Systematic Studies on the Protocol and Criteria for Selecting a Covalent Docking Tool


Abstract
:1. Introduction
- (1)
- classification of warhead chemotypes and the targets in receptors,
- (2)
- provision of a standard unbiased benchmark data set to test covalent docking tools objectively, and
- (3)
- elaboration of criteria used to measure the performance of covalent docking tools used in a warhead-receptor complex.
2. Results
2.1. Covalent Binding Reaction Classification
2.2. Evaluation of Covalent Docking Tools
- Precision. P(i,x), the precision for the ith docking tool (i ∈ MOE, GOLD, CovDock, ICM-Pro) using the xth RMSD measurement x ∈ (Best Scored Pose, Best Sampled Pose), measured for each covalent docking tool demonstrates different docking performances for different reaction classes and warheads. P(i,x) can be calculated with formula (1) as follows:where m is the total number of warhead types or the total number of receptor types, and n is the total number of co-crystal structures for a given warhead type, or a given receptor type. RMSD i,x,j,k is the RMSD value between the crystallographic ligand and the docked ligand pose produced by the ith docking tool with xth measurement for the kth co-crystal structure of the jth warhead type (the jth receptor type). Thus, a lower P(i,x) is better.
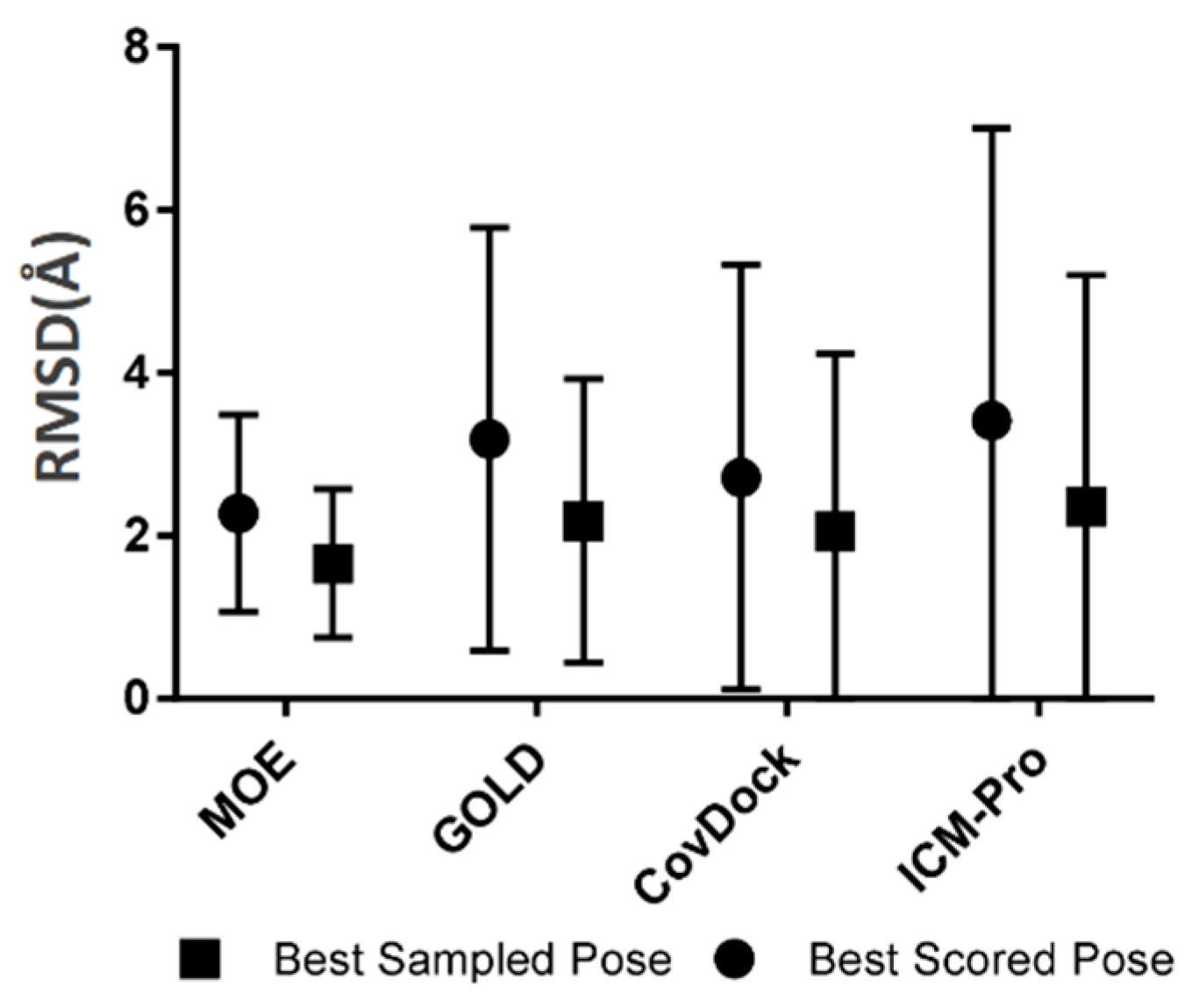
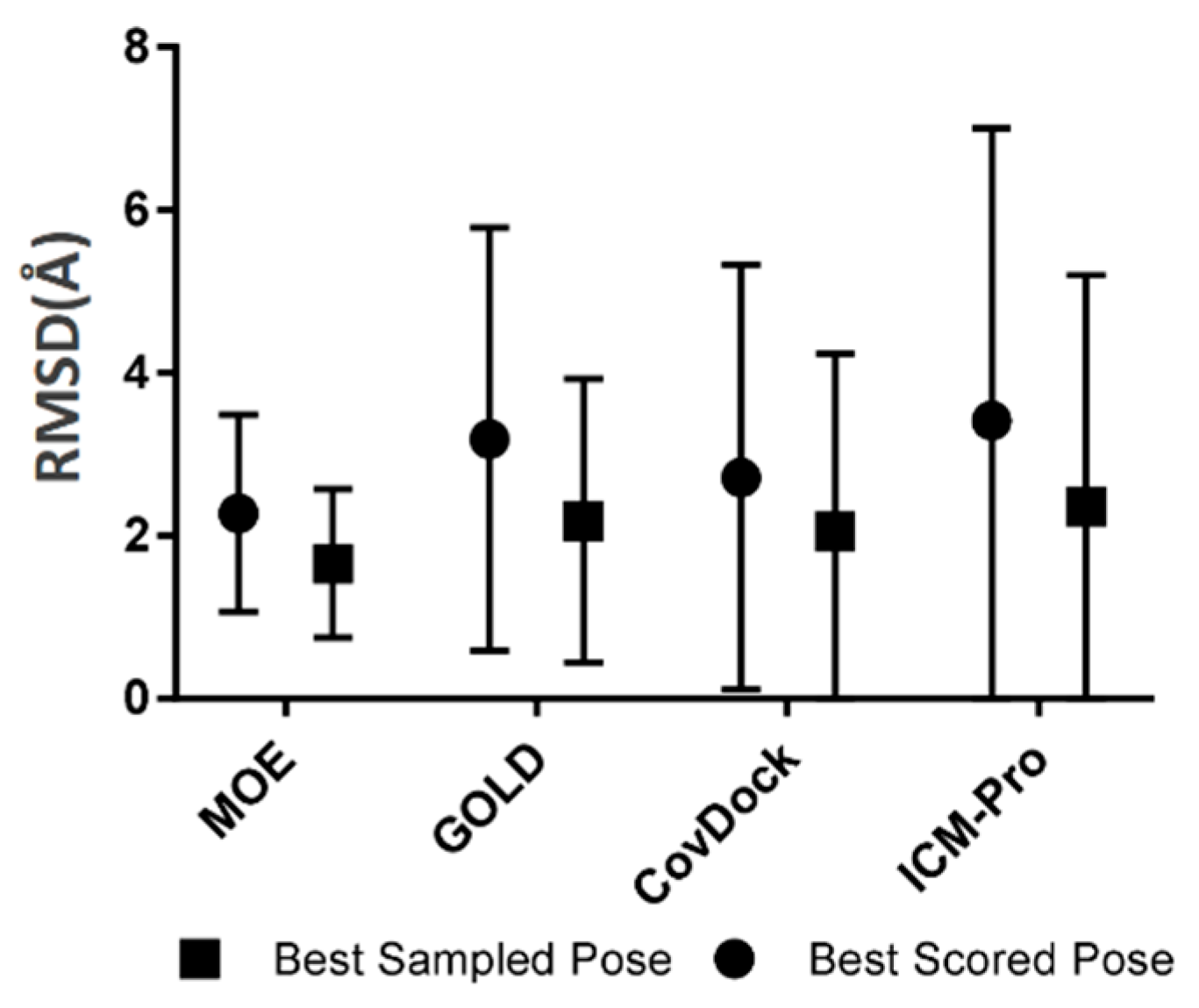
- Robustness. The robustness of a covalent docking tool is measured by the standard deviation of all RMSDs between native ligand poses and the docked poses covering all tested covalent binding complexes in the BCDE data set. The robustness analysis results for the four docking tools are depicted in Figure 4.
