Conformational Studies of Glucose Transporter 1 (GLUT1) as an Anticancer Drug Target


Abstract
:1. Introduction
2. Results and Discussion
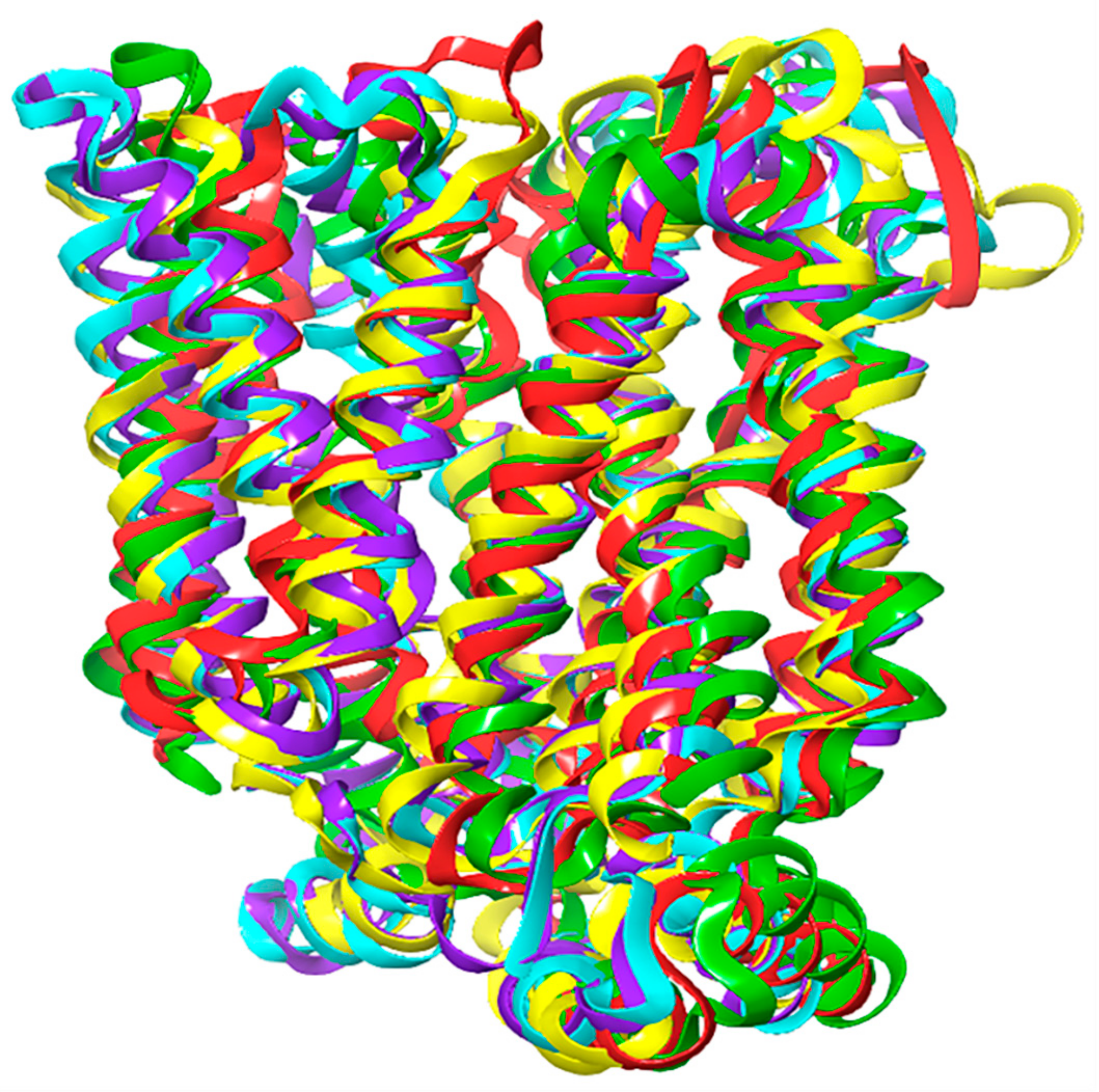
2.1. Homology Modeling of GLUT1
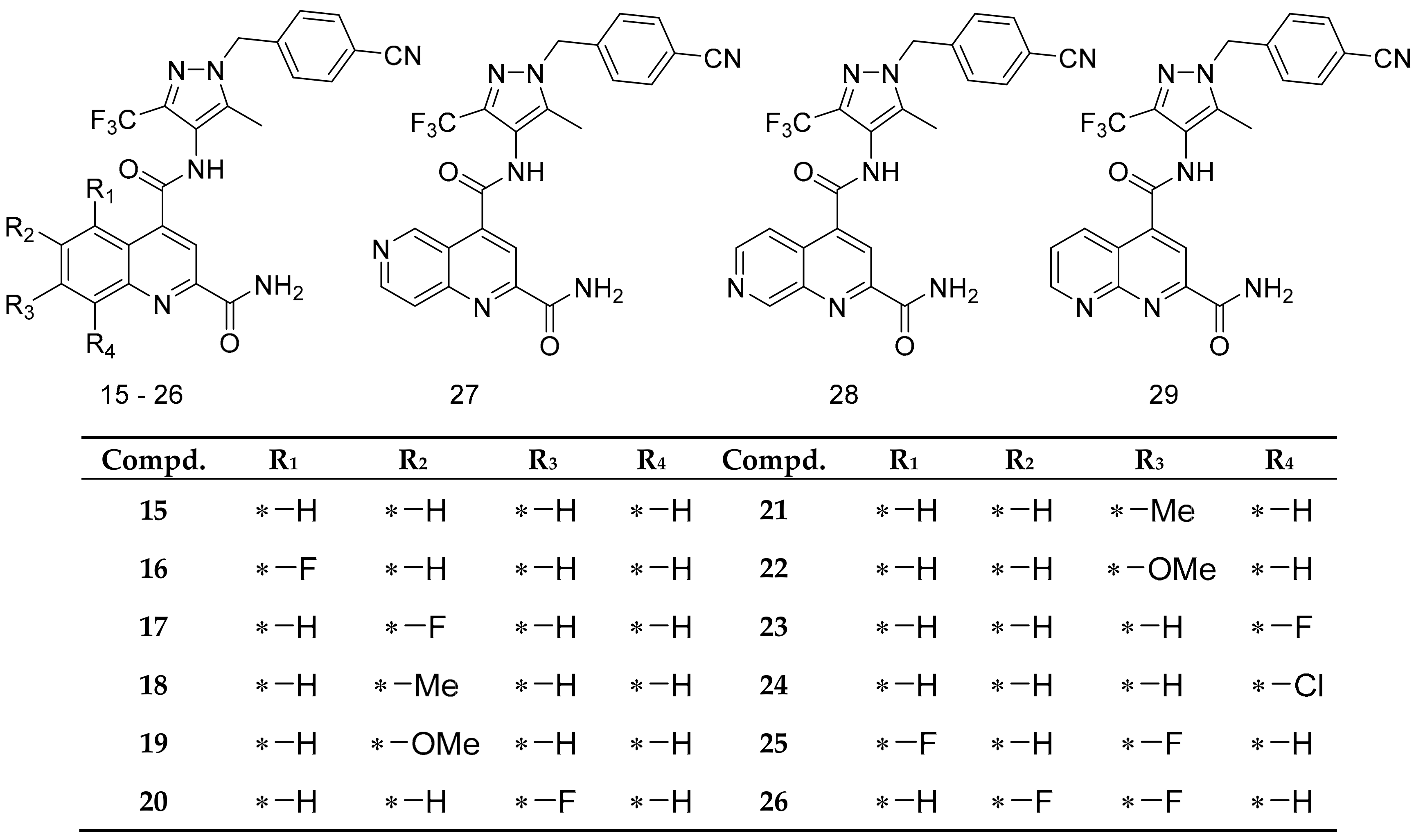
2.2. Docking Scores and Validation
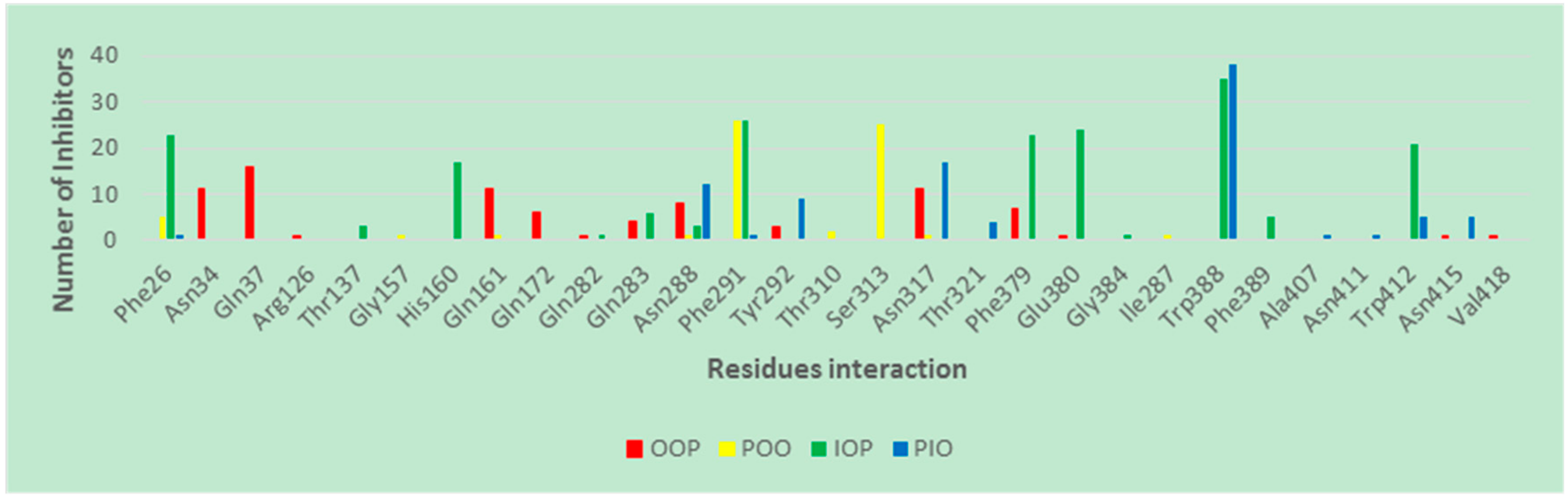
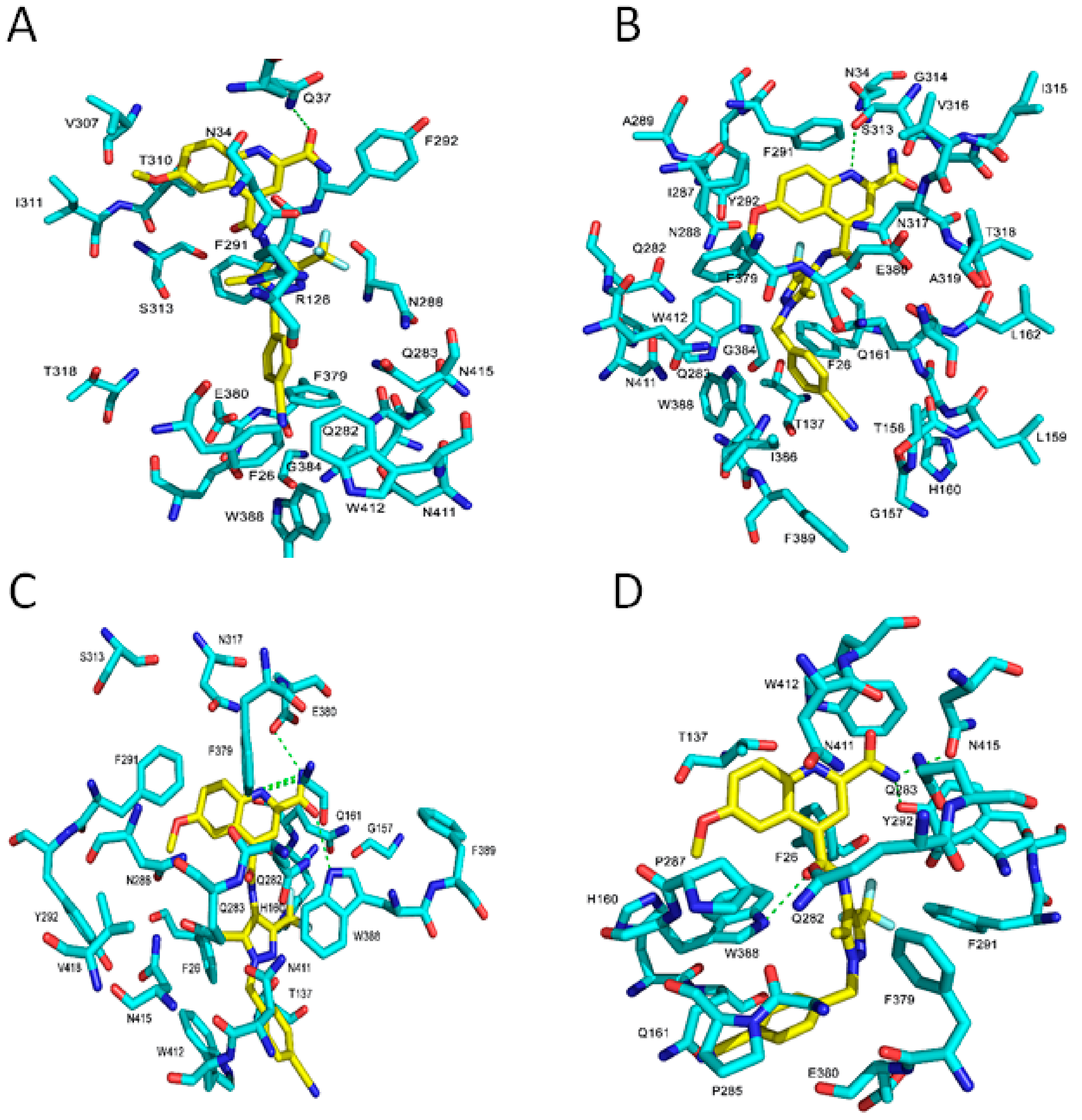
2.3. Binding Interaction of GLUT1 Inhbitors
3. Materials and Methods
3.1. Homology Modeling and Preparation of Model Proteins
3.2. Ligands Sources, Prepration, and Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mueckler, M.; Thorens, B. The SLC2 (GLUT) family of membrane transporters. Mol. Asp. Med. 2013, 34, 121–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pao, S.S.; Paulsen, I.T.; Saier, M.H., Jr. Major facilitator superfamily. Microbiol. Mol. Biol. Rev. 1998, 62, 1–34. [Google Scholar] [PubMed]
- Yan, N. Structural advances for the major facilitator superfamily (MFS) transporters. Trends Biochem. Sci. 2013, 38, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Deng, D.; Sun, P.; Yan, C.; Ke, M.; Jiang, X.; Xiong, L.; Ren, W.; Hirata, K.; Yamamoto, M.; Fan, S.; et al. Molecular basis of ligand recognition and transport by glucose transporters. Nature 2015, 526, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Deng, D.; Xu, C.; Sun, P.; Wu, J.; Yan, C.; Hu, M.; Yan, N. Crystal structure of the human glucose transporter GLUT1. Nature 2014, 510, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Rumsey, S.C.; Kwon, O.; Xu, G.W.; Burant, C.F.; Simpson, I.; Levine, M. Glucose transporter isoforms GLUT1 and GLUT3 transport dehydroascorbic acid. J. Biol. Chem. 1997, 272, 18982–18989. [Google Scholar] [CrossRef] [PubMed]
- Ung, P.M.; Song, W.; Cheng, L.; Zhao, X.; Hu, H.; Chen, L.; Schlessinger, A. Inhibitor Discovery for the Human GLUT1 from Homology Modeling and Virtual Screening. ACS Chem. Biol. 2016, 11, 1908–1916. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.Q.; Keating, A.F. Functional properties and genomics of glucose transporters. Curr. Genom. 