
Formulation Strategies for Enhancing the Bioavailability of Silymarin: The State of the Art


Abstract
1. Introduction
2. Silymarin: Source and Physicochemical Properties
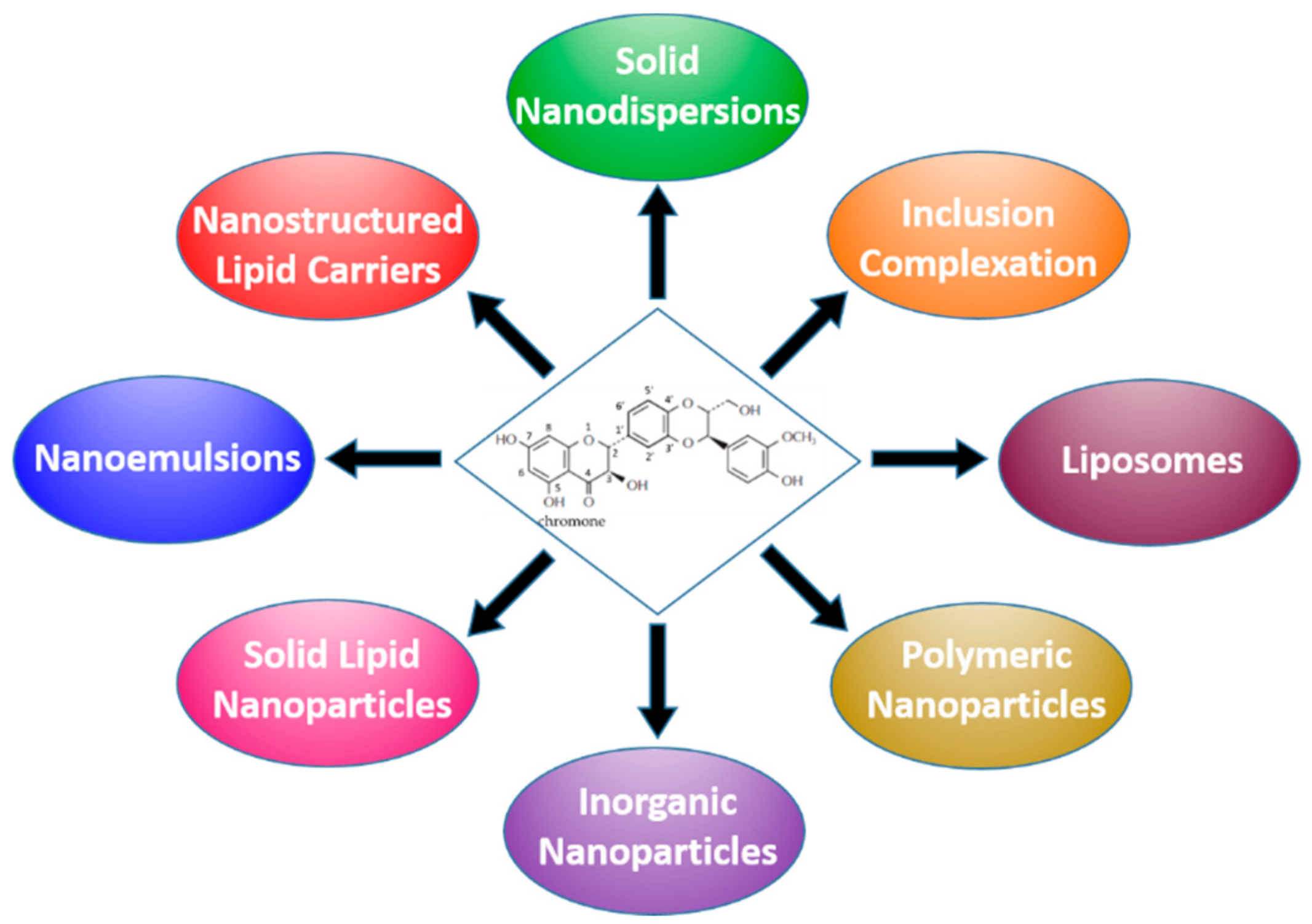
3. Formulation Strategies Designed to Improve the Bioavailability of Silymarin
3.1. Nanocrystals, Nanosuspensions and Solid Dispersions
3.2. Complexes with Cyclodextrins and Phospholipids
3.3. Lipid-based Formulations
3.3.1. Micro- and NanoEmulsions
3.3.2. Liposomes
3.3.3. Solid-Lipid Nanoparticles (SLNs), Nanostructured Lipid Carriers (NLCs)
3.4. Polymer-based Nanocarriers
3.4.1. Inclusion in Polymeric Matrices
3.4.2. Dendrimers and Polymeric NPs
3.5. Nanostructured Materials Based on Inorganic Compounds
4. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AUC | area under the plasma drug concentration-t curve |
| β-CD | β-cyclodextrin |
| Brij 78 or Brij S20 | polyoxyethylene 20 stearyl ether |
| BS | Bile Salts |
| Cmax | maximum plasma drug concentration |
| CA | Caprylic Acid |
| Capryol 90 | propylene glycol monocaprylate |
| CHOL | cholesterol |
| CMCHS | carboxymethylchitosan |
| CP | cetyl palmitate |
| Cremophor® EL | polyoxy-35-castor oil |
| Cremophor® RH40 | polyoxyl 40 hydrogenated castor oil |
| DCP | dicetylphosphate |
| DDs | Drug Delivery Systems |
| D-GaIN | D-galactosamine |
| DON | Deoxynivalenol |
| DPPC | DiPalmitoylPhosphatidylCholine |
| DSPE | DiStearoylPhosphatidylEthanolamine |
| EE | Entrapment Efficiency |
| EES | Emulsification Evaporation Solidification |
| ESD | Emulsification/Solvent Diffusion |
| ESE | Emulsification/Solvent Evaporation |
| EVO | extra virgin olive |
| GA | glycyrrhizic acid |
| GDS | GlycerylDiStearate |
| Geleol® | mono-, di- and triesters of palmitic and stearic acids |
| GIT | gastrointestinal tract |
| GMO | GlycerylMonoOleate |
| GMS | GlycerylMonoStearate |
| HCO-X® | PEG-X Hydrogenated Castor Oil, (X = 40, 50) |
| HP-β-CD | 2-hydroxypropyl-β-cyclodextrin |
| HPH | High Pressure Homogenization |
| HPMC (E50LV) | HydroxyPropyl MethylCellulose |
| HSPC | Soya Hydrogenated L-α-PhosphatidylCholine |
| i.p. | intraperitoneal |
| IPM (Estol) | isopropyl myristate |
| isoSIL | isosilybin |
| i.v. | intravenous |
| Labrafac® CC | Medium Chain Triglycerides (MCT) |
| Labrafil® | transesterified ethoxylated vegetable oils |
| Labrasol® | caprylocaproyl polyoxylglycerides (macrogolglycerides) |
| MCT | Medium chain triglycerides |
| NaCMC | Sodium CarboxyMethylCellulose |
| NAFLD | NonAlcoholic Fatty Liver Disease |
| NLCs | Nanostructured Lipid Carriers |
| NPs | nanoparticles |
| OA | oleic acid |
| P188 | Poloxamer 188 |
| P407 | Poloxamer 407 |
| PAMAM | polyamidoamine |
| PC | L-α-PhosphatidylCholine |
| PCL | poly-ε-caprolactone |
| PDI | polydispersity index |
| PEG | polyethyleneglycol |
| PGA | poly-γ-glutamic acid |
| PLGA | poly(d,l-lactic-co-glycolic acid) |
| PPC | Polyene PhosphatidylCholine |
| Precirol® ATO 5 | Glyceryl distearate/palmitostearate |
| PVA | Polyvinyl alcohol |
| PVP | polyvinylpyrrolidone |
| RAMEB | randomly methylated-β-cyclodextrin |
| RPE | reverse phase evaporation |
| SA | stearic acid |
| SC | Sodium Cholate |
| SCF-CO2 | SuperCritical Fluid of carbon dioxide |
| SDC | Sodium DeoxyCholate |
| SEDS | Solution-Enhanced Dispersion Supercritical fluids |
| SEDDS | Self Emulsifying Drug Delivery System |
| Sefsol® 218 | propylene glycol monocaprylic ester |
| SGC | Sodium GlycoCholate |
| SGF/SIF | simulated gastric fluid (pH 1.2)/simulated intestinal fluid (pH 7.4) |
| SIL | Silybin or silybinin |
| SILcr | silycristin |
| SILdi | silydianin |
| Sito-G | β-sitosterol β-d-glucoside |
| SLM | silymarin extract |
| SLNs | Solid Lipid Nanoparticles |
| SLS | Sodium Lauryl Sulfate |
| Solutol® HS 15 | PEG (15)-hydroxystearate |
| SPC | Soya L-α-PhosphatidylCholine |
| SPG | Shirasu Porous Glass membrane emulsification |
| SPMM | Na cholate/phospholipid mixed micelles |
| STC | Sodium TauroCholate |
| SUV | small unilamellar vesicles |
| TFD | Thin-Film Dispersion |
| TNF-α | Tumour Necrosis Factor-α |
| TPGS | D-α-Tocopheryl PEG 1000 Succinate |
| TPP | TriPolyPhosphate |
| Transcutol® | diethylene glycol monoethyl ether |
| Triacetin | glycerol triacetate |
| Tween 20 | polyoxyethylene sorbitan monolaurate (polysorbate 20) |
| Tween 80 | polyoxyethylene sorbitan monooleate (polysorbate 80) |
| TXF | taxifolin |
References
- Abenavoli, L.; Izzo, A.A.; Milić, N.; Cicala, C.; Santini, A.; Capasso, R. Milk thistle (Silybum marianum): A concise overview on its chemistry, pharmacological, and nutraceutical uses in liver diseases. Phytother. Res. 2018, 32, 2202–2213. [Google Scholar] [CrossRef] [PubMed]
- Bijak, M. Silybin, a major bioactive component of milk thistle (Silybum marianum L. Gaernt.)—Chemistry, Bioavailability, and Metabolism. Molecules 2017, 22, 1942. [Google Scholar] [CrossRef] [PubMed]
- Dressman, J.B.; Vertzoni, M.; Goumas, K.; Reppas, C. Estimating drug solubility in the gastrointestinal tract. Adv. Drug. Deliv. Rev. 2007, 59, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; del Pilar Rodriguez-Torres, M.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71–103. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.A.; Al Qaraghuli, M.M.; Alsaadi, M.; Alzahrani, A.R.; Niwasabutra, K.; Ferro, V.A. Delivering natural products and biotherapeutics to improve drug efficacy. Ther. Deliv. 2017, 8, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jiang, H.; Xu, C.; Gu, L. A review: Using nanoparticles to enhance absorption and bioavailability of phenolic phytochemicals. Food Hydrocoll. 2015, 43, 153–164. [Google Scholar] [CrossRef]
- Agarwal, R.; Agarwal, C.; Ichikawa, H.; Singh, R.P.; Agarwal, B.B. Anticancer potential of silymarin: From bench to bed side. Anticancer Res. 2006, 26, 4457–4498. [Google Scholar]
