 + CD3OD, 400 MHz, δH): 3.16 (s, 1Н, ≡СН), 4.20 (s, 2Н, СН2), 6.26 (d, J = 9.6 Hz, 1Н, Н-4), 6.83 (s, 1Н, Н-8), 7.76 (d, J = 9.6 Hz, 1Н, Н-3), 8.10 (s, 1Н, Н-5). 13C-NMR (CDCl3+CD3OD, 100 MHz, δC): 45.8, 71.0, 78.8, 103.9, 111.4, 112.6, 113.8, 129.3, 144.0, 157.1, 161.2, 162.4, 167.1 (С=O). Anal. calcd for C13H9NO4: C, 64.20; H, 3.73; N, 5.76; found: С 64.33; Н 3.77; N 5.65.
4.2.1. General Method for the Synthesis of Compounds 4a–c
A solution of the appropriate azidobenzoic acid 7a–с (35 mg, 0.14 mmol) in CH2Cl2 (10 mL) was mixed with a solution of sodium ascorbate (42 mg, 15 mol%, 0.021 mmol) and СuSO4·5H2O (1.2 mg, 5 mol%, 0.007 mmol) in Н2О (5 mL) and 6-(N-propynyl)carboxamidocoumarin (6, 20 mg, 0.14 mmol) was added while stirring. The reaction mixture was stirred for 3 h at room temperature and additional for 1 h at 40 °С. After the completion of the reaction, the mixture was diluted with Н2О (10 mL), the product was extracted with CH2Cl2 (4 × 10 mL), the combined extracts were dried over anhydrous MgSO4, and the solvent was removed at reduced pressure. The product was purified by column chromatography (eluent CHCl3).
2-(4-((7-Hydroxy-2-oxo-2H-chromene-6-carboxamido)methyl)-1H-1,2,3-triazol-1-yl)benzoic acid (4a). Colorless powder, m.p. 201–202 °C (ethanol). Yield 50%. IR (KBr, ν, cm−1): 3367, 3257, 3066, 2922, 2852, 2666, 1740, 1691, 1596, 1575, 1485, 1446, 1392, 1304, 1265, 1148, 1078, 1020, 908, 829, 796, 752, 687, 640. UV (EtOH) λmax, (lgε): 246 (3.45), 301 (2.76), 331 (3.51), 357 (3.23) nm. 1H-NMR (CDCl3+CD3OD, 400 MHz, δH): 4.93 (s, 2Н, СН2), 6.22 (d, J = 9.8 Hz, 1Н, Н-3), 6.82 (s, 1Н, Н-8), 7.13 (dd, J = 7.6, 7.2 Hz, 1Н, Н-4′′), 7.18 (1Н, d, J = 8.0 Hz, 1Н, Н-6′′), 7.48 (dd, J = 8.0, 7.2 Hz, 1Н, Н-5′′), 7.63 (d, J = 9.8 Hz, 1Н, Н-4), 7.78 (s, 1Н, Н-5), 7.88 (d, J = 7.6 Hz, 1Н, Н-3′′), 7.98 (s, 1Н, Н-5′). 13C-NMR (CDCl3+CD3OD, 100 MHz, δC): 44.5, 104.3, 111.8, 113.4, 119.8, 121.3, 123.0, 124.1, 125.9, 129.5, 130.7, 132.1, 135.9, 142.3, 143.8, 158.5, 160.5, 163.8, 167.6, 169.2. Anal. calcd for C20H14N4O6: C, 59.12; H, 3.47; N, 13.79; found: C, 58.88; H, 3.77; N, 14.14.
3-(4-((7-Hydroxy-2-oxo-2H-chromene-6-carboxamido)methyl)-1H-1,2,3-triazol-1-yl)benzoic acid (4b). Yellowish powder, m.p. 186–187 °C (hexane). Yield 64%. IR (KBr, ν, cm−1): 3427, 3095, 3062, 2923, 2852, 2665, 1738, 1687, 1631, 1600, 1581, 1456, 1305, 1236, 1153, 1113, 1097, 1056, 1008, 931, 912, 893, 800, 795, 754, 698, 673. UV (EtOH) λmax, (lgε): 225 (3.66), 246 (3.53), 301 (3.23), 336 (3.22), 347 (3.18), 357 (3.23) nm. 1H-NMR (CDCl3, 400 MHz, δH): 4.97 (s, 2Н, СН2), 6.30 (d, J = 9.8 Hz, 1Н, Н-3), 6.87 (s, 1Н, Н-8), 7.45 (d, J = 8.2 Hz, 1Н, Н-6′′), 7.47 (dd, J = 8.2, 7.6 Hz, 1Н, Н5′′), 7.62 (d, J = 9.8 Hz, 1Н, Н-4), 7.77 (s, 1Н, Н-5), 7.88 (d, J = 7.6 Hz, 1Н, Н-4′′), 8.01 (s, 2Н, Н-5′, 2′′), 11.19 (br.s, 3H, OH, NH). 13C-NMR (CDCl3, 100 MHz, δC): 44.2, 104.9, 111.9, 114.2, 120.4, 121.7, 123.4, 124.3, 126.9, 127.1, 130.0, 130.7, 135.7, 140.8, 143.1, 158.9, 160.2, 161.0 (С-7), 164.2, 170.8. Anal. calcd for C20H14N4O6: C, 59.12; H, 3.47; N, 13.79; found: C, 59.55; H, 3.49; N, 13.43.
4-(4-((7-Hydroxy-2-oxo-2H-chromene-6-carboxamido)methyl)-1H-1,2,3-triazol-1-yl)benzoic acid (4c). Grayish powder, m.p. 203–204 °C (hexane). Yield 60%. IR (KBr, ν, cm−1): 3433, 3062, 2958, 2924, 2852, 2673, 1734, 1676, 1630, 1600, 1579, 1448, 1427, 1392, 1288, 1236, 1215, 1143, 1113, 1076, 946, 904, 858, 827, 796, 768, 732. UV (EtOH) λmax, (lgε): 225 (3.66), 246 (3.78), 270 (3.65), 293 (3.53), 324 (3.36), 351 (3.21) nm. 1H-NMR (CDCl3, 400 MHz, δH): 4.95 (s, 2Н, СН2), 6.28 (d, J = 9.8 Hz, 1Н, Н-3), 6.87 (s, 1Н, Н-8), 7.10 (d, J = 8.4 Hz, 2Н, Н-2′′,6′′), 7.61 (d, J = 9.8 Hz, 1Н, Н-4), 7.98 (s, 1Н, Н-5), 8.02 (s, 1Н, Н-5′), 8.03 (br.s, 1Н, NН), 8.10 (d, J = 8.4 Hz, 2Н, Н-3′′,5′′), 11.19 (br.s, 2Н, ОН). 13C-NMR (CDCl3, 100 MHz, δC): 42.5, 104.9, 111.9, 114.1, 118.9 (2C), 121.8, 125.3, 125.7, 127.6, 130.7, 132.1 (2С), 143.0, 145.7, 158.9, 160.1, 164.2, 169.5, 170.7. Anal. calcd for C20H14N4O6: C, 59.12; H, 3.47; N, 13.79; found: C, 59.16; H, 3.14; N, 13.42.
3-Bromopeuruthenicin (11). A solution of 1.0 g (4.5 mmol) of peuruthenicin (2) and 2.48 g (9 mmol) of dioxane dibromide in CH2Cl2 (10 mL) was stirred at rt for 16 h. Then the reaction mixture was filtered, and the filtrate was evaporated. 3-Bromopeuruthenicin (11, yield 86%, 1.14 g) was finally purified by crystallization from chloroform. White solid; m.p. 187–188 °C (ether). IR (KBr, ν, cm−1): 3480, 3350, 3116, 3050, 2956, 2853, 1720, 1675, 1621, 1616, 1598, 1440, 1311, 1293, 1231, 1163, 1124, 1089, 1030, 989, 964, 918, 845, 783, 750, 729, 719. UV (EtOH) λmax, (lgε): 244 (4.19), 314 (4.02), 334 (4.10), 400 (3.39) nm. 1H-NMR (600 MHz, CDCl3, δH): 3.99 (s, 3H, OCH3), 6.85 (s, 1H, H-8), 7.97 (s, 2H, H-4,5), 11.21 (s, 1H, OH). 13C-NMR (150 MHz, CDCl3, δC): 52.9, 104.8, 108.8, 110.5, 112.5, 129.9, 143.8, 156.4, 157.9, 164.3, 169.2. HRMS (ESI), m/z (Irel, %): 298(66), 269(15.9), 240(24), 220(18), 188(31), 160(16), 131(17), 103(19), 79(12); calcd for C11H7BrO5 [M]: 297.9471; found: 297.9468; Anal. Calcd. for C11H7BrO5: С, 44.18; Н, 2.36; Br, 26.72. Found: С, 44.08; Н, 2.16; Br, 26.70.