2007, 8, 113–128. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Barron, C.C.; Bilan, P.J.; Tsakiridis, T.; Tsiani, E. Facilitative glucose transporters: Implications for cancer detection, prognosis and treatment. Metabolism 2015, 65, 124–139. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, T.; Oda, Y.; Seino, Y.; Yamamoto, T.; Inagaki, N.; Yano, H.; Imura, H.; Shigemoto, R.; Kikuchi, H. Distribution of the glucose transporters in human brain tumors. Cancer Res. 1992, 52, 3972–3979. [Google Scholar] [PubMed]
- Brown, R.S.; Wahl, R.L. Overexpression of Glut-1 glucose transporter in human breast cancer. An immunohistochemical study. Cancer 1993, 72, 2979–2985. [Google Scholar] [CrossRef]
- Sasaki, H.; Shitara, M.; Yokota, K.; Hikosaka, Y.; Moriyama, S.; Yano, M.; Fujii, Y. Overexpression of GLUT1 correlates with kras mutations in lung carcinomas. Mol. Med. Rep. 2012, 5, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Nagase, Y.; Takata, K.; Moriyama, N.; Aso, Y.; Murakami, T.; Hirano, H. Immunohistochemical localization of glucose transporters in human renal cell carcinoma. J. Urol. 1995, 153, 798–801. [Google Scholar] [CrossRef]
- Cai, Y.; Zhai, J.J.; Feng, B.B.; Duan, X.Z.; He, X.J. Expression of glucose transporter protein 1 and p63 in serous ovarian tumor. J. Obstet. Gynaecol. Res. 2014, 40, 1925–1930. [Google Scholar] [CrossRef]
- Reinicke, K.; Sotomayor, P.; Cisterna, P.; Delgado, C.; Nualart, F.; Godoy, A. Cellular distribution of Glut-1 and Glut-5 in benign and malignant human prostate tissue. J. Cell. Biochem. 2012, 113, 553–562. [Google Scholar] [CrossRef]
- Shen, Y.; Arbman, G.; Olsson, B.; Sun, X.F. Overexpression of GLUT1 in colorectal cancer is independently associated with poor prognosis. Int. J. Biol. Markers 2011, 26, 166–172. [Google Scholar] [CrossRef]
- Sheu, J.J.; Guan, B.; Tsai, F.J.; Hsiao, E.Y.-T.; Chen, C.-M.; Seruca, R.; Wang, T.-L.; Shih, I.-M. Mutant BRAF induces DNA strand breaks, activates DNA damage response pathway, and up-regulates glucose transporter-1 in nontransformed epithelial cells. Am. J. Pathol. 2012, 180, 1179–1188. [Google Scholar] [CrossRef]