- Javed, S.; Kohli, K.; Ali, M. Reassessing bioavailability of silymarin. Altern. Med. Rev. 2011, 16, 239–249. [Google Scholar]
- Chen, M.-W.; Tan, W.; Wang, S.-P.; Zhong, Z.-F.; Wang, Y.-T. Advances in the nanoparticle drug delivery systems of silymarin. J. Chin. Pharm. Sci. 2011, 20, 442–446. [Google Scholar] [CrossRef]
- Theodosiou, E.; Purchartová, K.; Stamatis, H.; Kolisis, F.; Křen, V. Bioavailability of silymarin flavonolignans: Drug formulations and biotransformation. Phytochem. Rev. 2013, 13, 1–18. [Google Scholar] [CrossRef]
- Bonifácio, B.V.; Silva, P.B.; Ramos, M.A.; Negri, K.M.; Bauab, T.M.; Chorilli, M. Nanotechnology-based drug delivery systems and herbal medicines: A review. Int. J. Nanomedicine 2014, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, L.; Wang, Q.; Zhang, D. Recent Advances in the Nanotechnology-Based Drug Delivery of Silybin. J. Biomed. Nanotechnol. 2014, 10, 543–558. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Zhuang, J.; Lu, Y.; Dong, X.; Zhao, W.; Wu, W. In vivo fate of lipid-based nanoparticles. Drug Discov. Today 2017, 22, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Tran, S.; De Giovanni, P.-J.; Piel, B.; Rai, P. Cancer nanomedicine: A review of recent success in drug delivery. Clin. Trans. Med. 2017, 6, 44–64. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Ukidve, A.; Krishnan, V.; Mitragotri, S. Effect of physicochemical and surface properties on in vivo fate of drug nanocarriers. Adv. Drug Deliv. Rev. 2019. [Google Scholar] [CrossRef] [PubMed]
- Kropacova, K.; Misurova, E.; Hakova, H. Protective and therapeutic effect of silymarin on the development of latent liver damage. Radiats. Biol. Radioecol. 1998, 38, 411–415. [Google Scholar] [PubMed]
- Ding, T.; Tian, S.; Zhang, Z.; Gu, D.; Chen, Y.; Shi, Y.; Sun, Z. Determination of active components in silymarin by RP-LC and LC/MS. J. Pharm. Biomed. Anal. 2001, 26, 155–161. [Google Scholar] [CrossRef]
- Lu, C.; Lu, Y.; Chen, J.; Zhang, W.; Wu, W. Synchronized and sustained release of multiple components in silymarin from erodible glyceryl monostearate matrix system. Eur. J. Pharm. Biopharm. 2007, 66, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Luper, S. A review of plants used in the treatment of liver diseases: Part 1. Altern. Med. Rev. 1998, 3, 410–421. [Google Scholar] [PubMed]
- Lee, D.Y.-W.; Liu, Y. Molecular Structure and Stereochemistry of Silybin A, Silybin B, Isosilybin A, and Isosilybin B, Isolated from Silybum marianum (Milk Thistle). J. Nat. Prod. 2003, 66, 1171–1174. [Google Scholar] [CrossRef]
- Kvasnička, F.; Bíba, B.; Ševčík, R.; Voldřich, M.; Krátká, J. Analysis of the active components of silymarin. J. Chromatogr. A 2003, 990, 239–245. [Google Scholar] [CrossRef]
- Napolitano, J.G.; Lankin, D.C.; Graf, T.N.; Friesen, J.B.; Chen, S.-N.; McAlpine, J.B.; Oberlies, N.H.; Pauli, G.F. HiFSA Fingerprinting applied to isomers with near-identical NMR spectra: The silybin/isosilybin case. J. Org. Chem. 2013, 78, 2827–2839. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.L.; Narayan, M.; Barrett, J.S. Analysis and comparison of active constituents in commercial standardized silymarin extracts by liquid chromatography-electrospray ionization mass spectrometry. J. Chromatogr. B 2007, 845, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Gažák, R.; Walterová, D.; Křen, V. Silybin and silymarin—New and emerging applications in medicine. Curr. Med. Chem. 2007, 14, 315–338. [Google Scholar] [CrossRef] [PubMed]
- Loguercio, C.; Festi, D. Silybin and the liver: From basic research to clinical practice. World J. Gastroenterol. 2011, 17, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Wang, C.; Li, B.B.; Zhang, A.H. Metabolic fingerprinting to understand therapeutic effects and mechanisms of silybin on acute liver damage in rat. Pharmacogn. Mag. 2015, 43, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Flaig, T.W.; Gustafson, D.L.; Su, L.J.; Zirrolli, J.A.; Crighton, F.; Harrison, G.S.; Pierson, A.S.; Agarwal, R.; Glodé, L.M. A phase I and pharmacokinetic study of silybin-phytosome in prostate cancer patients. Invest. New Drugs. 2007, 25, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Raina, K.; Deep, G.; Chan, D.; Agarwal, R. Silibinin suppresses growth of human prostate carcinoma PC-3 orthotopic xenograft via activation of extracellular signal-regulated kinase ½ and inhibition of signal transducers and activators of transcription signaling. Clin. Cancer. Res. 2009, 15, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Abenavoli, L.; Capasso, R.; Milic, N.; Capasso, F. Milk thistle in liver diseases: Past, present, future. Phytother. Res. 2010, 24, 1423–1432. [Google Scholar] [CrossRef]
- Parveen, R.; Baboota, S.; Ali, J.; Ahuja, A.; Vasudev, S.S.; Ahmad, S. Oil based nanocarrier for improved oral delivery of silymarin: In vitro and in vivo studies. Int. J. Pharm. 2011, 413, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Gažák, R.; Svobodová, A.; Psotová, J.; Sedmera, P.; Přikrylová, V.; Walterová, D.; Křen, V. Oxidised derivatives of silybin and their antiradical and antioxidant activity. Bioorgan. Med. Chem. 2004, 12, 5677–5687. [Google Scholar] [CrossRef] [PubMed]
- Bai, T.-C.; Yan, G.-B.; Hu, J.; Zhang, H.-L.; Huang, C.-G. Solubility of silybin in aqueous poly(ethylene glycol) solution. Int. J. Pharm. 2006, 308, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Bai, T.-C.; Zhu, J.-J.; Hu, J.; Zhang, H.-L.; Huang, C.-G. Solubility of silybin in aqueous hydrochloric acid solution. Fluid Phase Equilibr. 2007, 254, 204–210. [Google Scholar] [CrossRef]
- Saller, R.; Melzer, J.; Reichling, J.; Brignoli, R.; Meier, R. An updated systematic review of the pharmacology of silymarin. Forsch Komplementarmed. 2007, 14, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Shakeel, F.; Anwer, M.K.; Shazly, G.A.; Jamil, S. Measurement and correlation of solubility of bioactive compound silymarin in five different green solvents at 298.15 K to 333.15 K. J. Mol. Liq. 2014, 195, 255–258. [Google Scholar] [CrossRef]