2.3. Profiling Covalent Docking Tools
- Pose deviation and success and failure counts: For a co-crystal structure, a docking tool can generate two RMSD values, Best Scored Pose, and Best Sampled Pose, denoted S1 and S2, respectively. Also, (S1–S2) is denoted as P-deviation, representing the difference between the two measurements of docking accuracies. For a given docking tool, if (S1 ≤ 2.0 Å) then it is considered as a success count (S-count); If (S1 > 2.0 Å) and (S2 > 2.0 Å) then it is considered as a failure count (F-count). The P-deviations, S-counts, and F-counts for the four evaluated docking tools have been computed and are listed in Table S13.
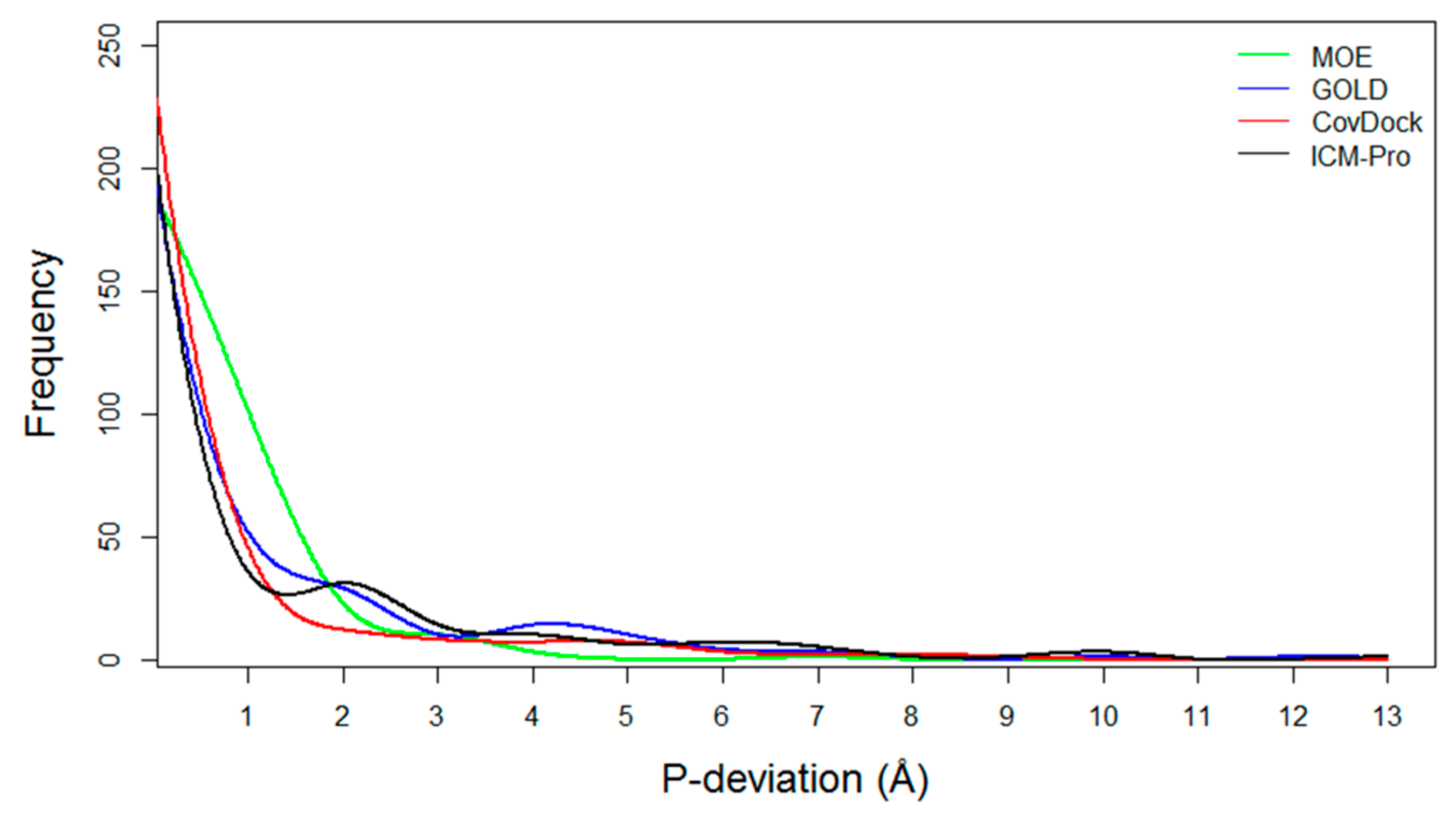
- P-deviation distributions. These distributions for all docking tools can be used to measure docking error ranges of the tools as shown in Figure 5 (the data are listed in Table S14). MOE (green) and CovDock (red) are closer to Gaussian distribution. GOLD (blue) and ICM-Pro (black) have two significant waves around 2Å and 4.5 Å suggesting that these tools have extraordinary errors around the 2Å and 4.5 Å ranges.