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.; Lee, L.A.; Dang, C.V. Deregulation of glucose transporter 1 and glycolytic gene expression by c-myc. J. Biol. Chem. 2000, 275, 21797–21800. [Google Scholar] [CrossRef]
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Wincewicz, A.; Sulkowska, M.; Koda, M.; Sulkowski, S. Clinicopathological significance and linkage of the distribution of HIF-1alpha and GLUT-1 in human primary colorectal cancer. Pathol. Oncol. Res. 2007, 13, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.B.; Hay, N. Akt, hexokinase, mTOR: Targeting cellular energy metabolism for cancer therapy. Drug Discov. Today Dis. Mech. 2005, 2, 239–246. [Google Scholar] [CrossRef]
- Xu, R.H.; Pelicano, H.; Zhou, Y.; Carew, J.S.; Feng, L.; Bhalla, K.N.; Keating, M.J.; Huang, P. Inhibition of glycolysis in cancer cells: A novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005, 65, 613–621. [Google Scholar] [PubMed]
- Gerhart, D.Z.; Broderius, M.A.; Borson, N.D.; Drewes, L.R. Neurons and microvessels express the brain glucose transporter protein GLUT3. Proc. Natl. Acad. Sci. USA 1992, 89, 733–737. [Google Scholar] [CrossRef]
- Mantych, G.J.; James, D.E.; Chung, H.D.; Devaskar, S.U. Cellular localization and characterization of glut 3 glucose transporter isoform in human brain. Endocrinology 1992, 131, 1270–1278. [Google Scholar] [CrossRef]
- Patching, S.G. Glucose transporters at the blood-brain barrier: Function, regulation and gateways for drug delivery. Mol. Neurobiol. 2017, 54, 1046–1077. [Google Scholar] [CrossRef]
- Simpson, I.A.; Dwyer, D.; Malide, D.; Moley, K.H.; Travis, A.; Vannucci, S.J. The facilitative glucose transporter GLUT3: 20 years of distinction. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E242–E253. [Google Scholar] [CrossRef] [Green Version]
- Salas, M.; Obando, P.; Ojeda, L.; Ojeda, P.; Pérez, A.; Vargas-Uribe, M.; I Rivas, C.; Vera, J.C.; Reyes, A.M. Resolution of the direct interaction with and inhibition of the human GLUT1 hexose transporter by resveratrol from its effect on glucose accumulation. Am. J. Physiol. Cell Physiol. 2013, 305, C90–C99. [Google Scholar] [CrossRef] [Green Version]