- Pérez-Sánchez, A.; Cuyàs, E.; Ruiz-Torres, V.; Agulló-Chazarra, L.; Verdura, S.; González-Álvarez, I.; Bermejo, M.; Joven, J.; Micol, V.; Bosch-Barrera, J.; et al. Intestinal Permeability Study of Clinically Relevant Formulations of Silibinin in Caco-2 Cell Monolayers. Int. J. Mol. Sci. 2019, 20, 1606. [Google Scholar] [CrossRef]
- Wu, J.W.; Lin, L.C.; Hung, S.C.; Chi, C.W.; Tsai, T.H. Analysis of silibinin in rat plasma and bile for hepatobiliary excretion and oral bioavailability application. J. Pharm. Biomed. Anal. 2007, 45, 635–641. [Google Scholar] [CrossRef]
- Gažák, R.; Purchartová, K.; Marhol, P.; Zivná, L.; Sedmera, P.; Valentová, K.; Kato, N.; Matsumura, H.; Kaihatsu, K.; Křen, V. Antioxidant and antiviral activities of silybin fatty acid conjugates. Eur. J. Med. Chem. 2010, 45, 1059–1067. [Google Scholar] [CrossRef]
- Cufí, S.; Bonavia, R.; Vazquez-Martin, A.; Corominas-Faja, B.; Oliveras-Ferraros, C.; Cuyàs, E.; Martin-Castillo, B.; Barrajón-Catalán, E.; Visa, J.; Segura-Carretero, A.; et al. Silibinin meglumine, a water-soluble form of milk thistle silymarin, is an orally active anti-cancer agent that impedes the epithelial-to-mesenchymal transition (EMT) in EGFR-mutant non-small-cell lung carcinoma cells. Food Chem. Toxicol. 2013, 60, 360–368. [Google Scholar] [CrossRef]
- Kurkin, V.A.; Ryzhov, V.M.; Biryukova, O.V.; Mel’nikova, N.B.; Selekhov, V.V. Interaction of milk-thistle-fruit flavanonols with Langmuir monolayers of lecithin and bilayers of liposomes. Pharm. Chem. J. 2009, 43, 101–109. [Google Scholar] [CrossRef]
- Leone, F.; Cavalli, R. Drug nanosuspensions: A ZIP tool between traditional and innovative pharmaceutical formulations. Expert Opin. Drug Deliv. 2015, 12, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-B.; Shen, Z.-G.; Wang, J.-X.; Zhang, H.-X.; Zhao, H.; Chen, J.-F.; Yun, J. Micronization of silybin by the emulsion solvent diffusion method. Int. J. Pharm. 2009, 376, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-H.; Lin, C.-C.; Hsu, W.-C.; Chung, C.-Y.; Lin, C.-C.; Jassey, A.; Chang, S.-P.; Tai, C.-J.; Tai, C.-J.; Shields, J.; et al. Highly bioavailable silibinin nanoparticles inhibit HCV infection. Gut 2017, 66, 1853–1861. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, D.; Liu, Z.; Liu, G.; Duan, C.; Jia, L.; Feng, F.; Zhang, X.; Shi, Y.; Zhang, Q. In vitro and in vivo evaluation of silybin nanosuspensions for oral and intravenous delivery. Nanotechnology 2010, 21, 155104–155115. [Google Scholar] [CrossRef] [PubMed]
- Siepmann, J.; Siepmann, F. Mathematical modeling of drug dissolution. Int. J. Pharm. 2013, 453, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, L.; Liu, Z.; Zhang, D.; Zhang, Q. In vivo evaluation of silybin nanosuspensions targeting liver. J. Biomed Nanotechnol. 2012, 8, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Hwang Du, H.; Kim, Y.; Cho, K.H.; Poudel, B.K.; Choi, J.Y.; Kim, D.-W.; Shin, Y.-J.; Bae, O.-N.; Yousaf, A.M.; Yong, C.S.; et al. A novel solid dispersion system for natural product-loaded medicine: Silymarin-loaded solid dispersion with enhanced oral bioavailability and hepatoprotective activity. J. Microencapsul. 2014, 31, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Wei, X.; Wu, B.; Chen, J.; Lu, Y.; Wu, W. Enhanced dissolution of silymarin/polyvinylpyrrolidone solid dispersion pellets prepared by a one-step fluid-bed coating technique. Powder Technol. 2008, 182, 72–80. [Google Scholar] [CrossRef]
- Xie, Y.; Lu, Y.; Qi, J.; Li, X.; Zhang, X.; Han, J.; Jin, S.; Yuan, H.; Wu, W. Synchronized and controlled release of multiple components in silymarin achieved by the osmotic release strategy. Int. J. Pharm. 2013, 441, 111–120. [Google Scholar] [CrossRef]
- Sansone, F.; Esposito, T.; Lauro, M.R.; Picerno, P.; Mencherini, T.; Gasparri, F.; De Santis, S.; Chieppa, M.; Cirillo, C.; Aquino, R.P. Application of spray drying particle engineering to a high-functionality/low-solubility milk thistle extract: Powders production and characterization. Molecules 2018, 23, 1716. [Google Scholar] [CrossRef]
- Cui, G.-J.; Xu, L.-M.; Zhou, Y.; Zhang, J.-J.; Wang, J.-X.; Chen, J.-F. Microfluidic fabrication of silybin nanodispersion with high dissolution rate and tunable sizes. Chem. Eng. J. 2013, 222, 512–519. [Google Scholar] [CrossRef]
- Sahibzada, M.U.K.; Sadiq, A.; Khan, S.; Faidah, H.S. Fabrication, characterization and in vitro evaluation of silibinin nanoparticles: An attempt to enhance its oral bioavailability. Drug. Des. Devel. Ther. 2017, 11, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Wang, Y.; Zhang, D.; Liu, Z.; Duan, C.; Jia, L.; Wang, F.; Liu, Y.; Liu, G.; Hao, L.; et al. In vitro antitumor activity of silybin nanosuspension in PC-3 cells. Cancer Lett. 2011, 307, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Yi, T.; Liu, C.; Zhang, J.; Wang, F.; Wang, J.; Zhang, J. A new drug nanocrystal self-stabilized Pickering emulsion for oral delivery of silybin. Eur. J. Pharm. Sci. 2017, 96, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zhao, Y.; Feng, N.; Zhang, Y.; Liu, Y.; Dang, B. Improved dissolution and bioavailability of silymarin delivered by a solid dispersion prepared using supercritical fluids. Asian J. Pharm. Sci. 2015, 10, 194–202. [Google Scholar] [CrossRef]