3-Azidopeuruthenicin (12). To a stirred solution of 25 mg (0.84 mmol) of 3-bromopeuruthenicin (11) in DMF at room temperature was added NaN3 (82 mg, 1.26 mmol). The reaction mixture was heated at 40 °C for 14h. After consumption of the bromocoumarin, the mixture was cooled to room temperature, quenched with eq. NaCl (10 mL), and the products and were extracted with CH2Cl2 (5 × 5 mL). The combined organic phases were dried over anhydrous MgSO4, after filtration, the solvent was removed under reduced pressure and was further dried in a freeze drier at oil pump vacuum. (P = 1 mm). The product (0.15 g, 68%) was obtained as a yellow solid, m.p. 161–162 °C. IR (KBr, ν, cm−1): 3455, 3390, 3357, 3299, 3004, 2971, 2923, 2858, 2121, 1727, 1621, 1467, 1434, 1375, 1309, 1290, 1263, 1193, 975, 869, 728. UV (EtOH) λmax, (lgε): 223(4.51), 243(4.4), 329(4.2), 373(3.97). 1H-NMR (CDCl3, 400 MHz, δH): 3.99 (s, 3H, OCH3), 6.83 (s, 1H, H-8), 7.58 (s, 1Н, H-4), 8.00 (s, 1H, H-5), 11.19 (s, 1H, OH). 13C- NMR (CDCl3, 100 MHz, δC): 52.7, 104.8, 109.9, 111.9, 114.0, 120.6, 142.9, 158.8, 159.9, 164.2, 169.4. Anal. Calcd for C11H7N3O5: С, 50.58; Н, 2.70; N, 16.09; found С, 51.04; Н, 3.12; N, 15.74.
4.2.2. General Method for the N-Propargylation of Amino Acid Methyl Esters
Propargyl bromide (10.5 mmol, 80% in toluene solution) was added to a solution of the appropriate amino acid ether hydrochlorides (10 mmol) and potassium carbonate (21 mmol) in dry DMF at room temperature under argon. The mixture was stirred for 1–3 days (TLC control), the solvent was evaporated under reduced pressure and the residue partitioned between water (30 mL) and CH2Cl2 (30 mL). The organic phase was separated and the aqueous phase was extracted with CH2Cl2 (3 × 10 mL). The organic phases were combined, dried (MgSO4), filtered and the solvent evaporated under reduced pressure. The residue was purified by column chromatography to give secondary amines 13, 14, 15.
Methyl 9-(prop-2-ynylamino)nonanoate (13). Yellowish oil. 1H-NMR (CDCl3, 400 MHz, δH): 1.22 (m, 4H, CH2-11′,12′), 1.39 (2H, CH2-13′), 1.52 (2H, CH2-10′), 2.12 (4H, CH2-9′,14′), 2.21 (m, 2H, CH2-15′), 2.58 (2H, CH2-8′), 3.32 (s, 2H, CH2-6′), 3.57 (s, 3H, OCH3).13C-NMR (CDCl3, 100 MHz, δC): 25.2, 27.4, 29.3, 29.5, 30.0, 34.32, 38.40, 48.90 (CH2-12′,9′,5′), 51.62 (OCH3), 71.38, 73.02 (C≡CH). Anal. calcd for C13H23NO2: С, 69.29; Н, 10.29; N, 6.22; found С, 68.74; Н, 10.10; N, 5.94.
Methyl 7-hydroxy-3-(4-((9-methoxy-9-oxononylamino)methyl)-1H-1,2,3-triazol-1-yl)-2-oxo-2H-chromene-6-carboxylate (8). A solution of 3-azidopeuruthenicin (12, 50 mg, 0.19 mmol) in methylene chloride (10 mL) and a solution of sodium ascorbate (15 mol%, 0.255 mmol) and СuSO4 × 5H2O (5 mol%, 0.085 mmol) in water (10 mL) were mixed and the terminal alkyne 13 (63 mg, 0.28 mmol) was added with stirring. The reaction mixture was stirred for 3 h at room temperature and then for 1 h at 40 °С. The cooled mixture was diluted with water (10 mL) and the product was extracted with methylene chloride (4 × 10 mL). The combined extracts were dried over MgSO4, and the solvent was removed in vacuo. The residue was purified by column chromatography to give 73 mg (67%) of compound 8 as a yellow oil. IR (KBr, ν, cm−1): 3465, 3396, 3118, 3047, 2954, 2923, 2852, 1751, 1735, 1673, 1623, 1602, 1573, 1469, 1438, 1367, 1288, 1230, 1162, 1124, 1089, 1031, 962, 943, 865, 792. UV (EtOH) λmax, (lgε): 242 (4.49), 299 (4.17), 313 (4.26), 334 (4.34), 390 (3.39) nm. 1H-NMR (CDCl3, 400 MHz, δH): 1.25 (m, 4H, CH2-11′,12′), 1.66 (2H, CH2-13′), 2.18 (2H, CH2-10′), 2.40 (4H, CH2-9′,14′), 2.47 (m, 2H, CH2-15′), 2.72 (2H, CH2-8′), 3.65 (s, 3H, OCH3), 3.99 (s, 3H, OCH3), 4.02 (s, 2H, CH2-6′), 6.87 (s, 1H, H-8), 7.97 (s, 1Н, H-4), 7.98 (s, 1H, H-5), 8.01 (s, 1H, H-5′), 11.25 (br.s, 2H, OH, NH). 13C-NMR (CDCl3, 100 MHz, δC): 29.7, 30.8, 31.9, 35.5, 39.6, 40.1, 40.5, 45.1, 52.9, 61.9, 104.7, 108.7, 110.5, 112.5, 121.4, 124.9, 142.2, 143.8, 156.4, 157.8, 164.3, 169.2, 174.4. Anal. calcd. for C24H30N4O7: C, 59.25; H, 6.22; N, 11.52; found: C, 59.02; H, 6.12; N, 11.27.
4.2.3. Preparation of 3-(Triazolyl)coumarins 9, 10
A stirred solution of 3-azidopeuruthenicin (12, 90 mg, 0.35 mmol) in MeCN (15 mL) was successively treated with alkyne 14 or 15 (1.2 equiv.), CuI (0.1 equiv.) and Et3N (1.1 equiv.) and the reaction mixture was stirred at room temperature until complete consumption of the starting material. Then, water (5 mL) was added and the products were extracted with CH2Cl2 (3 × 10 mL). The combined organic phases were dried (MgSO4), filtered and concentrated in vacuo. The residue was purified by column chromatography, eluting with chloroform, chloroform:ethanol 100:1.
Methyl 7-hydroxy-3-(4-((1-methoxy-1-oxobutan-2-ylamino)methyl)-1H-1,2,3-triazol-1-yl)-2-oxo-2H-chromene-6-carboxylate (9). Yield 56%. Yellow oil. IR (KBr, ν, cm−1): 3463, 3398, 3307, 3050, 2954, 2923, 2852, 1733, 1677, 1625, 1575, 1465, 1440, 1367, 1309, 1292, 1232, 1220, 1164, 1124, 1089, 1012, 968, 865, 794, 948. UV (EtOH) λmax, (lgε): 207 (4.69), 244 (4.29), 300 (3.95), 315 (4.02), 336 (4.1), 387 (3.41) nm. 1H-NMR (CDCl3+CD3OD, 400 MHz, δH): 0.81 (t, 3H, J = 7.0 Нz, CH3-10′), 31.55 (m, 2H, CH2-9′), 3.27 (m, 1H, H-8′), 3.62 (s, 3H, OCH3), 3.98 (s, 3H, OCH3), 3.90 (m, 2H, H-6′), 6.76 (s, 1H, H-8), 7.90 (s, 1Н, H-4), 7.91 (s, 1H, H-5), 8.09 (s, 1H, H-5′). 13C-NMR (CDCl3, 100 MHz, δC): 9.5, 25.8, 36.3, 51.6, 51.8, 60.7, 104.4, 108.2, 110.3, 112.3, 121.3, 124.8, 141.0, 144.1, 156.5, 157.5, 164.0, 168.9, 174.8. Anal. calcd. for C19H20N4O7: C, 54.81; H, 4.84; N, 13.46; found, %: C, 55.13; H, 5.02; N, 13.59.