- Martin, H.J.; Kornmann, F.; Fuhrmann, G.F. The inhibitory effects of flavonoids and antiestrogens on the Glut1 glucose transporter in human erythrocytes. Chem. Biol. Interact. 2003, 146, 225–235. [Google Scholar] [CrossRef]
- Cho, S.J.; Moon, J.S.; Lee, C.M.; Choi, A.M.; Stout-Delgado, H.W. Glucose Transporter 1-Dependent Glycolysis Is Increased during Aging-Related Lung Fibrosis, and Phloretin Inhibits Lung Fibrosis. Am. J. Respir. Cell Mol. Biol. 2017, 56, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, K.; Finer-Moore, J.S.; Pedersen, B.P.; Caboni, L.; Waight, A.; Hillig, R.C.; Bringmann, P.; Heisler, I.; Müller, T.; Siebeneicher, H.; et al. Mechanism of inhibition of human glucose transporter GLUT1 is conserved between cytochalasin B and phenylalanine amides. Proc. Natl. Acad. Sci. USA 2016, 113, 4711–4716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Cao, Y.; Zhang, W.; Bergmeier, S.; Qian, Y.; Akbar, H.; Colvin, R.; Ding, J.; Tong, L.; Wu, S.; et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth In Vitro and In Vivo. Mol. Cancer Ther. 2012, 11, 1672–1682. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.A.; Sutphin, P.D.; Nguyen, P.; Turcotte, S.; Lai, E.W.; Banh, A.; Reynolds, G.E.; Chi, J.-T.; Wu, J.; Solow-Cordero, D.E.; et al. Targeting GLUT1 and the warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci. Transl. Med. 2011, 3, 94ra70. [Google Scholar] [CrossRef] [PubMed]
- Siebeneicher, H.; Bauser, M.; Buchmann, B.; Heisler, I.; Müller, T.; Neuhaus, R.; Rehwinkel, H.; Telser, J.; Zorn, L. Identification of novel GLUT inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 1732–1737. [Google Scholar] [CrossRef] [PubMed]
- Siebeneicher, H.; Cleve, A.; Rehwinkel, H.; Neuhaus, R.; Heisler, I.; Müller, T.; Bauser, M.; Buchmann, B. Identification and optimization of the first highly selective GLUT1 inhibitor BAY-876. ChemMedChem 2016, 11, 2261–2271. [Google Scholar] [CrossRef] [PubMed]