- Braga Carneiro, S.; Costa Duarte, F.Í.; Heimfarth, L.; Siqueira Quintans, J.S.; Quintans-Júnior, L.J.; da Veiga Júnior, V.F.; Neves de Lima, Á.A. Cyclodextrin-drug inclusion complexes: In vivo and in vitro approaches. Int. J. Mol. Sci. 2019, 20, 642. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Biswas, S.; Ghosh, T. Preparation and evaluation of silymarin β-cyclodextrin molecular inclusion complexes. J. Young Pharmacists 2011, 3, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Kellici, T.F.; Ntountaniotis, D.; Leonis, G.; Chatziathanasiadou, M.; Chatzikonstantinou, A.V.; Becker-Baldus, J.; Glaubitz, C.; Tzakos, A.G.; Viras, K.; Chatzigeorgiou, P.; et al. Investigation of the interactions of silibinin with 2-hydroxypropyl-β-cyclodextrin through biophysical techniques and computational methods. Mol. Pharmaceutics 2015, 12, 954–965. [Google Scholar] [CrossRef] [PubMed]
- Gharbia, S.; Balta, C.; Herman, H.; Rosu, M.; Váradi, J.; Bácskay, I.; Vecsernyés, M.; Gyöngyösi, S.; Fenyvesi, F.; Voicu, S.N.; et al. Enhancement of silymarin anti-fibrotic effects by complexation with hydroxypropyl (HPBCD) and randomly methylated (RAMEB) β-cyclodextrins in a mouse model of liver fibrosis. Front. Pharmacol. 2018, 9, 883–900. [Google Scholar] [CrossRef]
- Lu, M.; Qiu, Q.; Luo, X.; Liu, X.; Sun, J.; Wang, C.; Lin, X.; Deng, Y.; Song, Y. Phyto-phospholipid complexes (phytosomes): A novel strategy to improve the bioavailability of active constituents. Asian J. Pharm. Sci. 2018. [Google Scholar] [CrossRef]
- Yanyu, X.; Yunmei, S.; Zhipeng, C.; Qineng, P. The preparation of silybin–phospholipid complex and the study on its pharmacokinetics in rats. Int. J. Pharm. 2006, 307, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Semalty, A.; Semalty, M.; Rawat, M.S.M.; Franceschi, F. Supramolecular phospholipids–polyphenolics interactions: The PHYTOSOME® strategy to improve the bioavailability of phytochemicals. Fitoterapia 2010, 81, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhu, Y.; Wang, L.; Peng, M.; Tong, S.S.; Cao, X.; Qiu, H.; Xu, X. Enhancement of oral bioavailability of the poorly water-soluble drug silybin by sodium cholate/phospholipid-mixed micelles. Acta Pharm. Sin. 2010, 31, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yu, J.; Tong, S.S.; Wang, L.; Peng, M.; Cao, X.; Xu, X. Preparation and in vitro evaluation of povidone-sodium cholate-phospholipid mixed micelles for the solubilization of poorly soluble drugs. Arch. Pharm. Res. 2010, 33, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.L.; Sun, X.; Liu, J.; Gong, T.; Zhang, Z.R. Mixed micelles loaded with silybin-polyene phosphatidylcholine complex improve drug solubility. Acta Pharmacol. Sin. 2011, 32, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Panapisal, V.; Charoensri, S.; Tantituvanont, A. Formulation of microemulsion systems for dermal delivery of silymarin. Pharm. Sci. Tech. 2012, 13, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Abrol, S.; Trehan, A.; Katare, O.P. Formulation, characterization, and in vitro evaluation of silymarin-loaded lipid microspheres. Drug Deliv. 2004, 11, 185–191. [Google Scholar] [CrossRef]
- Singh, B.; Bandopadhyay, S.; Kapil, R.; Singh, R.; Katare, O. Self-Emulsifying Drug Delivery Systems (SEDDS): Formulation development, characterization, and applications. Crit. Rev. Ther. Drug Carrier Syst. 2009, 26, 427–521. [Google Scholar] [CrossRef] [PubMed]
- Zanchetta, B.; Chaud, M.V.; Santana, M.H.A. Self-Emulsifying Drug Delivery Systems (SEDDS) in Pharmaceutical Development. J. Adv. Chem. Eng. 2015, 5, 130–136. [Google Scholar] [CrossRef]
- Wu, W.; Wang, Y.; Que, L. Enhanced bioavailability of silymarin by self-microemulsifying drug delivery system. Eur. J. Pharm. Biopharm. 2006, 63, 288–294. [Google Scholar] [CrossRef]
- Woo, J.S.; Kim, T.-S.; Park, J.-H.; Chi, S.-C. Formulation and biopharmaceutical evaluation of silymarin using SMEDDS. Arch. Pharm. Res. 2007, 30, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yuan, Q.; Huang, Y.; Zhou, Y.; Liu, Y. Development of silymarin self-microemulsifying drug delivery system with enhanced oral bioavailability. AAPS Pharm. Sci. Tech. 2010, 11, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Ye, X.; Shang, X.; Peng, X.; Bao, Q.; Liu, M.; Guo, M.; Li, F. Enhanced oral bioavailability of silybin by a supersaturatable self-emulsifying drug delivery system (S-SEDDS). Colloid Surface A 2012, 396, 22–28. [Google Scholar] [CrossRef]
- Adhikari, M.; Arora, R. Nano-silymarin provides protection against γ-radiation-induced oxidative stress in cultured human embryonic kidney cells. Mutat. Res.-Gen. Tox. En. 2015, 792, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.Y.; Hwang, D.H.; Yousaf, A.M.; Kim, D.W.; Shin, Y.J.; Bae, O.N.; Kim, Y.I.; Kim, J.O.; Yong, C.S.; Choi, H.G. Silymarin-loaded solid nanoparticles provide excellent hepatic protection: Physicochemical characterization and in vivo evaluation. Int. J. Nanomed. 2013, 8, 3333–3343. [Google Scholar] [CrossRef]
- Calligaris, S.; Comuzzo, P.; Bot, F.; Lippe, G.; Zironi, R.; Anese, M.; Nicoli, M.C. Nanoemulsions as delivery systems of hydrophobic silybin from silymarin extract: Effect of oil type on silybin solubility, in vitro bioaccessibility and stability. LWT—Food Sci. Technol. 2015, 63, 77–84. [Google Scholar] [CrossRef]
- Nagi, A.; Iqbal, B.; Kumar, S.; Sharma, S.; Ali, J.; Baboota, S. Quality by design based silymarin nanoemulsion for enhancement of oral bioavailability. J. Drug Deliv. Sci. Tec. 2017, 40, 35–44. [Google Scholar] [CrossRef]
- Murgia, S.; Fadda, P.; Colafemmina, G.; Angelico, R.; Corrado, L.; Lazzari, P.; Monduzzi, M.; Palazzo, G. Characterization of the Solutol® HS15/water phase diagram and the impact of the Δ9-tetrahydrocannabinol solubilisation. J. Colloid Interf. Sci. 2013, 390, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Piazzini, V.; Rosseti, C.; Bigagli, E.; Luceri, C.; Bilia, A.R.; Bergonzi, M.C. Prediction of permeation and cellular transport of silybum marianum extract formulated in a nanoemulsion by using PAMPA and caco-2 cell models. Planta Med. 2017, 83, 1184–1193. [Google Scholar] [CrossRef] [PubMed]