(S)-Methyl 7-hydroxy-3-(4-((1-methoxy-1-oxo-3-phenylpropan-2-ylamino)methyl)-1H-1,2,3-triazol-1-yl)-2-oxo-2H-chromene-6-carboxylate (10). Yield 62% (by method for snthesis of compound 8–34%). Yellow oil. [α]D +12.04 (с 1.00, CHCl3). IR (ν, cm−1): 3459, 3395, 3290, 3049, 2954, 2925, 2854, 1737, 1673, 1623, 1602, 1573, 1537, 1490, 1438, 1367, 1288, 1232, 1162, 1124, 1087, 962, 943, 865, 792. UV (EtOH) λmax, (lgε): 244 (4.22), 312 (3.94), 334 (4.02), 399 (3.26) nm. 1H-NMR (CDCl3, 400 MHz, δH): 2.90-3.03 (m, 2H, H-9′), 3.36 (dd, J = 16.3, 1.8 Hz, 2H, H-6′), 3.65 s (3H, OCH3), 3.73 (dd, J = 6.4, 7.0 Hz, 1H, H-8′), 3.98 (s, 3H, OCH3), 6.84 s (1H, H8), 7.16-7.18 (m, 2H, o-Ph), 7.19-7.22 (m, 1H, p-Ph), 7.26-7.32 (m, 2H, m-Ph), 7.93 s (1Н, H-4), 7.95 (s, 1H, H-5), 8.08 (s, 1H, H-5′), 11.13 (br s, 2H, OH, NH). 13C-NMR (CDCl3, 100 MHz, δC): 36.7, 39.3, 51.8, 52.8, 61.0, 104.9, 108.4, 110.3, 112.4, 120.5 (C-4), 125.1, 126.9, 129.1 (2C), 129.5 (2C), 132.5, 141.3, 143.8, 156.1, 157.7, 164.2, 169.2, 174.2. Anal. calcd. for C24H22N4O7: C, 60.25; H, 4.63; N, 11.71; found: C, 60.16; H, 4.96; N, 11.55.
4.2.4. General Procedure for the Sonogashira Cross-coupling Reactions of 3-Bromopeuruthenicin 11 with Aryl Alkynes 17, 19–24 or 2-methylbut-3-yn-2-ol 31
To a solution of 3-bromopeurutenicin (11, 100 mg, 0.34 mmol) and terminal alkynes 17, 19–24 or 31 (0.54 mmol) in benzene (5 mL) was added CuI (6 mg, 10 mol%), Pd(PPh3)2Cl2 (12 mg, 5 mol%), and Et3N (0.076 mL, 0.44 mmol; 1.3 (eq. to 11) under argon. The reaction mixture was stirred at 80 °C for 12–14 h (TLC). The mixture was cooled and 5 mL of water was added. The separated water layer was extracted with СН2Cl2 (5 × 4 mL). The combined organic extracts was washed with water, dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was subjected to column chromatography on silica gel. Eluting with chloroform and crystallization from ether afforded the corresponding 3-(R)-alkynylcoumarins 18, 25–30, 32.
Methyl 7-hydroxy-2-oxo-3-(phenylethynyl)-2H-chromene-6-carboxylate (18). Yield 65% (70 mg). Yellow oil. IR (ν, cm−1): 3430, 3350, 3052, 2954, 2925, 2852, 2112 (С≡С), 1736, 1682, 1616, 1571, 1442, 1367, 1290, 1224, 1160, 1128, 1091, 966, 873, 792, 750. UV (EtOH) λmax, (lgε): 237 (4.14), 272 (3.86), 280 (3.88), 315 (3.78), 335 (3.83) nm. 1H-NMR (CDCl3, 400 MHz, δH): 3.85 (s, 3H, OCH3), 6.79 (s, 1H, H-8), 7.48–7.56 (m, 5H, H-Ph), 7.92 s(1H, H-4), 7.93 (s, 1H, H5), 11.21 (1H, OH). 13C-NMR (CDCl3, 100 MHz, δC): 52.1, 83.3, 95.2, 108.1, 112.0, 113.1, 114.7, 129.8 (2C), 130.0, 130.8, 132.8, 133.6 (2C), 144.4, 157.6, 158.4, 164.6, 169.7. Anal. Calcd for C19H12O5: 320.0745; found: 320.0743.
Methyl 3-((4-fluorophenyl)ethynyl)-7-hydroxy-2-oxo-2H-chromene-6-carboxylate (25). Yield 68% (78 mg), yellowish powder, m.p. 167–168 °C (ether). IR (KBr, ν, cm−1): 3425, 2933, 2836, 2145, 1733, 1673, 1608, 1517, 1463, 1450, 1373, 1346, 1257, 1228, 1124, 1012, 939, 873, 852, 840, 792, 753. UV (EtOH) λmax, (lgε): 218 (4.56), 225 (4.54), 245 (4.64), 305 (4.3), 329 (4.41) nm. 1H-NMR (CDCl3, 400 MHz, δH): 3.91 (s, 3H, OCH3), 6.77 (s, 1H, H-8), 6.93 (dd, J = 8.2, 7.2 Нz, 2H, H-3′,5′), 7.40 (dd, J = 8.2, 3.4 Нz, 2H, H-2′,6′), 7.74 (s, 1H, H-4), 7.92 (s, 1H, H-5), 11.26 (1H, OH). 13C-NMR (CDCl3, 100 MHz, δC): 53.3, 82.0, 95.7, 104.7, 110.7, 111.2, 112.1, 118.9 (2C), 128.6, 130.5, 131.8 (2C), 143.4, 158.0, 160.1(C-4′, JC-F 259.1 Hz), 161.3, 164.4, 169.4. Anal. calcd for C19H11FO5: С, 67.46; Н, 3.28; F, 5.62; found: С, 68.08; Н, 3.50; F, 5.12.
Methyl 7-hydroxy-2-oxo-3-(p-tolylethynyl)-2H-chromene-6-carboxylate (26). Yield 61% (70 mg). brownish powder, m.p. 132–133 °C (ether). IR (KBr, ν, cm−1): 3430, 3299, 3259, 3049, 2956, 2852, 2121, 1753, 1735, 1697, 1677, 1594, 1516, 1436, 1367, 1321, 1294, 1234, 1203, 1162, 1087, 960, 848, 794, 748, 734. UV (EtOH) λmax, (lgε): 237 (4.41), 251 (4.21), 272 (4.13), 281 (4.16), 314 (4.04), 338 (4.11) nm. 1H-NMR (CDCl3, 400 MHz, δH) 2.36 (s, 3H, CH3), 4.00 (s, 3H, OCH3), 6.88 (s, 1H, H-8), 7.15 (d, J = 8.2 Нz, 2H, H3′,5′), 7.45 (d, J = 8.2 Нz, 2H, H-2′,6′), 7.81 (s, 1H, H-4), 8.00 (s, 1H, H-5), 11.24 (1H, OH). 13C-NMR (CDCl3, 100 MHz, δC): 21.7, 52.9, 82.4, 95.9, 104.8, 110.5, 111.2, 112.1, 119.00, 129.2 (2C), 130.5, 131.8 (2C), 139.3, 143.9, 158.0, 158.8, 164.4, 169.5. Anal. calcd for C20H14O5: С 71.85, Н 4.22, found: С 71.36, Н 3.94.
Methyl 3-((4-ethylphenyl)ethynyl)-7-hydroxy-2-oxo-2H-chromene-6-carboxylate (27). Yield 52% (61 mg) brownish powder, m.p. 130–131 °C (ether). IR (KBr, ν, cm−1): 3380, 3139, 3049, 2929, 2856, 2185, 1736, 1673, 1625, 1579, 1481, 1439, 1389, 1351, 1288, 1230, 1162, 1126, 1091, 1037, 962, 946, 865, 825, 750, 736. UV (EtOH) λmax, (lgε): 225 (4.1), 249 (3.67), 259 (3.59), 335 (4.18) nm. 1H-NMR (CDCl3, 400 MHz, δH): 1.21 (t, J = 6.8 Нz, 3H, Et), 2.62 (q, J = 6.8 Нz, 2H, Et), 3.97 (s, 3H, OCH3), 6.85 (s, 1H, H-8), 7.13 (dd, J = 8.4 Нz, 2H, H-3′,5′), 7.40 (d, J = 8.4 Нz, 2H, H-2′,6′), 7.96 (s, 1H, H4), 7.97 (s, 1H, H5), 11.21 (1H, OH). 13C-NMR (CDCl3, 100 MHz, δC): 17.3, 28.2, 53.5, 81.9, 97.3, 105.6, 112.1, 113.5, 114.8, 122.9, 129.2, 129.9 (2C), 132.6 (2C), 144.2, 144.5, 158.8, 159.5, 165.2, 170.2. Anal. calcd for C21H16O5: С, 72.41; Н, 4.63; found: С, 72.18; Н, 4.90.
Methyl 7-hydroxy-3-((4-methoxyphenyl)ethynyl)-2-oxo-2H-chromene-6-carboxylate (28). Yield 58% (70 mg) brownish powder, m.p. 134–135 °C (ether). IR (KBr, ν, cm−1): 3430, 3303, 3216, 3049, 3014, 2956, 2129, 1732, 1677, 1594, 1516, 1491, 1436, 1367, 1321, 1294, 1267, 1234, 1203, 1163, 1124, 1088, 960, 865, 849, 794, 748, 734. UV (EtOH) λmax, (lgε): 237 (4.18), 265 (3.78), 277 (3.91), 280 (3.92), 324 (3.84), 335 (3.88) nm. 1H-NMR (CDCl3, 400 MHz, δH): 3.91 (s, 3H, OCH3), 3.96 (s, 3H, OCH3), 6.86 (s, 1H, H-8), 7.13 (d, J = 8.6 Нz, 2H, H-3′,5′), 7.41 (d, J = 8.6 Нz, 2H, H-2′,6′), 7.97 (s, 1H, H-4), 7.98 (s, 1H, H-5), 11.18 (1H, OH). 13C-NMR (CDCl3, 100 MHz, δC): 53.2, 53.6, 81.6, 95.4, 105.1, 111.1, 111.7, 112.8, 118.7, 119.4 (2C), 129.5, 133.3 (2C), 144.1, 157.9, 158.7, 161.6, 164.7, 169.8. Anal. calcd for C20H14O6: С, 68.57; Н, 4.03; found: С, 68.88; Н, 3.90.