- DeFelice, L.J. Transporter structure and mechanism. Trends Neurosci. 2004, 27, 352–359. [Google Scholar] [CrossRef]
- Shimamura, T.; Weyand, S.; Beckstein, O.; Rutherford, N.G.; Hadden, J.M.; Sharples, D.; Sansom, M.S.P.; Iwata, S.; Henderson, P.J.F.; Cameron, A.D.; et al. Molecular basis of alternating access membrane transport by the sodium-hydantoin transporter Mhp1. Science 2010, 328, 470–473. [Google Scholar] [CrossRef]
- Wisedchaisri, G.; Park, M.S.; Iadanza, M.G.; Zheng, H.; Gonen, T. Proton-coupled sugar transport in the prototypical major facilitator superfamily protein XylE. Nat. Commun. 2014, 5, 4521. [Google Scholar] [CrossRef] [Green Version]
- Radestock, S.; Forrest, L.R. The alternating-access mechanism of MFS transporters arises from inverted-topology repeats. J. Mol. Biol. 2011, 407, 698–715. [Google Scholar] [CrossRef]
- Majumdar, D.S.; Smirnova, I.; Kasho, V.; Nir, E.; Kong, X.; Weiss, S.; Kaback, H.R. Single-molecule FRET reveals sugar-induced conformational dynamics in LacY. Proc. Natl. Acad. Sci. USA 2007, 104, 12640–12645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smirnova, I.; Kasho, V.; Kaback, H.R. Lactose permease and the alternating access mechanism. Biochemistry 2011, 50, 9684–9693. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zeng, X.; Yan, C.; Sun, X.; Gong, X.; Rao, Y.; Yan, N. Crystal structure of a bacterial homologue of glucose transporters GLUT1-4. Nature 2012, 490, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Quistgaard, E.M.; Löw, C.; Moberg, P.; Trésaugues, L.; Nordlund, P. Structural basis for substrate transport in the GLUT-homology family of monosaccharide transporters. Nat. Struct. Mol. Biol. 2013, 20, 766–768. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.; Bergner, A.; Schwede, T. Modelling three-dimensional protein structures for applications in drug design. Drug Discov. Today 2014, 19, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Hevener, K.E.; Zhao, W.; Ball, D.M.; Babaoglu, K.; Qi, J.; White, S.W.; Lee, R.E. Validation of molecular docking programs for virtual screening against dihydropteroate synthase. J. Chem. Inf. Model. 