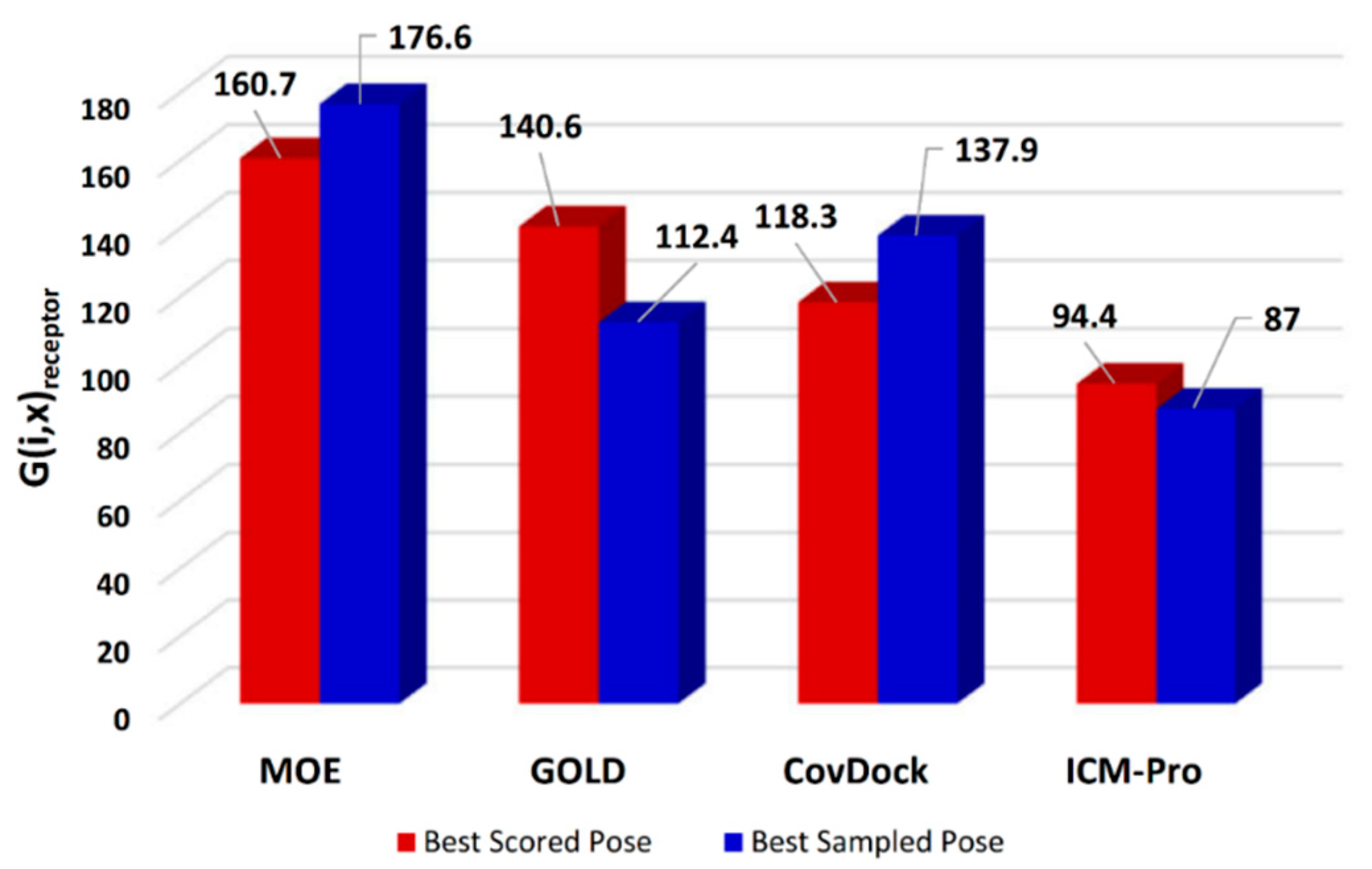