Methyl 3-((4-acetylamino-3-(methoxycarbonyl)phenyl)ethynyl)-7-hydroxy-2-oxo-2H-chromene-6-carboxylate (29). Yield 45% (66 mg) yellowish powder, m.p. 154–155 °C (ethanol). IR (KBr, ν, cm−1): 3430, 3299, 3259, 3216, 3049, 2956, 2925, 2852, 2208, 1753, 1735, 1697, 1677, 1620, 1594, 1515, 1491, 1437, 1367, 1321, 1294, 1234, 1203, 1162, 1124, 1088, 960, 920, 866, 849, 795, 748, 735. UV (EtOH) λmax, (lgε): 237 (4.28), 272 (4.00), 280 (4.01), 316 (3.92), 335 (3.96) nm. 1H-NMR (CDCl3, 400 MHz, δH): 2.19 (s, 3H, CH3-CO), 3.88 (s, 3H, OCH3), 3.95 (s, 3H, OCH3), 6.82 (s, 1H, H-8), 7.55 (dd, J = 8.8, 2.0 Нz, 1H, H-6′), 7.94 (s, 1H, H-4), 7.95 (s, 1H, H-5), 8.09 (d, J = 2.0 Нz, 1H, H-2′), 8.58 (d, J = 8.8 Нz, 1H, H-5′), 11.16 (br.s, 2H, NH, OH). 13C-NMR (CDCl3+CD3OD, 100 MHz, δC): 25.2, 52.5, 52.8, 71.3, 82.2, 104.6, 108.5, 112.4, 116.2, 120.0, 130.0, 130.5, 133.7, 134.6, 137.7, 141.2, 143.9, 156.5, 157.7, 166.4, 167.9, 169.1, 169.3. Anal. calcd for C23H17NO8: С, 63.45; Н, 3.94; N, 3.22; found: С, 63.18; Н, 3.92; N, 3.12.
Methyl 7-hydroxy-2-oxo-3-(pyridin-2-ylethynyl)-2H-chromene-6-carboxylate (30). Yield 55% (60 mg) brown oil. IR (ν, cm−1): 3444, 2971, 2929, 2856, 2133, 1735, 1674, 1626, 1579, 1481, 1438, 1388, 1352, 1300, 1230, 1163, 1126, 1092, 1038, 962, 947, 865, 825, 750, 736, 723. UV (EtOH) λmax, (lgε): 227 (4.25), 238 (4.15), 294 (3.93), 306 (3.92), 351 (4.13) nm. 1H-NMR (CDCl3, 400 MHz, δH): 3.76 (s, 3H, OCH3), 6.72 (s, 1H, H-8), 7.18 (ddd, J = 7.4, 5.2, 1.4 Нz, 1H, H-4′), 7.31 (dd, J = 6.6, 1.4 Нz, 1H, H-6′), 7.54 (ddd, J = 7.4, 6.6, 1.8 Нz, 1H, H-5′), 7.69 (s, 1H, H-4), 7.77 (s, 1H, H-5), 8.49 (ddd, J = 7.2, 1.8 Нz, 1H, H-3′), 11.20 (1H, OH). 13C-NMR (CDCl3, 100 MHz, δC): 52.9, 82.6, 94.8, 105.0, 112.8, 115.1, 116.6, 123.5, 129.4, 130.4, 134.1, 138.1, 141.6, 144.4, 156.9, 158.1, 164.4, 169.7. Anal. calcd for C18H11NO5: С, 67.29; Н, 3.45; N, 4.36; found: С, 67.08; Н, 3.38; N, 4.22.
Methyl 7-hydroxy-3-(3-hydroxy-3-methylbut-1-ynyl)-2-oxo-2H-chromene-6-carboxylate (32). Yield 66% (68 mg), m.p.114–115 °C (ethanol). IR (KBr, ν, cm−1): 3282, 3221, 2981, 2932, 2868, 2250, 1741, 1679, 1612, 1600, 1584, 1462, 1446, 1363, 1296, 1269, 1210, 1170, 1120, 1096, 1058, 998, 964, 954, 900, 889, 842, 792, 735, 723. UV (EtOH) λmax, (lgε): 223 (4.04), 246 (3.84), 338 (3.67), 365 (3.57) nm. 1H-NMR (CDCl3, 400 MHz, δH): 1.34 (s, 6H, 2 × CH3), 3.95 (s, 3H, OCH3), 6.78 (s, 1H, H-8), 7.76 (br. s, 1H, H-4), 7.93 (s, 1H, H-5). 3C-NMR (CDCl3, 100 MHz, δc): 30.9, 31.0, 52.7, 65.3, 66.2, 84.1, 104.8, 109.9, 110.5, 114.0, 130.6, 143.1, 157.8, 160.0, 164.1, 169.4. Anal. calcd for C16H14O6: С, 63.57; Н, 4.67; found: С, 63.22; Н, 4.44. HRMS, m/z calcd.: 302.0785; found 302.0783 [M].
Methyl 7-hydroxy-2-oxo-3-((trimethylsilyl)ethynyl)-2H-chromene-6-carboxylate (34). A sealed 10 mL glass tube containing 3-bromopeuruthenicin (11, 100 mg (0.34 mmol), toluene (3 mL), trimethylsilylacetylene (33, 0.1 mL, 0.68 mmol), CuI (3 mg, 5 mol %), Pd(PPh3)2Cl2 (23 mg, 10 mol. %), and Et3N (0.061 mL, 1.3 eq.) was placed in the cavity of a microwave reactor and irradiated for 2 h at 100 °C and power 50 W in an Anton Paar Microwave 50 reactor. After cooling to 25 °C, the tube was removed from the reactor. Then the mixture was cooled and 5 mL of water was added. The separated water layer was extracted with СН2Cl2 (5 × 4 mL). The combined organic extracts was washed with water, dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was crystallized from ether, yield 64% (68 mg), m.p. 104–107 °C. IR (KBr, ν, cm−1): 3437, 2924, 2852, 2223, 1735, 1672, 1314, 1597, 1508, 1425, 1388, 1328, 1236, 1222, 1159, 1107, 908, 839, 752, 694, 680. UV (EtOH) λmax, (lgε): 325 (3.97), 304 (3.85), 241 (4.01) nm. 1H-NMR (CDCl3, 400 MHz, δH) 0.20 (s, 9H, SiMe3), 3.93 (s, 3H, OCH3), 6.76 (s, 1H, H-8), 7.74 (s, 1H, H-4), 7.91 (s, 1H, H-5), 10.97 (s, 1H, OH). 13C-NMR (CDCl3, 100 MHz, δC): 8.6, 52.7, 73.8, 81.5, 101.6, 110.2, 110.4, 111.7, 128.4, 145.1, 158.0, 158.3, 164.45, 169.2. Anal. calcd for C16H16O5Si: С, 60.74; Н, 5.10; Si, 8.88; found: С, 60.55; Н, 5.18; Si, 8.54.
Methyl 3-ethynyl-7-hydroxy-2-oxo-2H-chromene-6-carboxylate (16). To a solution of compound 34 (60 mg, 0.00019 mol) in methanol (3 mL) were added CsF (14 mg, 0.00095 mol) and TEBA (5 mg). The mixture was stirred at rt for 10 h in an argon flow (TLC). Then 10 mL of water was added and the mixture was extracted with methylene chloride (5 × 4 mL). The combined extract was washed with water, dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was subjected to column chromatography on silica gel. Eluting with chloroform and crystallization from ether gave compound 16, yield 85%, m.p. 84- 88 oC. IR (KBr, ν, cm−1): 3463, 2956, 2923, 2854, 2102, 1741, 1677, 1621, 1581, 1494, 1444, 1416, 1367, 1295, 1234, 1186, 1157, 1120, 1099, 1024, 952, 914, 856, 721. UV (EtOH) λmax, (lgε): 225 (4.1), 246 (3.96), 321 (3.84), 344 (3.92). 1H-NMR (CDCl3, 400 MHz) 2.16 (s, 1H, CH), 3.99 (s, 3H, OCH3), 6.86 (s, 1H, H-8), 7.83 (s, 1H, H-4), 8.00 (s, 1H, H-5), 11.29 (s, 1H, OH). 13C- NMR (CDCl3, 100 MHz, δC): 52.3, 75.4, 82.2, 105.1, 112.6, 114.0, 114.8, 128.5, 143.3, 159.4, 159.7, 164.7, 168.9. Anal. calcd for C13H8O5 С 63.94, Н 3.30 found: С 63.65, Н 3.84.