2009, 49, 444–460. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Glen, R.C. A discussion of measures of enrichment in virtual screening: Comparing the information content of descriptors with increasing levels of sophistication. J. Chem. Inf. Model. 2005, 45, 1369–1375. [Google Scholar] [CrossRef]
- Katagiri, H.; Asano, T.; Ishihara, H.; Lin, J.L.; Inukai, K.; Shanahan, M.F.; Tsukuda, K.; Kikuchi, M.; Yazaki, Y.; Oka, Y. Role of tryptophan-388 of GLUT1 glucose transporter in glucose-transport activity and photoaffinity-labelling with forskolin. Biochem. J. 1993, 291, 861–867. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.C.; Strube, M.; Leingang, K.; Keller, K.; Mueckler, M.M. Amino acid substitutions at tryptophan 388 and tryptophan 412 of the HepG2 (Glut1) glucose transporter inhibit transport activity and targeting to the plasma membrane in xenopus oocytes. J. Biol. Chem. 1992, 267, 7770–7776. [Google Scholar]
- Kasahara, T.; Kasahara, M. Tryptophan 388 in putative transmembrane segment 10 of the rat glucose transporter Glut1 is essential for glucose transport. J. Biol. Chem. 1998, 273, 29113–29117. [Google Scholar] [CrossRef]
- Kumar, H.; Kasho, V.; Smirnova, I.; Finer-Moore, J.S.; Kaback, H.R.; Stroud, R.M. Structure of sugar-bound LacY. Proc. Natl. Acad. Sci. USA 2014, 111, 1784–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iancu, C.V.; Zamoon, J.; Woo, S.B.; Aleshin, A.; Choe, J. Crystal structure of a glucose/H+ symporter and its mechanism of action. Proc. Natl. Acad. Sci. USA 2013, 110, 17862–17867. [Google Scholar] [CrossRef] [PubMed]
- Lešnik, S.; Miller, B.T.; Štular, T.; Woodcock, H.L.; Brooks, B.R.; Janežič, D.; Konc, J. ProBiS-CHARMMing: Web interface for prediction and optimization of ligands in protein binding sites. J. Chem. Inf. Model. 2015, 55, 2308–2314. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- The Molecular Operating Environment (MOE). Chemical Computing Group Inc.: Montreal, QC, Canada, 2018.