- Van Hoogevest, P.; Leigh, M.; Fahr, A. Liposomes as intravenous solubilizers for poorly water-soluble drugs. In Drug Delivery Strategies for Poorly Water-Soluble Drugs. Douroumis, D., Fahr, A., Eds.; John Wiley & Sons: Chichester, West Sussex, UK, 2013; Chapt. 2; pp. 37–66. [Google Scholar]
- Riaz, M.K.; Riaz, M.A.; Zhang, X.; Lin, C.; Wong, K.H.; Chen, X.; Zhang, G.; Lu, A.; Yang, Z. Surface Functionalization and Targeting Strategies of Liposomes in Solid Tumor Therapy: A Review. Int. J. Mol. Sci. 2018, 19, 195. [Google Scholar] [CrossRef] [PubMed]
- Maheshwari, H.; Agarwal, R.; Patil, C.; Katare, O.P. Preparation and pharmacological evaluation of silibinin liposomes. Arzneimittelforschung 2003, 53, 420–427. [Google Scholar] [CrossRef] [PubMed]
- El-Samaligy, M.S.; Afifi, N.N.; Mahmoud, E.A. Increasing bioavailability of silymarin using a buccal liposomal delivery system: Preparation and experimental design investigation. Int. J. Pharm. 2006, 308, 140–148. [Google Scholar] [CrossRef] [PubMed]
- El-Samaligy, M.S.; Afifi, N.N.; Mahmoud, E.A. Evaluation of hybrid liposomes-encapsulated silymarin regarding physical stability and in vivo performance. Int. J. Pharm. 2006, 319, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.-Y.; Song, Y.-M.; Chen, Z.-P.; Ping, Q.-N. Preparation of silymarin proliposome: A new way to increase oral bioavailability of silymarin in beagle dogs. Int. J. Pharm. 2006, 319, 162–168. [Google Scholar] [CrossRef]
- Payne, N.I.; Timmis, P.; Ambrose, C.V.; Warel, M.D. Proliposomes: A novel solution to an old problem. J. Pharm. Sci. 1986, 75, 325–329. [Google Scholar] [CrossRef]
- Tong, S.S.; Chu, C.; Wei, Y.; Wang, L.; Gao, X.Z.; Xu, X.M.; Yu, J. Preparation and effects of 2,3-dehydrosilymarin, a promising and potent antioxidant and free radical scavenger. J. Pharm. Pharmacol. 2011, 63, 238–244. [Google Scholar] [CrossRef]
- Chu, C.; Tong, S.S.; Xu, Y.; Wang, L.; Fu, M.; Ge, Y.R.; Yu, J.N.; Xu, X.M. Proliposomes for oral delivery of dehydrosilymarin: Preparation and evaluation in vitro and in vivo. Acta Pharmacol. Sin. 2011, 32, 973–980. [Google Scholar] [CrossRef]
- Elmowafy, M.; Viitala, T.; Ibrahim, H.M.; Abu-Elyazid, S.K.; Samy, A.; Kassem, A.; Yliperttula, M. Silymarin loaded liposomes for hepatic targeting: In vitro evaluation and HepG2 drug uptake. Eur. J. Pharm. Sci. 2013, 50, 161–171. [Google Scholar] [CrossRef]
- Ochi, M.M.; Amoabediny, G.; Rezayat, S.M.; Akbarzadeh, A.; Ebrahimi, B. In vitro co-delivery evaluation of novel pegylated nano-liposomal herbal drugs of silibinin and glycyrrhizic acid (nano-phytosome) to hepatocellular carcinoma cells. Cell J. 2016, 18, 135–148. [Google Scholar] [CrossRef]
- Kumar, N.; Rai, A.; Reddy, N.D.; Raj, P.V.; Jain, P.; Deshpande, P.; Mathew, G.; Kutty, N.G.; Udupa, N.; Rao, C.M. Silymarin liposomes improves oral bioavailability of silybin besides targeting hepatocytes, and immune cells. Pharmacol. Rep. 2014, 66, 788–798. [Google Scholar] [CrossRef]
- Angelico, R.; Ceglie, A.; Sacco, P.; Colafemmina, G.; Ripoli, M.; Mangia, A. Phyto-liposomes as nanoshuttles for water-insoluble silybin-phospholipid complex. Int. J. Pharm. 2014, 471, 173–181. [Google Scholar] [CrossRef]
- Ripoli, M.; Angelico, R.; Sacco, P.; Ceglie, A.; Mangia, A. Phytoliposome-based silibinin delivery system as a promising strategy to prevent hepatitis C virus infection. J. Biomed. Nanotechnol. 2016, 12, 770–780. [Google Scholar] [CrossRef]
- Yang, G.; Zhao, Y.; Zhang, Y.; Dang, B.; Liu, Y.; Feng, N. Enhanced oral bioavailability of silymarin using liposomes containing a bile salt: Preparation by supercritical fluid technology and evaluation in vitro and in vivo. Int. J. Nanomed. 2015, 10, 6633–6644. [Google Scholar] [CrossRef]
- Mohsen, A.M.; Asfour, M.H.; Salama, A.A.A. Improved hepatoprotective activity of silymarin via encapsulation in the novel vesicular nanosystem bilosomes. Drug Dev. Ind. Pharm. 2017, 43, 2043–2054. [Google Scholar] [CrossRef]
- Lian, R.; Lu, Y.; Qi, J.; Tan, Y.; Niu, M.; Guan, P.; Hu, F.; Wu, W. Silymarin glyceryl monooleate/poloxamer 407 liquid crystalline matrices: Physical characterization and enhanced oral bioavailability. AAPS Pharm. Sci. Tech. 2011, 12, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Mady, F.; Essa, H.; El-Ammawi, T.; Abdelkader, H.; Hussein, A. Formulation and clinical evaluation of silymarin pluronic-lecithin organogels for treatment of atopic dermatitis. Drug Des. Dev. Ther. 2016, 10, 1101–1110. [Google Scholar] [CrossRef][Green Version]
- Joseph, S.; Bunjes, H. Solid Lipid Nanoparticles for Drug Delivery. In Drug Delivery Strategies for Poorly Water-Soluble Drugs; Douroumis, D., Fahr, A., Eds.; John Wiley & Sons: Chichester, West Sussex, UK, 2013; Chapt. 4; pp. 103–149. [Google Scholar]
- Gill, B.; Singh, J.; Sharma, V.; Hari Kumar, S.L. Emulsomes: An emerging vesicular drug delivery system. Asian J. Pharm. 2012, 6, 87–94. [Google Scholar] [CrossRef]
- Ghasemiyeh, P.; Mohammadi-Samani, S. Solid lipid nanoparticles and nanostructured lipid carriers as novel drug delivery systems: Applications, advantages and disadvantages. Res. Pharm. Sci. 2018, 13, 288–303. [Google Scholar] [CrossRef]
- Khan, S.; Baboota, S.; Ali, J.; Khan, S.; Narang, R.S.; Narang, J.K. Nanostructured lipid carriers: An emerging platform for improving oral bioavailability of lipophilic drugs. Int. J. Pharma. Investig. 2015, 5, 182–191. [Google Scholar] [CrossRef]
- Xu, P.; Yin, Q.; Shen, J.; Chen, L.; Yu, H.; Zhang, Z.; Li, Y. Synergistic inhibition of breast cancer metastasis by silibinin-loaded lipid nanoparticles containing TPGS. Int. J. Pharm. 2013, 454, 21–30. [Google Scholar] [CrossRef]