Methyl 7-hydroxy-3-[1-(2-isopropyl-3,7-dioxo-2,3-dihydro-7H-furo[3,2-g]chromen-2-yl)-1H-1,2,3-triazol-4-yl]-2-oxo-2H-chromene-6-carboxylate (36). A solution of 2-azidooreoselone (35, 100 mg, 0.35 mmol) and 3-ethynylpeurutenicin (16, 85 mg, 0.35 mmol) in CH2Cl2 (10 mL) was mixed with a solution of sodium ascorbate (10 mg, 15 mol%) and СuSO4·5H2O (4 mg, 5 mol%) in Н2О (5 mL). The reaction mixture was stirred for 3 h at rt and additional for 1 h at 40 °С. After the completion of the reaction, the mixture was diluted with Н2О (10 mL), the product was extracted with CH2Cl2 (4 × 10 mL), the combined extracts were dried over anhydrous MgSO4, filtered, and the solvent was removed at reduced pressure. Yield 70% (0.130 g), yellowish solid. IR (KBr, ν, cm−1): 3469, 3049, 2983, 2956, 2883, 2850, 1739, 1675, 1629, 1556, 1467, 1438, 1390, 1353, 1284, 1132, 1162, 1128, 1093, 1025, 964, 944, 869, 827, 736. UV (EtOH) λmax, (lgε): 217 (4.57), 249 (4.59), 294 (4.26), 307 (4.27), 337 (4.27), 381 (3.76), 393 (3.36) nm. 1H-NMR (CDCl3, 400 MHz, δH): 0.89 (d, J = 7.0 Нz, 3H, CH3), 1.11 (d, J = 7.0 Нz, 3H, CH3), 3.30 (m, 1H, CH(Me)2), 3.99 (s, 3H, OCH3), 6.41 (d, J = 9.6 Нz, 1H, H-6), 6.88 (s, 1H, H-8′), 7.24 (s, 1H, H-9), 7.71 (d, J = 9.6 Нz, 1H, H-5), 7.87 (s, 1H, H-4), 7.97 (s, 1H, H-4′), 7.98 (s, 1H, H-5′), 8.13 (s, 1H, H-1′′), 11.29 (s, 1H, OH). 13C-NMR (CDCl3, 100 MHz, δC): 16.2, 16.3, 30.1, 53.3, 96.5, 101.4, 105.2, 110.7, 111.8, 112.9, 115.7, 117.3, 117.5, 125.7, 128.0, 130.3, 142.9, 143.5, 144.3, 158.5, 159.3, 162.1, 164.7, 165.8, 169.6, 172.4, 194.7. Anal. calcd for C27H19N3O9: С, 61.25; Н, 3.62; N, 7.94; found: С, 61.01; Н, 3.54; N, 7.62.
4.2.5. General Method for the Preparation of Monoalkynes 38a–c
A solution of 2-azidooreoselone (35, 1.7 mmol) in methylene chloride (10 mL) and a solution of sodium ascorbate (15 mol%, 0.255 mmol) and СuSO4∙5H2O (5 mol%, 0.085 mmol) in water (10 mL) were mixed and the appropriate terminal alkyne 39–41 (0.85 mmol) was added with stirring. The reaction mixture was stirred for 5 h at 40 °С. The cooled mixture was diluted with water (10 mL) and the product was extracted with methylene chloride (4 × 10 mL). The combined extracts were dried over MgSO4, filtered, the solvent was removed to give the desired products 38a–с.
2-Isopropyl-2-(4-(pent-4-ynyl)-1H-1,2,3-triazol-1-yl)-2H-furo[3,2-g]chromene-3,7-dione (38a). Yield 66% (0.420 g), brownish oil. IR (ν, cm−1): 3120, 2981, 2932, 2867, 2140, 1741, 1678, 1600, 1584, 1461, 1446, 1381, 1363, 1295, 1269, 1210, 1169, 1120, 1058, 964, 953, 909, 889, 792, 749. UV (EtOH) λmax, (lgε): 231 (3.91), 282 (3.58), 320 (3.52), 348 (3.43) nm. 1H-NMR (CDCl3, 400 MHz, δH): 0.89 (d, J = 7.0 Hz, 3Н, СН3), 0.95 (d, J = 7.0 Hz, 3Н, СН3), 1.40 (m, 2H, CH2-7′), 1.85 (s, 1H, H-10′), 2.06 (m, 2H, CH2-6′), 2.55 (m, 2H, CH2-8′), 3.06 (m, 1H, CH(CH3)2), 6.28 (d, J = 9.6 Нz, 1H, H-6), 6.99 (s, 1H, H-9), 7.39 (s, 1H, H-4), 7.67 (d, J = 9.6 Нz, 1H, H-5), 7.82 (s, 1H, H-5′). 13C-NMR (CDCl3, 100 MHz, δC): 15.4, 15.8, 19.5, 28.1, 28.2, 33.7, 69.2, 82.3, 98.4, 100.4, 115.4, 115.8, 117.0, 126.7, 127.5, 137.4, 145.9, 161.2, 164.0, 173.6, 191.5. Anal. calcd for C21H19N3O4: С, 66.83; Н, 5.07; N, 11.13; found: С, 67.01; Н, 5.12; N, 10.88.
2-(4-(Hex-5-ynyl)-1H-1,2,3-triazol-1-yl)-2-isopropyl-2H-furo[3,2-g]chromene-3,7-dione (38b). Yield 62% (0.415 g). Yellowish powder, m.p. 111–112 °C (ether). IR (KBr, ν, cm−1): 2971, 2828, 2856, 2115, 1735, 1673, 1625, 1579, 1481, 1438, 1388, 1351, 1230, 1126, 1091, 1037, 997, 962, 946, 865, 825, 750. UV (EtOH) λmax, (lgε): 221 (4.12), 255 (4.47), 292 (3.98), 308 (3.89), 351 (4.03) nm. 1H-NMR (CDCl3, 400 MHz, δH): 0.89 (d, J = 7.0 Hz, 3Н, СН3), 1.03 (m, 2H, CH2-8′), 1.12 (d, J = 7.0 Hz, 3Н, СН3), 1.24 (m, 2H, CH2-7′), 1.92 (s, 1H, H-11′), 2.14 (m, 2H, CH2-6′), 2.30 (m, 1H, CH(СH3)2), 3.40 (m, 2H, CH2-9′ ), 6.35 (d, J = 9.6 Нz, 1H, H-6), 7.02 (s, 1H, H-9), 7.43 (s, 1H, H-4), 7.69 (d, J = 9.6 Нz, 1H, H-5), 7.84 (s, 1H, H-5′). 13C- NMR (CDCl3, 100 MHz, δC): 14.9, 15.3, 17.8, 27.6, 27.7, 27.73, 33.2, 68.7, 81.8, 97.9, 100.6, 114.9, 115.3, 116.1, 125.4, 126.9, 136.8, 144.7, 162.4, 165.6, 173.1, 191.0. Anal. calcd for C22H21N3O4: С, 67.51; Н, 5.41; N, 10.74; found: С, 67.17; Н, 5.21; N, 10.72.
2-Isopropyl-2-(4-(oct-7-ynyl)-1H-1,2,3-triazol-1-yl)-2H-furo[3,2-g]chromene-3,7-dione (38c). Yield 74% (0.527 g). Yellowish powder, m.p. 121–122 °C (ether). IR (KBr, ν, cm−1): 3143, 3084, 2976, 2935, 2858, 2113, 1735, 1627, 1581, 1481, 1419, 1390, 1352, 1286, 1226, 1128, 1093, 1039, 997, 947, 866, 825, 739. UV (EtOH) λmax, (lgε): 221 (4.16), 255 (4.5), 294 (4.00), 307 (3.94), 339 (4.05), 351 (4.06) nm. 1H-NMR (CDCl3, 400 MHz, δH): 0.94 (d, J = 7.0 Hz, 3Н, СН3), 0.97 (d, J = 7.0 Hz, 3Н, СН3), 1.27 (m, 2H, CH2-8′), 1.34 (m, 2H, CH2-9′),1.46 (m, 2H, CH2-10′), 1.55 (m, 2H, (m, 2H, CH2-7′), 1.89 (s, 1H, H-13′), 2.12 (m, 2H, CH2-6′), 2.60 (m, 2H, CH2-11′), 3.09 (m, 1H, CH(CH3)2), 6.32 (d, J = 9.6 Нz, 1H, H-6), 7.04 (s, 1H, H-9), 7.44 (s, 1H, H-4), 7.72 (d, J = 9.6 Нz, 1H, H-5), 7.87 (s, 1H, H-5′). 13C-NMR (CDCl3, 100 MHz, δC): 15.1, 15.5, 18.1, 27.9, 28.0, 28.4, 28.67, 28.73, 33.5, 68.1, 84.4, 98.1, 100.8, 115.1, 115.5, 116.3, 125.7, 128.4, 137.7, 143.1, 158.5, 161.4, 171.4, 191.6. Anal. calcd for C24H25N3O4: С, 68.72; Н, 6.01; N, 10.02; [M] 419; found: С, 69.08; Н, 6.06; N, 10.27; М = 419.