- Bergeret, F.; Gavalda, S.; Chalut, C.; Malaga, W.; Quémard, A.; Pedelacq, J.-D.; Daffé, M.; Guilhot, C.; Mourey, L.; Bon, C. Biochemical and structural study of the atypical acyltransferase domain from the mycobacterial polyketide synthase Pks13. J. Biol. Chem. 2012, 287, 33675–33690. [Google Scholar] [CrossRef]
- Kiburu, I.N.; LaRonde-LeBlanc, N. Interaction of Rio1 kinase with toyocamycin reveals a conformational switch that controls oligomeric state and catalytic activity. PLoS ONE 2012, 7, e37371. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Darden, T.A.; Cheatham, T.E., III; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; et al. Amber 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Schrödinger Suite 2019-1. Protein Preparation Wizard, Maestro, MacroModel, LigPrep, EPik, Glide Grid Generation, and Glide; Schrödinger, LLC: New York, NY, USA, 2019. [Google Scholar]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- NCI Open Database Compounds, Release 3; National Cancer Institute, National Institutes of Health: Bethseda, MD, September 2003. Available online: http://Cactus.Nci.Nih.Gov/Download/Nci (accessed on 18 August 2008).
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Guariento, S.; Bruno, O.; Fossa, P.; Cichero, E. New insights into PDE4B inhibitor selectivity: CoMFA analyses and molecular docking studies. Mol. Divers. 2016, 20, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Franchini, S.; Manasieva, L.I.; Sorbi, C.; Battisti, U.M.; Fossa, P.; Cichero, E.; Denora, N.; Iacobazzi, R.M.; Cilia, A.; Pirona, L.; et al. Synthesis, biological evaluation and molecular modeling of 1-oxa-4-thiaspiro- and 1,4-dithiaspiro [4.5] decane derivatives as potent and selective 5-HT1A receptor agonists. Eur. J. Med. Chem. 2017, 125, 435–452. [Google Scholar] [CrossRef]
- Sabbah, D.A.; Zhong, H.A. Modeling the protonation states of β-secretase binding pocket by molecular dynamics simulations and docking studies. J. Mol. Graph. Model. 2016, 68, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, D.A.; Vennerstrom, J.L.; Zhong, H. Docking studies on isoform-specific inhibition of phosphoinositide-3-kinases. J. Chem. Inf. Model. 2010, 50, 1887–1898. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | IC50 (uM) a | ∆Gexp b | 5EQG | ΔΔG(5EQG) | HM_4GBZ | HM_4ZWC | HM_4JA3 |
|---|---|---|---|---|---|---|---|
| 1 | 0.11 | −9.49 | −9.39 | −0.10 | NA c | −5.84 | NA c |
| 2 | 0.267 | −8.97 | −10.61 | 1.64 | −11.63 | −11.11 | −10.53 |
| 3 | 0.14 | −9.35 | −11.54 | 2.19 | −9.46 | −10.26 | −10.84 |
| 4 | 0.11 | −9.49 | −9.53 | 0.04 | −9.05 | −8.88 | −6.64 |
| 5 | 0.006 | −11.22 | −9.39 | −1.83 | −7.22 | −7.71 | −7.96 |
| 6 | 0.004 | −11.46 | −9.24 | −2.22 | −7.45 | −9.11 | −8.23 |
| 7 | 0.003 | −11.63 | −9.45 | −2.18 | −9.77 | −10.23 | −8.49 |
| 8 | 0.008 | −11.05 | −9.68 | −1.37 | −9.61 | −10.16 | −8.35 |
| 9 | 0.087 | −9.63 | −9.43 | −0.20 | −7.39 | −8.75 | −8.73 |
| 10 | 0.002 | −11.87 | −9.24 | −2.63 | −8.83 | −8.10 | −8.17 |
| 11 | 0.004 | −11.46 | −10.62 | −0.84 | −10.34 | −9.01 | −10.23 |
| 12 | 0.032 | −10.22 | −10.30 | 0.08 | −9.93 | −8.74 | −10.12 |
| 13 | 0.005 | −11.32 | −10.96 | −0.36 | −9.36 | −9.18 | −9.68 |