- Shangguan, M.; Qi, J.; Lu, Y.; Wu, W. Comparison of the oral bioavailability of silymarin-loaded lipid nanoparticles with their artificial lipolysate counterparts: Implications on the contribution of integral structure. Int. J. Pharm. 2015, 489, 195–202. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Hou, S.; Lu, W.; Zhu, L.; Feng, J. Preparation, pharmacokinetics and body distribution of silymarin-loaded solid lipid nanoparticles after oral administration. J. Biomed. Nanotechnol. 2007, 3, 195–202. [Google Scholar] [CrossRef]
- Cengiz, M.; Kutlu, H.M.; Burukoglu, D.D.; Ayhancı, A. A comparative study on the therapeutic effects of silymarin and silymarin-loaded solid lipid nanoparticles on d-GaIN/TNF-α-induced liver damage in Balb/c mice. Food Chem. Toxicol. 2015, 77, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Q.; Liu, J.; Li, X.L.; Jasti, B.R. Preparation and characterization of solid lipid nanoparticles containing silibinin. Drug Deliv. 2007, 14, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Piazzini, V.; Lemmi, B.; D’Ambrosio, M.; Cinci, L.; Luceri, C.; Bilia, A.R.; Bergonzi, M.C. Nanostructured lipid carriers as promising delivery systems for plant extracts: The case of silymarin. Appl. Sci. 2018, 8, 1163. [Google Scholar] [CrossRef]
- Ma, Y.; He, H.; Xia, F.; Li, Y.; Lu, Y.; Chen, D.; Qi, J.; Lu, Y.; Zhang, W.; Wu, W. In vivo fate of lipid-silybin conjugate nanoparticles: Implications on enhanced oral bioavailability. Nanomedicine 2017, 13, 2643–2654. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; He, H.; Fan, W.; Li, Y.; Zhang, W.; Zhao, W.; Qi, J.; Lu, Y.; Dong, X.; Wu, W. In vivo fate of biomimetic mixed micelles as nanocarriers for bioavailability enhancement of lipid-drug conjugates. ACS Biomater. Sci. Eng. 2017, 3, 2399–2409. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, Z. Preparation and performance evaluation of emulsomes as a drug delivery system for silybin. Arch. Pharm. Res. 2015, 38, 2193–2200. [Google Scholar] [CrossRef]
- Jia, L.; Zhang, D.; Li, Z.; Duan, C.; Wang, Y.; Feng, F.; Wang, F.; Liu, Y.; Zhang, Q. Nanostructured lipid carriers for parenteral delivery of silybin: Biodistribution and pharmacokinetic studies. Colloid Surface B 2010, 80, 213–218. [Google Scholar] [CrossRef]
- Jia, L.; Zhang, D.; Li, Z.; Feng, F.; Wang, Y.; Dai, W.; Duan, C.; Zhang, Q. Preparation and characterization of silybin-loaded nanostructured lipid carriers. Drug Deliv. 2010, 17, 11–18. [Google Scholar] [CrossRef]
- Shangguan, M.; Lu, Y.; Qi, J.; Han, J.; Tian, Z.; Xie, Y.; Hu, F.; Yuan, H.; Wu, W. Binary lipids-based nanostructured lipid carriers for improved oral bioavailability of silymarin. J. Biomater. Appl. 2014, 28, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, S.; Garg, T.; Murthy, R.S.R.; Rath, G.; Goyal, A.K. Development, optimization and evaluation of long chain nanolipid carrier for hepatic delivery of silymarin through lymphatic transport pathway. Int. J. Pharm. 2015, 485, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, B.; Ali, J.; Baboota, S. Silymarin loaded nanostructured lipid carrier: From design and dermatokinetic study to mechanistic analysis of epidermal drug deposition enhancement. J. Mol. Liq. 2018, 255, 513–529. [Google Scholar] [CrossRef]
- Chen, C.-H.; Chen, C.-J.; Elzoghby, A.O.; Yeh, T.-S.; Fang, J.-Y. Self-assembly and directed assembly of lipid nanocarriers for prevention of liver fibrosis in obese rats: A comparison with the therapy of bariatric surgery. Nanomedicine 2018, 13, 1551–1566. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Williams, R.O. Polymeric nanomedicines for poorly soluble drugs in oral delivery systems: An update. J. Pharm. Investig. 2018, 48, 61–75. [Google Scholar] [CrossRef]
- Sonali, D.; Tejal, S.; Vaishali, T.; Tejal, G. Silymarin-solid dispersions: Characterization and influence of preparation methods on dissolution. Acta Pharm. 2010, 60, 427–443. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.-H.; Yu, H.; Dong, B.; Hadinoto, K. A supersaturating delivery system of silibinin exhibiting high payload achieved by amorphous nano-complexation with chitosan. Eur. J. Pharm. Sci. 2016, 89, 163–171. [Google Scholar] [CrossRef]
- Zhao, X.; Deng, Y.; Zhang, Y.; Zu, Y.; Bolin, L.; Wu, M.; Zu, C.; Wu, W. Silymarin nanoparticles through emulsion solvent evaporation method for oral delivery with high antioxidant activities, bioavailability, and absorption in the liver. RSC Adv. 2016. [Google Scholar] [CrossRef]
- Gohulkumar, M.; Gurushankar, K.; Rajendra Prasad, N.; Krishnakumar, N. Enhanced cytotoxicity and apoptosis-induced anticancer effect of silibinin-loaded nanoparticles in oral carcinoma (KB) cells. Mater. Sci. Eng. C Mater. Biol. Appl. 2014, 41, 274–282. [Google Scholar] [CrossRef]
- Younis, N.; Shaheen, M.A.; Abdallah, M.H. Silymarin-loaded Eudragit® RS100 nanoparticles improved the ability of silymarin to resolve hepatic fibrosis in bile duct ligated rats. Biomed. Pharmacother. 2016, 81, 93–103. [Google Scholar] [CrossRef]
- El-Nahas, A.E.; Allam, A.N.; Abdelmonsif, D.A.; El-Kamel, A.H. Silymarin-loaded Eudragit nanoparticles: Formulation, characterization, and hepatoprotective and toxicity evaluation. AAPS Pharm. Sci. Tech. 2017, 18, 3076–3086. [Google Scholar] [CrossRef]
- Yousaf, A.M.; Malik, U.R.; Shahzad, Y.; Mahmood, T.; Hussain, T. Silymarin-laden PVP-PEG polymeric composite for enhanced aqueous solubility and dissolution rate: Preparation and in vitro characterization. J. Pharm. Anal. 2019, 9, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Elkin, I.; Banquy, X.; Barrett, C.J.; Hildgen, P. Non-covalent formulation of active principles with dendrimers: Current state-of-the-art and prospects for further development. J. Cont. Rel. 2017, 264, 288–305. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wu, Z.; Gao, W.; Chen, Q.; Yu, B. Polyamidoamine dendrimers as potential drug carriers for enhanced aqueous solubility and oral bioavailability of silybin. Drug Dev. Ind. Pharm. 2011, 37, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Diaz, C.; Guzmán, J.L.; Jiménez, V.A.; Alderete, J.B. Partially PEGylated PAMAM dendrimers as solubility enhancers of Silybin. Pharm. Dev. Technol. 2018, 23, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Sui, W.; Yin, C.; Kong, X. Micellar solubilization and in vitro release of silymarin in the self-aggregates of an amphiphilic derivative of chitosan. Macromol. Symp. 2010, 297, 147–153. [Google Scholar] [CrossRef]