4.2.6. Sonogashira Cross-Coupling Reactions of Bromide 11 with Alkynes 38a–c
To a solution of 3-bromopeurutenicin (11, 100 mg, 0.34 mmol) and a terminal alkyne 38a–c (0.54 mmol) in benzene (5 mL) was added CuI (6 mg, 10 mol%), Pd(PPh3)2Cl2 (12 mg, 5 mol%), and Et3N (0.076 mL, 0.44 mmol; 1.3 equiv) under argon. The reaction mixture was stirred at 80 °C for 14 h (TLC). The mixture was cooled and 5 mL of water was added. The separated water layer was extracted with СН2Cl2 (5 × 4 mL). The combined organic extracts was washed with water, dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was subjected to column chromatography on silica gel (eluent—chloroform).
Methyl 7-hydroxy-3-(5-(1-(2-isopropyl-3,7-dioxo-3,7-dihydro-2H-furo[3,2-g]chromen-2-yl)-1H-1,2,3-triazol-4-yl)-pent-1-ynyl)-2-oxo-2H-chromene-6-carboxylate (37a). Yield 52% (0.105 g). Brownish oil. IR (ν, cm−1): 3350, 3200, 2981, 2932, 2867, 2254, 1741, 1678, 1620, 1600, 1580, 1461, 1446, 1381, 1363, 1295, 1269, 1210, 1169, 1120, 1096, 1058, 964, 953, 889, 792, 735, 723. UV (EtOH) λmax, (lgε): 234 (4.41), 256 (4.44), 273 (4.31), 310 (4.09), 339 (4.14), 354 (4.08) nm. 1H-NMR (CDCl3, 400 MHz, δH): 0.99 (d, J = 7.0 Hz, 3Н, СН3), 1.02 (d, J = 7.0 Hz, 3Н, СН3), 1.60 (m, 2H, CH2-7′), 2.18 (m, 2H, CH2-6′), 2.65 (m, 2H, CH2-8′), 3.13 (m, 1H, CH(CH3)2), 3.97 (s, 3H, OCH3), 6.37 (d, J = 9.8 Hz, 1H, H-6), 6.84 (s, 1H, H-8′′), 7.08 (s, 1H, H-9), 7.48 (d, 1H, J = 9.8 Hz, H-5), 7.76 (s, 1H, H-4), 7.71, 7.74 (both s, 2H, H-4′′,5′′), 7.97 (br.s, 2H, H-5′, OH). 13C-NMR (CDCl3, 100 MHz, δC): 15.0, 15.4, 20.0, 23.5, 32.4, 34.7, 55.7, 70.0, 85.7, 98.1, 99.9, 104.3, 107.2, 110.2, 112.3, 115.1, 116.0, 116.3, 125.5, 128.2, 130.7, 139.7, 142.9, 143.8, 156.1, 157.5, 158.5, 161.4, 162.0, 169.0, 171.4, 192.5. Anal. calcd for C32H25N3O9: С, 64.54; Н, 4.23; N, 7.06; [M] 598; found: С, 64.58, Н, 4.36, N, 7.07; М = 595.
Methyl 7-hydroxy-3-(6-(1-(2-isopropyl-3,7-dioxo-3,7-dihydro-2H-furo[3,2-g]chromen-2-yl)-1H-1,2,3-triazol-1-yl)-hex-1-ynyl)-2-oxo-2H-chromene-6-carboxylate (37b). Yield 57% (0.118 g). Yellowish powder, m.p. 126–127 °C (ether). IR (KBr, ν, cm−1): 3388, 3139, 3049, 2929, 2856, 2235, 1735, 1673, 1626, 1579, 1481, 1439, 1388, 1352, 1288, 1230, 1163, 1126, 1091, 1037, 997, 962, 946, 865, 825, 750, 737. UV (EtOH) λmax, (lgε): 234 (4.39), 256 (4.43), 273 (3.81), 309 (4.09), 339 (4.13), 352 (3.62) nm. 1H-NMR (CDCl3, 400 MHz, δH): 0.98 (d, J = 7.0 Hz, 3Н, СН3), 1.04 (d, J = 7.0 Hz, 3Н, СН3), 1.44–1.53 (m, 4H, CH2-7′,8′), 2.14 (m, 2H, CH2-6′), 2.64 (m, 2H, CH2-9′), 3.13 (m, 1H, CH(CH3)2), 3.95 (s, 3H, OCH3), 6.35 (d, J = 9.8 Hz, 1H, H-6), 6.92 (s, 1H, H-8′′), 7.07 (s, 1H, H-9), 7.64 (d, J = 9.8 Hz, 1H, H-5), 7.71, 7.74 (both s, 2H, H-4′′,5′′), 7.88 (s, 1H, H-4), 7.97 (s, 1H, H5′). 13C-NMR (CDCl3, 100 MHz, δC): 15.6, 16.0, 19.4, 32.5, 33.0, 36.7, 25.7, 54.2, 70.6, 88.5, 98.7, 100.6, 104.9, 108.8, 110.8, 112.8, 115.7, 116.6, 116.9, 126.1, 128.8, 132.3, 141.1, 143.5, 144.4, 156.6, 158.1, 159.1, 162.0, 162.6, 169.5, 172.0, 192.4. Anal. calcd for C33H27N3O9: С, 65.02; Н, 4.46; N, 6.89; [M] 609; found: С, 65.08; Н, 4.06; N, 6.37; M = 605.
Methyl 7-hydroxy-3-(8-(1-(2-isopropyl-3,7-dioxo-3,7-dihydro-2H-furo[3,2-g]chromen-2-yl)-1H-1,2,3-triazol-4-yl)-oct-1-ynyl)-2-oxo-2H-chromene-6-carboxylate (37c). Brownish powder, m.p. 152–153 °C (ether). Yield 55% (0.119 g). IR (KBr, ν, cm−1): 3440, 3141, 3049, 2971, 2929, 2856, 2213, 1735, 1673, 1625, 1579, 1481, 1388, 1351, 1288, 1230, 1126, 1091, 1037, 962, 946, 865, 825, 750. UV (EtOH) λmax, (lgε): 234 (4.63), 251 (4.67), 256 (4.67), 309 (4.33), 339 (4.37), 350 (4.34) nm. 1H-NMR (CDCl3, 400 MHz, δH): 0.91 (d, J = 7.0 Hz, 3Н, СН3), 0.94 (d, J = 7.0 Hz, 3Н, СН3), 1.23, 1.29 (both m, 4H, CH2-8′,9′), 1.42 (m, 2H, CH2-10′), 1.51 (m, 2H, CH2-7′), 2.07 (m, 2H, CH2-6′), 2.56 (m, 2H, CH2-11′), 3.07 (m, 1H, CH(CH3)2), 3.92 (s, 3H, OCH3), 6.31 (d, J = 9.8 Hz, 1H, H-6), 6.78 (s, 1H, H-8′′), 7.03 (s, 1H, H-9), 7.46 (s, 1H, H-4), 7.68 (d, J = 9.8 Hz, 1H, H-5), 7.83 (s, 1H, H-5′). 7.89, 7.90 (both s, 2H, H-4′′,5′′). 13C-NMR (CDCl3, 100 MHz, δC): 15.2, 15.6, 19.0, 25.2, 29.9, 30.7, 31.2, 33.6, 36.3, 52.7, 70.2, 87.4, 98.2, 101.0, 104.5, 108.4, 110.4, 112.4, 115.2, 115.6, 116.5, 125.7, 128.2, 131.8, 141.3, 143.0, 144.0, 156.2, 157.7, 158.9, 161.6, 162.2, 169.2, 171.5, 191.8. Anal. calcd for C35H31N3O9: С, 65.93; Н, 4.90; N 6.59; [M] 637; found: С, 65.38; Н, 4.66; N, 6.87; М = 635.
4.3. Antibacterial Activity Assay
Method 1. Compounds 4a–c, 11, 26, 29, 30, 37a–c, 42a–c and parent compounds 1, 2, 3 were tested for their in vitro antibacterial activity against Staphylococcus aureus 209р ATCC 6538-P and strains from the collection of Department of Microbiology, Immunology and Virology, Novosibirsk State Medical University: Staphylococcus aureus С-18 - clinical isolated strain, Staphylococcus aureus “Viotko” and Actinomyces viscosus U-18. Cultivation of bacterial cultures was carried out on agar and broth media Muller-Hinton in aerobic conditions at a temperature of 37 °C. Cultivation time for Staphylococcus aureus was 1–2 days, for Actinomyces viscosus U – 2–3 days. Analysis of antibacterial activity was performed using the method of serial macrodilutions in a liquid medium in the total volume of 1.0 mL [24,38]. All compounds were primarily dissolved in 0.05 mL of 96% ethyl alcohol and brought to the desired concentration with 0.9% sodium chloride solution (0.2 mL) and nutrient broth. To assess antibacterial activity, a number of double dilutions of the studied substances starting from 1000 µg/mL were used. Doses of substances above 1000 µg/mL were not considered because of their low solubility. The introduced dose of daily cultures of bacteria was determined using the standard of turbidity according to McFarland and was controlled by seeding on a dense nutrient medium. It gave (2.75 ± 0.85) 103 colony-forming units (CFU). For the minimum inhibitory concentration (MIC), the smallest dose of the substance was taken, completely suppressing the growth of bacteria. The absence of signs of growth in the liquid medium was controlled by seeding to the surface of a agar medium with subsequent incubation under standard conditions. As a negative control, the test culture was introduced into 1 mL of broth and cultivated under the same conditions, followed by sowing on an agar nutrient medium and taking into account the growth of bacteria. Based on the results of triplicate repeated experiments, the mean value of the MIC and the standard error of measurement (M ± SEM) were calculated. The primary data of antibacterial activity are given in Supplementary Materials.