| 14 | 0.026 | −10.35 | −10.38 | 0.03 | −8.99 | −9.10 | −9.42 |
| 15 | 0.006 | −11.22 | −9.49 | −1.73 | −9.47 | −9.17 | −9.49 |
| 16 | 0.003 | −11.63 | −11.47 | −0.16 | −10.22 | −9.26 | −9.34 |
| 17 | 0.005 | −11.32 | −9.34 | −1.98 | −10.36 | −9.51 | −10.99 |
| 18 | 0.007 | −11.12 | −10.29 | −0.83 | −10.88 | −9.02 | −9.32 |
| 19 | 0.0003 | −12.99 | −9.61 | −3.38 | −10.91 | −9.77 | −10.78 |
| 20 | 0.002 | −11.87 | −9.70 | −2.17 | −9.71 | −9.51 | −10.44 |
| 21 | 0.008 | −11.05 | −8.92 | −2.13 | −10.95 | −10.03 | −9.78 |
| 22 | 0.024 | −10.39 | −9.52 | −0.87 | −9.12 | −9.72 | −11.13 |
| 23 | 0.005 | −11.32 | −10.32 | −1.00 | −10.24 | −9.84 | −10.00 |
| 24 | 0.003 | −11.63 | −10.34 | −1.29 | −10.94 | −9.96 | −9.68 |
| 25 | 0.0005 | −12.69 | −11.37 | −1.32 | −10.46 | −9.04 | −10.77 |
| 26 | 0.002 | −11.87 | −7.42 | −4.45 | −10.11 | −9.39 | −11.14 |
| 27 | 0.014 | −10.71 | −10.82 | 0.11 | −10.20 | −10.56 | −9.65 |
| 28 | 0.01 | −10.91 | −10.49 | −0.42 | −9.20 | −9.42 | −9.88 |
| 29 | 0.045 | −10.02 | −9.33 | −0.69 | −10.25 | −9.61 | −9.26 |
| 30 | 0.004 | −11.46 | −9.48 | −1.98 | −10.02 | −10.45 | −10.28 |
| 31 | 0.76 | −8.35 | −9.12 | 0.77 | −4.47 | −9.53 | −8.30 |
| 32 | 0.007 | −11.12 | −9.25 | −1.87 | −9.83 | −9.23 | −10.18 |
| 33 | 0.074 | −9.73 | −9.54 | −0.19 | −5.26 | −10.13 | −10.32 |
| 34 | 0.92 | −8.23 | −10.62 | 2.39 | −9.99 | −9.42 | −10.60 |
| 35 | 0.34 | −8.82 | −11.77 | 2.95 | −12.23 | −11.43 | −12.81 |
| 36 | 0.0009 | −12.34 | −9.02 | −3.32 | −9.99 | −9.18 | −9.65 |
| 37 | 0.003 | −11.63 | −9.70 | −1.93 | −9.75 | −9.53 | −8.38 |
| 38 | 0.007 | −11.12 | −8.89 | −2.23 | −9.81 | −8.92 | −9.61 |
| 39 | 0.007 | −11.12 | −10.36 | −0.76 | −9.83 | −8.64 | −9.40 |
| 40 | 0.003 | −11.63 | −9.45 | −2.18 | −9.84 | −10.10 | −10.28 |
| 41 | 0.007 | −11.12 | −10.09 | −1.03 | −8.71 | −10.13 | −9.73 |
| 42 | 0.009 | −10.98 | −11.28 | 0.30 | −10.07 | −10.32 | −10.81 |
| 43 | 0.005 | −11.32 | −10.58 | −0.74 | −9.91 | −10.01 | −9.84 |
| 44 | 0.0009 | −12.34 | −9.59 | −2.75 | −9.20 | −9.38 | −10.89 |
| 5EQG (IOP) | 4GBZ (POO) | 4ZWC (OOP) | 4JA3 (PIO) | |
|---|---|---|---|---|
| ∆∆G | −0.97 | −1.37 | −1.44 | −1.16 |
| MAE | 1.45 | 1.75 | 1.82 | 1.66 |
| RMSE | 1.79 | 2.07 | 2.05 | 1.92 |
| Model | EF (1%) | EF (5%) | EF (10%) |
|---|---|---|---|
| 5EQG | 10.45 | 8.51 | 7.76 |
| 4GBZ | 4.24 | 3.30 | 4.63 |
| 4ZWC | 8.35 | 8.05 | 7.06 |
| 4JA3 | 6.37 | 8.02 | 6.72 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almahmoud, S.; Wang, X.; Vennerstrom, J.L.; Zhong, H.A. Conformational Studies of Glucose Transporter 1 (GLUT1) as an Anticancer Drug Target. Molecules 2019, 24, 2159. https://doi.org/10.3390/molecules24112159
Almahmoud S, Wang X, Vennerstrom JL, Zhong HA. Conformational Studies of Glucose Transporter 1 (GLUT1) as an Anticancer Drug Target. Molecules. 2019; 24(11):2159. https://doi.org/10.3390/molecules24112159
Chicago/Turabian StyleAlmahmoud, Suliman, Xiaofang Wang, Jonathan L. Vennerstrom, and Haizhen A. Zhong. 2019. "Conformational Studies of Glucose Transporter 1 (GLUT1) as an Anticancer Drug Target" Molecules 24, no. 11: 2159. https://doi.org/10.3390/molecules24112159