- El-Sherbiny, I.M.; Abdel-Mogib, M.; Dawidar, A.-A.M.; Elsayed, A.; Smyth, H.D.C. Biodegradable pH-responsive alginate-poly (lactic-co-glycolic acid) nano/micro hydrogel matrices for oral delivery of silymarin. Carbohyd. Polym. 2011, 83, 1345–1354. [Google Scholar] [CrossRef]
- Snima, K.S.; Arunkumar, P.; Jayakumar, R.; Lakshmanan, V.-K. Silymarin Encapsulated Poly(D,L-lactic-co-glycolic acid) Nanoparticles: A Prospective Candidate for Prostate Cancer Therapy. J. Biomed. Nanotechnol. 2014, 10, 559–570. [Google Scholar] [CrossRef]
- Xie, Y.; Yi, Y.; Hu, X.; Shangguan, M.; Wang, L.; Lu, Y.; Qi, J.; Wu, W. Synchronous microencapsulation of multiple components in silymarin into PLGA nanoparticles by an emulsification/solvent evaporation method. Pharm. Dev. Technol. 2016, 21, 672–679. [Google Scholar] [CrossRef]
- Bonepally, C.R.; Gandey, S.J.; Bommineni, K.; Gottumukkala, K.M.; Aukunuru, J. Preparation, characterisation and in vivo evaluation of silybin nanoparticles for the treatment of liver fibrosis. Trop. J. Pharm. Res. 2013, 12, 1–6. [Google Scholar] [CrossRef]
- Pooja, D.; Bikkina, D.J.B.; Kulhari, H.; Nikhila, N.; Chinde, S.; Raghavendra, Y.M.; Sreedhar, B.; Tiwari, A.K. Fabrication, characterization and bioevaluation of silibinin loaded chitosan nanoparticles. Int. J. Biol. Macromol. 2014, 69, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Guhagarkar, S.A.; Shah, D.; Patel, M.D.; Sathaye, S.S.; Devarajan, P.V. Polyethylene sebacate-silymarin nanoparticles with enhanced hepatoprotective activity. J. Nanosci. Nanotechnol. 2015, 15, 4090–4093. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; He, S.; Ma, X.; Hong, T.; Li, Z.; Park, K.; Wang, W. Silymarin-loaded nanoparticles based on stearic acid-modified bletilla striata polysaccharide for hepatic targeting. Molecules 2016, 21, 265. [Google Scholar] [CrossRef]
- Lee, J.-S.; Hong, D.Y.; Kim, E.S.; Lee, H.G. Improving the water solubility and antimicrobial activity of silymarin by nanoencapsulation. Colloid Surface B 2017, 154, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahhab, M.A.; El-Nekeety, A.A.; Salman, A.S.; Abdel-Aziem, S.H.; Mehaya, F.M.; Hassan, N.S. Protective capabilities of silymarin and inulin nanoparticles against hepatic oxidative stress, genotoxicity and cytotoxicity of Deoxynivalenol in rats. Toxicon 2018, 142, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Hao, X.; Liang, X.; Zhang, Q.; Zhang, C.; Zhou, G.; Shen, S.; Jia, G.; Zhang, J. Inorganic nanomaterials as carriers for drug delivery. J. Biomed. Nanotechnol. 2016, 12, 770–780. [Google Scholar] [CrossRef]
- Cao, X.; Deng, W.W.; Fu, M.; Wang, L.; Tong, S.S.; Wei, Y.W.; Xu, Y.; Su, W.Y.; Xu, X.M.; Yu, J.-N. In vitro release and in vitro–in vivo correlation for silybin meglumine incorporated into hollow-type mesoporous silica nanoparticles. Int. J. Nanomed. 2012, 7, 753–762. [Google Scholar] [CrossRef]
- Cao, X.; Deng, W.; Fu, M.; Zhu, Y.; Liu, H.; Wang, L.; Zeng, J.; Wei, Y.; Xu, X.; Yu, J. Seventy-two-hour release formulation of the poorly soluble drug silybin based on porous silica nanoparticles: In vitro release kinetics and in vitro/in vivo correlations in beagle dogs. Eur. J. Pharm. Sci. 2013, 48, 64–71. [Google Scholar] [CrossRef]
- Cao, X.; Fu, M.; Wang, L.; Liu, H.; Deng, W.; Qu, R.; Su, W.; Wei, Y.; Xu, X.; Yu, J. Oral bioavailability of silymarin formulated as a novel 3-day delivery system based on porous silica nanoparticles. Acta Biomater. 2012, 8, 2104–2112. [Google Scholar] [CrossRef]
- Dolatabadi, J.E.N.; Omidi, Y.; Losic, D. Carbon nanotubes as an advanced drug and gene delivery nanosystem. Curr. Nanosci. 2011, 7, 297–314. [Google Scholar] [CrossRef]
- Tan, J.M.; Karthivashan, G.; Arulselvan, P.; Fakurazi, S.; Hussein, M.Z. Characterization and in vitro sustained release of silibinin from pH responsive carbon nanotube-based drug delivery system. J. Nanomater. 2014, 2014. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, H.; Yang, S.; Zhou, B.; You, F.; Yan, X. Nanostructured calcium phosphate carriers for deliver of poor water-soluble drug silybin. Mater. Lett. 2015, 143, 252–255. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, M.; Zhang, Y.; Zeng, J.; Omari-Siaw, E.; Yu, J.; Xu, X. In vitro release and bioavailability of silybin from micelle-templated porous calcium phosphate microparticles. AAPS Pharm. Sci. Tech. 2016, 17, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimnezhad, Z.; Zarghami, N.; Keyhani, M.; Amirsaadat, S.; Akbarzadeh, A.; Rahmati, M.; Taheri, Z.M.; Nejati-Koshki, K. Inhibition of hTERT gene expression by silibinin-loaded PLGA-PEG-Fe3O4 in T47D breast cancer cell line. BioImpacts 2013, 3, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Di Iorio, E.; Colombo, C.; Cheng, Z.; Capitani, G.; Mele, D.; Ventruti, G.; Angelico, R. Characterization of magnetite nanoparticles synthetized from Fe(II)/nitrate solutions for arsenic removal from water. J. Environ. Chem. Eng. 2019, 7, 102986. [Google Scholar] [CrossRef]
- Khalkhali, M.; Sadighian, S.; Rostamizadeh, K.; Khoeini, F.; Naghibi, M.; Bayat, N.; Hamidi, M. Simultaneous diagnosis and drug delivery by silymarin-loaded magnetic nanoparticles. Nanomed. J. 2015, 2, 223–230. [Google Scholar] [CrossRef]
- Fazio, E.; Scala, A.; Grimato, S.; Ridolfo, A.; Grassi, G.; Neri, F. Laser light triggered smart release of silibinin from a PEGylated–PLGA gold nanocomposite. J. Mater. Chem. B 2015, 3, 9023–9032. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Type of Formulation | Method of Preparation | Results | References |