Method 2. Compounds 36, 37а–с, 1 and 2 were tested for their in vitro antibacterial activity against Bacillus subtilis and Escherichia coli (JM109). This strain is most similar to wild type of E. coli in our bacterial collection. The known tumorogenic compounds 4-nitroquinolin-1-oxide (NQO) was used as the reference compound [39]. This procedure was maintained according to the standard broth microdilution method as recommended in guidelines of Clinical and Laboratory Standards Institute [24,40,41] and the minimum inhibitory concentration (MIC) of compounds was tested. In short, testing was performed in U-bottomed 96-well sterile plastic microdilution trays (Falcon 3077, Becton Dickinson and Co., Cockeysville, MD, USA) in cation (Ca2+ and Mg2+) adjusted Mueller-Hinton broth medium (Becton Dickinson and Co., Cockeysville, MD, USA). The concentration range of test compounds was started from 1000 µg/mL by using serial two fold dilution. Standardized initial inoculum was prepared by the direct colony suspension method to the final inoculum to 5 × 105 CFU/mL, as described (CLSI M7-A7). After inoculation of previously prepared microdilution trays with tested compounds, trays were incubated at 35 ± 2 °C overnight (18–20 h) in an ambient air incubator. E. coli JM109 served as quality control of MIC determination procedure, as well. The MIC was determined as the lowest concentration of tested compound that completely inhibits growth of the organism in the microdilution wells as detected by the unaided eye and comparing the amount of growth in the wells containing the tested agent with the amount of growth in the growth-control wells (no antimicrobial agent). All testing were done in triplicate.
4.4. Molecular Docking
Molecular modeling was carried out in the Schrodinger Maestro visualization environment using applications from the Schrodinger Small Molecule Drug Discovery Suite 2016-1 (Python, NY, USA) [42]. Three-dimensional structures of the derivatives were obtained empirically in the LigPrep application using the OPLS3 force field [43]. All possible tautomeric forms of compounds, as well as various states of polar protons of molecules in the pH range of 7.0 ± 2.0 were taken into account. The search area for docking was selected automatically, based on the size and physico-chemical properties of FAD. The extra precision (XP) algorithm of docking was applied. Docking was performed in comparison with FAD. The three-dimensional structures of FAD was obtained in the PubChem database and prepared in the LigPrep application. Non-covalent interactions of compounds in the binding site were visualized using Biovia Discovery Studio Visualizer (Biovia, San Diego, CA, USA) [44].
Supplementary Materials
Supplementary data associated with this article can be found in the online version, at Supporting information included the copies of NMR spectra (1H & 13C) of new compounds and also the primary data of antibacterial activity.
Author Contributions
Project administration, E.E.S., A.N.E., O.I.S., and T.G.T.; Supervision, E.E.S. and L.G.B.; Writing—original draft, E.E.S., A.V.L. and L.G.B.; Methodology, E.E.S., D.S.B., T.S.F; investigation, A.V.L., D.O.Z., L.G.B., D.S.B., I.V.S. and T.S.F; Writing—review & editing, E.E.S and L.G.B.
Funding
This work was performed under financial support in part from the Russian Science Foundation (Grants 17-73-10099 and 18-13-00361).
Acknowledgments
Authors would like to acknowledge the Multi-Access Chemical Research Center SB RAS for spectral and analytical measurements.
Conflicts of Interest
The authors declare that they have no conflict of interests.
References
- Bilal, M.; Rasheed, T.; Iqbal, H.M.N.; Hu, H.; Wang, W.; Zhang, X. Macromolecular agents with antimicrobial potentialities: A drive to combat antimicrobial resistance. Int. J. Biol. Macromol. 2017, 103, 554–574. [Google Scholar] [CrossRef] [PubMed]
- Detsi, A.; Kontogiorgis, C.; Hadjipavlou-Litina, D. Coumarin derivatives: An updated patent review (2015–2016). Expert. Opin. Ther. Pat. 2017, 27, 1201–1226. [Google Scholar] [CrossRef] [PubMed]
- Ashok, D.; Gundu, S.; Aamate, V.K.; Devulapally, M.G.; Bathini, R.; Manga, V. Dimers of coumarin-1,2,3-triazole hybrids bearing alkyl spacer: Design, microwave-assisted synthesis, molecular docking andevaluation as antimycobacterial and antimicrobial agents. J. Mol. Sructure 2018, 1157, 312–321. [Google Scholar] [CrossRef]
- Dheer, D.; Singh, V.; Shankar, R. Medicinal attributes of 1,2,3-triazoles: Current developments. Bioorg. Chem. 2017, 71, 30–54. [Google Scholar] [CrossRef] [PubMed]
- Chougala, B.M.; Samundeeswari, S.; Holiyachi, M.; Shastri, L.A.; Dodamani, S.; Jalapure, S.; Dixit, S.R.; Joshi, S.D.; Sunagar, V.A. Synthesis, characterization and molecular docking studies of substituted 4-coumarinylpyrano[2,3-c]pyrazole derivatives as potent antibacterial and anti-inflammatory agents. Eur. J. Med. Chem. 2017, 125, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B. Comprehensive review on the anti-bacterial activity of 1,2,3-triazole hybrids. Eur. J. Med. Chem. 2019, 168, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Kushwaha, K.; Kaushik, N.; Lata; Jain, S.C. Design and synthesis of novel 2H-chromen-2-one derivatives bearing 1,2,3-triazole moiety as lead antimicrobials. Bioorg. Med. Chem. Lett. 2014, 24, 1795–1801. [Google Scholar] [CrossRef] [PubMed]
- Ashok, D.; Bommidi, V.L.; Arram, G.; Sidda, R. Microwave-assisted synthesis of (E)-7-[(1-benzyl-1H-1,2,3-triazol-4-yl)methoxy]-8-(3-arylacryloyl)-4-methyl-2H-chromen-2-ones and their antimicrobial activity. Heterocycl. Commun. 2014, 20, 293–298. [Google Scholar]
- Zayane, M.; Rahmouni, A.; Daami-Remadi, M.; Mansour, M.B.; Romdhane, A.; Jannet, H.B. Design and synthesis of antimicrobial, anticoagulant, and anticholinesterase hybrid molecules from 4-methylumbelliferone. J. Enzyme Inhib. Med. Chem. 2016, 31, 1566–1575. [Google Scholar] [CrossRef]
- Ferreira, S.Z.; Carneiro, H.C.; Lara, H.A.; Alves, R.B.; Resende, J.M.; Oliveira, H.M.; Silva, L.M.; Santos, D.A.; Freitas, R.P. Synthesis of a New Peptide–Coumarin Conjugate: A Potential Agent against Cryptococcosis. ACS Med. Chem. Lett. 2015, 6, 271–275. [Google Scholar] [CrossRef]
- Kusanur, R.A.; Kulkarni, M.V. New 1,3-Dipolar Cycloadducts of 3-Asidoacetylcoumarins with DMAD and their antimicrobial activity. Indian J. Chem. 2005, 44, 591–594. [Google Scholar]
- Lipeeva, A.V.; Shults, E.E.; Makhneva, E.A.; Shakirov, M.M.; Tolstikov, G.A. Study of plant coumarins. 12. Synthesis of 2-(1,2,3-triazolyl)modified furocoumarins. Chem. Heterocycl. Compd. 2013, 49, 551–560. [Google Scholar] [CrossRef]
- Lipeeva, A.V.; Shults, E.E.; Shakirov, M.M.; Pokrovsky, M.A.; Pokrovsky, A.G. Synthesis and Cytotoxic Activity of a New Group of Heterocyclic Analogues of the Combretastatins. Molecules 2014, 19, 7881–7900. [Google Scholar] [CrossRef] [PubMed]
- Lipeeva, A.V.; Pokrovsky, M.A.; Baev, D.S.; Shakirov, M.M.; Bagryanskaya, I.Y.; Tolstikova, T.G.; Pokrovsky, A.G.; Shults, E.E. Synthesis of 1H-1,2,3-triazole linked aryl(arylamidomethyl)-dihydrofurocoumarin hybrids and analysis of their cytotoxicity. Eur. J. Med. Chem. 2015, 100, 119–128. [Google Scholar] [CrossRef]