|---|---|---|---|
| Nanocrystals, nanosuspensions | ESD | Rod-shaped NPs | Zhang et al. [42] |
| ESD | NPs < 200 nm | Liu et al. [43] | |
| HPH | NPs 637 and 132 nm | Whang et al. [44,46,53] | |
| HPH | Pickering emulsion | Yi et al. [54] | |
| Spray-drying | Dissolution studies | Hwang et al. [47] | |
| Spray-drying | Microparticles | Sansone et al. [50] | |
| Fluid-bed coating | Synchronized release | Wu et al. [48,49] | |
| Microfluidics | NP size 26–101 nm | Cui et al. [51] | |
| Antisolvent precip. | Dissolution studies | Sahibzada et al. [52] | |
| SEDS | Dissolution studies | Yang et al. [55] | |
| Inclusion complexes, phytosomes | Co-precipitation | Complex with β-CD | Ghosh et al. [57] |
| Freeze-drying | Complex with HP-CD | Kellici et al. [58] | |
| Kneading | HP-β-CD, RAMEB | Gharbia et al. [59] | |
| Solvent evaporation | Phospholipids | Yanyu et al. [61] | |
| Solvent evaporation | Phospholipids | Duan et al. [65] | |
| Mixed micelles | BS-phospholipids | Yu et al. [63] | |
| Mixed micelles | BS-phospholipids | Zhu et al. [64] | |
| Micro- and NanoEmulsions | Spontaneous emulsif. | Microemulsion | Panapisal et al. [66] |
| Low energy emulsif. | O/W emulsion | Abrol et al. [67] | |
| Low energy emulsif. | O/W emulsion | Parveen et al. [30] | |
| Low energy emulsif. | Nanoemulsion | Adhikari et al. [74] | |
| Low energy emulsif. | Nanoemulsion | Calligaris et al. [76] | |
| Low energy emulsif. | Nanoemulsion | Piazzini et al. [79] | |
| Membrane emulsif. | Nanoemulsion | Yang et al. [75] | |
| HPH | Nanoemulsion | Nagi et al. [77] | |
| SEDDS | Water titration | Wu et al. [70] | |
| SEDDS | Water titration | Woo et al. [71] | |
| SEDDS | Water titration | Li et al. [72] | |
| S-SEDDS | Supersaturated state | Wei et al. [73] | |
| Liposomes | Ethanol injection | Drug EE 95% | Maheshwari et al. [82] |
| RPE | Drug EE 69% | El-Samaligy et al. [83,84] | |
| TFD | Drug EE 55% | Kumar et al. [91] | |
| RPE | Phytosome | Angelico et al. [92,93] | |
| SEDS | Bile salt | Yang et al. [94] | |
| TFD | Bile salt | Mohsen et al. [95] | |
| PEGylated liposomes | TFD | Hepatic targeting | Elmowafy et al. [89] |
| PEGylated liposomes | TFD | Hepatic targeting | Ochi et al. [90] |
| Proliposomes | Film-deposition | Drug EE 93% | Xiao et al. [85] |
| Proliposomes | TFD-freeze drying | Drug EE 82% | Tong et al. [87,88] |
| Cubosomes | Melting/Congealing | Pluronic | Lian et al. [96] |
| Organogels | Mixed Solution | Lecithin/pluronic | Mady et al. [97] |
| Solid-Lipid Nanoparticles | TFD | Drug EE 99% | Xu et al. [102] |
| HPH | Lipolysis mechanism | Shangguan et al. [103] | |
| Cold/hot HPH | Drug EE 87% | He et al. [104] | |
| Hot HPH | NP size 165–200 nm | Cengiz et al. [105] | |
| Hot HPH | SIL-conjugates | Ma et al. [108,109] | |
| EES | Stealth SLNs | Zhang et al. [106] | |
| EES | Drug EE 92% | Piazzini et al. [107] | |
| Film hydration | SIL-emulsomes | Zhou et al. [110] | |
| Nanostructured Lipid Carriers | ESE | NP size 230 nm | Jia et al. [111,112] |
| ESE | NP size 126 nm | Iqbal et al. [115] | |
| ESE | NP size 225 nm | Chen et al. [116] | |
| Hot HPH | Drug EE 87% | Wu et al. [113] | |
| Emulsif./ultrasound | Drug EE 79% | Chaudhary et al. [114] | |
| Inclusion in polymeric matrices | Co-precipitation | Dissolution studies | Sonali et al. [118] |
| Complexation | Chitosan NPs | Nguyen et al. [119] | |
| ESE/freeze-drying | NP size 100 nm | Zhao et al. [120] | |
| Nanoprecipitation | Drug EE 79% | Gohulkumar et al. [121] | |
| Nanoprecipitation | Drug EE 89% | Younis et al. [122] | |
| Nanoprecipitation | Drug EE 83% | El-Nahas et al. [123] | |
| Solvent evaporation | Dissolution studies | Yousaf et al. [124] | |
| Dendrimers and polymeric NPs | PAMAM dendrimers | Solubility studies | Huang et al. [126] |
| PEG-PAMAM | Solubility studies | Diaz et al. [127] | |
| Polymeric micelles | Chitosan derivative | Sui et al. [128] | |
| ESE | PLGA | El-Sherbiny [129] | |
| ESE | PLGA | Snima et al. [130] | |
| ESE | PLGA | Xie et al. [131] | |
| ESE | PCL | Bonepally et al. [132] | |
| Ionic gelation | Chitosan-TPP | Pooja et al. [133] | |
| Nanoprecipitation | PE Sebacate NPs | Guhagarkar et al. [134] | |
| Ultrasonication | Polysaccharide NPs | Ma et al. [135] | |
| Ionic gelation | Chitosan/PGA | Lee et al. [136] | |
| Ionic gelation | Inulin NPs | Abdel-Wahhab et al. [137] | |
| Inorganic nanomaterials | Microemulsion | Mesoporous Si NPs | Cao et al. [139] |
| Ultrasonic corrosion | Porous Si NPs | Cao et al. [140,141] | |
| Drug conjugation | Carbon NT | Tan et al. [143] | |
| Precipitation | Calcium phosphate | Chen et al. [144] | |
| Precipitation | Calcium phosphate | Zhu et al. [145] | |
| Precipitation | PLGA-PEG-Fe3O4 | Ebrahimnezhad et al. [146] | |
| Coprecipitation | Chitosan-Fe3O4 | Khalkhali et al. [148] | |
| Emulsion-diffusion | PEG-PLGA-Au | Fazio et al. [149] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Costanzo, A.; Angelico, R. Formulation Strategies for Enhancing the Bioavailability of Silymarin: The State of the Art. Molecules 2019, 24, 2155. https://doi.org/10.3390/molecules24112155
Di Costanzo A, Angelico R. Formulation Strategies for Enhancing the Bioavailability of Silymarin: The State of the Art. Molecules. 2019; 24(11):2155. https://doi.org/10.3390/molecules24112155
Chicago/Turabian StyleDi Costanzo, Alfonso, and Ruggero Angelico. 2019. "Formulation Strategies for Enhancing the Bioavailability of Silymarin: The State of the Art" Molecules 24, no. 11: 2155. https://doi.org/10.3390/molecules24112155
APA StyleDi Costanzo, A., & Angelico, R. (2019). Formulation Strategies for Enhancing the Bioavailability of Silymarin: The State of the Art. Molecules, 24(11), 2155. https://doi.org/10.3390/molecules24112155