- Lipeeva, A.V.; Baev, D.S.; Dolgikh, M.P.; Tolstikova, T.G.; Shults, E.E. Rapid access to oxazine fused furocoumarins and in vivo and in silico studies of theirs biological activity. Med. Chem. 2017, 13, 625–632. [Google Scholar] [CrossRef]
- Lipeeva, A.V.; Khvostov, M.V.; Baev, D.S.; Shakirov, M.M.; Tolstikova, T.G.; Shults, E.E. Synthesis, in vivo anticoagulant evaluation and molecular docking studies of new groups of bicoumarins obtained from furocoumarin peucedanin. Med. Chem. 2016, 12, 674–683. [Google Scholar] [CrossRef]
- Mukusheva, G.K.; Lipeeva, A.V.; Zanimkhanova, P.Z.; Shults, E.E.; Gatilov, Y.V.; Shakirov, M.M.; Adekenov, S.M. The flavanone pinostrobin in the synthesis of coumarin-chalcone hybrids with a triazole linker. Chem. Heterocycl. Compd. 2015, 51, 146–152. [Google Scholar] [CrossRef]
- Lipeeva, A.V.; Bryzgalov, A.O.; Tolstikova, T.G.; Shults, E.E. Synthesis, Transformations and Characterization of 8-Aminomethyl Substituted Umbelliferones as Probable Anti-Arrhythmic Agents. Curr. Bioact. Compd. 2019, 15, 71–82. [Google Scholar] [CrossRef]
- Soine, T.O.; Zheveleva, A.; Mahandrus, M.M.; Erhardt, P.; Bubeva-Ivanova, L. Natural Coumarins VII: Isolation and Structure of a New Coumarin, Peuruthenicin, from Peucedanum ruthenicum M.B.J. Pharm. Sci. 1973, 62, 1879–1880. [Google Scholar] [CrossRef]
- Mal’kina, A.G.; Brandsma, L.; Vasilevsky, S.F.; Trofimov, B.A. An Improved Procedure for the Preparation of Aryl- and Hetarylacetylenes. Synthesis 1996, 5, 589–590. [Google Scholar] [CrossRef]
- Joy, M.N.; Bodke, Y.D.; Khader, K.K.A.; Sajit, A.M. A rapid approach for the copper, amine, and ligand-free Sonogashira coupling of 4-methyl-7-nonafluorobutylsulfonyloxy coumarins under microwave irradiation. Tetrahedron Lett. 2014, 55, 2355–2358. [Google Scholar] [CrossRef]
- Lei, Y.; Hu, T.; Wu, X.; Wu, Y.; Xiang, H.; Sun, H.; You, Q.; Zhang, X. Microwave-assisted copper- and palladium-catalyzed Sonogashira-type coupling of aryl bromides and iodides with trimethylsilylacetylene. Tetrahedron Lett. 2016, 57, 1100–1103. [Google Scholar] [CrossRef]
- Lipeeva, A.V.; Shults, E.E. A study of plant coumarins 16. Synthesis and transformations of 7-alkynylcoumarins. Chem. Heterocycl. Compd. 2017, 53, 1302–1309. [Google Scholar] [CrossRef]
- Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically. Available online: https://clsi.org/standarts/products/microbiology/documents/m07/ (accessed on 1 June 2019).
- Merk, D.; Lamers, C.; Weber, J.; Flesch, D.; Gabler, M.; Proschak, E.; Schubert-Zsilavecz, M. Anthranilic acid derivatives as nuclear receptor modulators—Development of novel PPAR selective and dual PPAR/FXR ligands. Bioorg. Med. Chem. 2015, 23, 499–514. [Google Scholar] [CrossRef]
- Kronenberger, T.; Windshügel, B.; Wrenger, C.; Honorio, K.M.; Maltarollo, V.G. On the relationship of anthranilic derivatives structure and the FXR (Farnesoid X receptor) agonist activity. J. Biomol. Struct. Dynamics. J. Biomol. Struct. Dyn. 2018, 36, 4378–4391. [Google Scholar] [CrossRef] [PubMed]
- Ambesi-Impiombato, A.; Bernardo, D.D. Computational Biology and Drug Discovery: From Single Target to Network Drugs. Curr. Bioinform. 2006, 1, 3–13. [Google Scholar] [CrossRef]
- Benson, T.E.; Harris, M.S.; Choi, G.H.; Cialdella, J.I.; Herberg, J.T.; Martin, J.P.; Baldwin, E.T. A Structural Variation for MurB: X-Ray Crystal Structure of Staphylococcus Aureus UDP-N-Acetylenolpyruvylglucosamine Reductase (MurB). Biochemistry 2001, 40, 2340–2350. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Ferguson, G.; McCrindle, R.; McAlees, A.J.; Parvez, M.T. Trans-Dichlorobis(triphenylphosphine)palladium(II). Acta Cryst. 1982, B38, 2679–2681. [Google Scholar] [CrossRef]
- Shults, E.E.; Petrova, T.N.; Shakirov, M.M.; Chernyak, E.I.; Pokrovskii, L.M.; Nekhoroshev, S.A.; Tolstikov, G.A. Coumarins from the roots of Peucedanum morisonii Bess. Chem. Sust. Dev. 2003, 11, 683–691. [Google Scholar]
- Osadchii, S.A.; Shul’ts, E.E.; Shakirov, M.M.; Tolstikov, G.A. Study of Plant Coumarins. 1. Transformations of peucedanin. Russ. Chem. Bull. 2006, 55, 375–379. [Google Scholar] [CrossRef]
- Mohamed, Z.H.; El-Koussi, N.A.; Mahfouz, N.M.; Youssef, A.F.; Jaleel, G.A.A.; Shouman, S.A. Cu (I) catalyzed alkyne-azide 1,3-dipolar cycloaddition (CuAAC): Synthesis of 17a-[1-(substituted phenyl)-1,2,3-triazol-4-yl]-19-nortestosterone-17b-yl acetates targeting progestational and antiproliferative activities. Eur. J. Med. Chem. 2015, 97, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Petrenko, N.I.; Petukhova, V.Z.; Shakirov, M.M.; Shul’ts, E.E.; Tolstikov, G.A. Transformations of higher terpenoids: IV. Synthesis and spectral parameters of glycyrrhetinic acid derivatives containing amino acid fragments. Russ. J. Org. Chem. 2000, 36, 982–995. [Google Scholar]
- Davies, S.G.; Sanganee, H.J.; Szolcsanyi, P. The ’SuperQuat’ (R)-4-Phenyl-5,5-Dimethyl Oxazolidin-2-one as an Effective Chiral Auxiliary for Conjugate additions: Asymmetric Synthesis of (-)-Aplysillamide B. Tetrahedron 1999, 55, 3337–3354. [Google Scholar] [CrossRef]
- Grigg, R.; Sridharan, V.; Thornton-Pett, M.; Wang, J.; Xu, J.; Zhang, J. Kinetic acidity of iminium ions. 2-Alkynyl- and 2,5-dialkynyl-pyrrolidines via the iminium ion route to azomethine ylides. Tetrahedron 2002, 58, 2627–2640. [Google Scholar] [CrossRef]
- Osadchii, S.A.; Shults, E.E.; Polukhina, E.V.; Shakirov, M.M.; Vasilevskii, S.F.; Stepanov, A.A.; Tolstikov, G.A. Study of alkaloids of the Siberian and Altai flora 14. Synthesis of alkaloid-based tertiary N-(3-arylprop-2-ynyl)amines. Russ. Chem. Bull. Int. Ed. 2007, 56, 1261–1267. [Google Scholar] [CrossRef]
- Mironov, A.N. The Guidelines for Preclinical Studies of Pharmaceuticals; Minsdrav: Moskwa, Russia, 2012; p. 944. (In Russian) [Google Scholar]
- Ikenaga, M.; Ichikawa-Ryo, H.; Sohei, K. The major cause of inactivation and mutation by 4-Nitroquinoline 1-Oxide in Escherichia coli: Excisable 4NQO-purine adducts. J. Mol. Biol. 1975, 92, 341–356. [Google Scholar] [CrossRef]
- Performance Standards for Antimicrobial Susceptibility Testing. Available online: https://clsi.org/standards/products/microbiology/documents/m100/ (accessed on 1 June 2019).
- Harbeck, R.J.; McCarter, Y.S.; Ortez, J.H.; Rankin, I.D.; Sautter, R.L.; Sharp, S.E.; Spiegel, C.A. Manual of Antimicrobial Susceptibility Testing; American Society for Microbiology: Washington, DC, USA, 2005. [Google Scholar]
- Schrodinger Small Molecule Drug Discovery Suite. Available online: https://www.schrodinger.com/suites/small-molecule-drug-discovery-suite (accessed on 1 June 2019).
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Discovery Studio Visualizer. Available online: https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/download-instructions/discovery-studio-visualizer-download-instructions-45.html (accessed on 1 June 2019).
Sample Availability: Samples of the compounds 1, 4a–c, 11, 16, 4a–c, 28, 29, 30, 36, 37a–c, 42a–c are available from the authors. |








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).