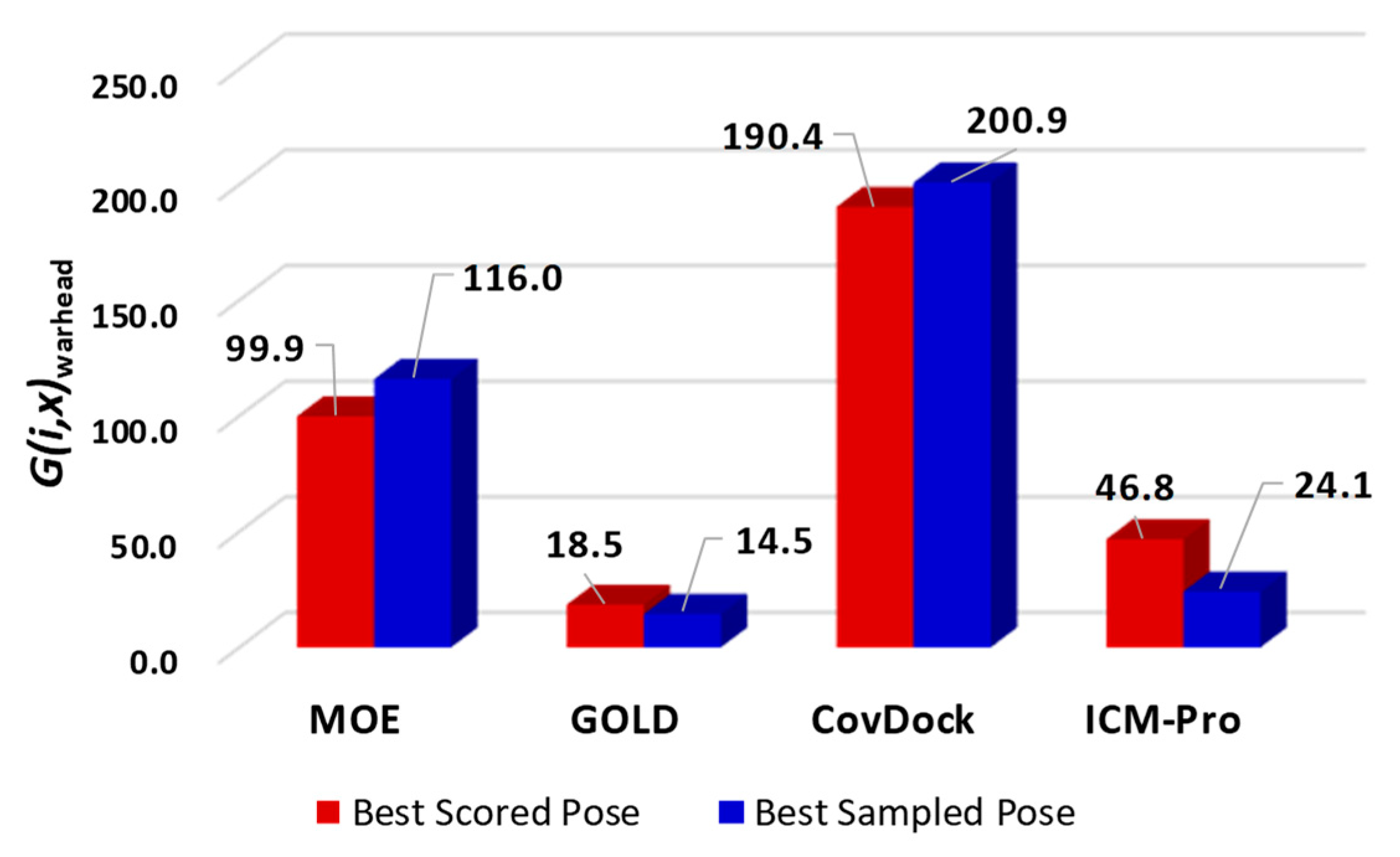
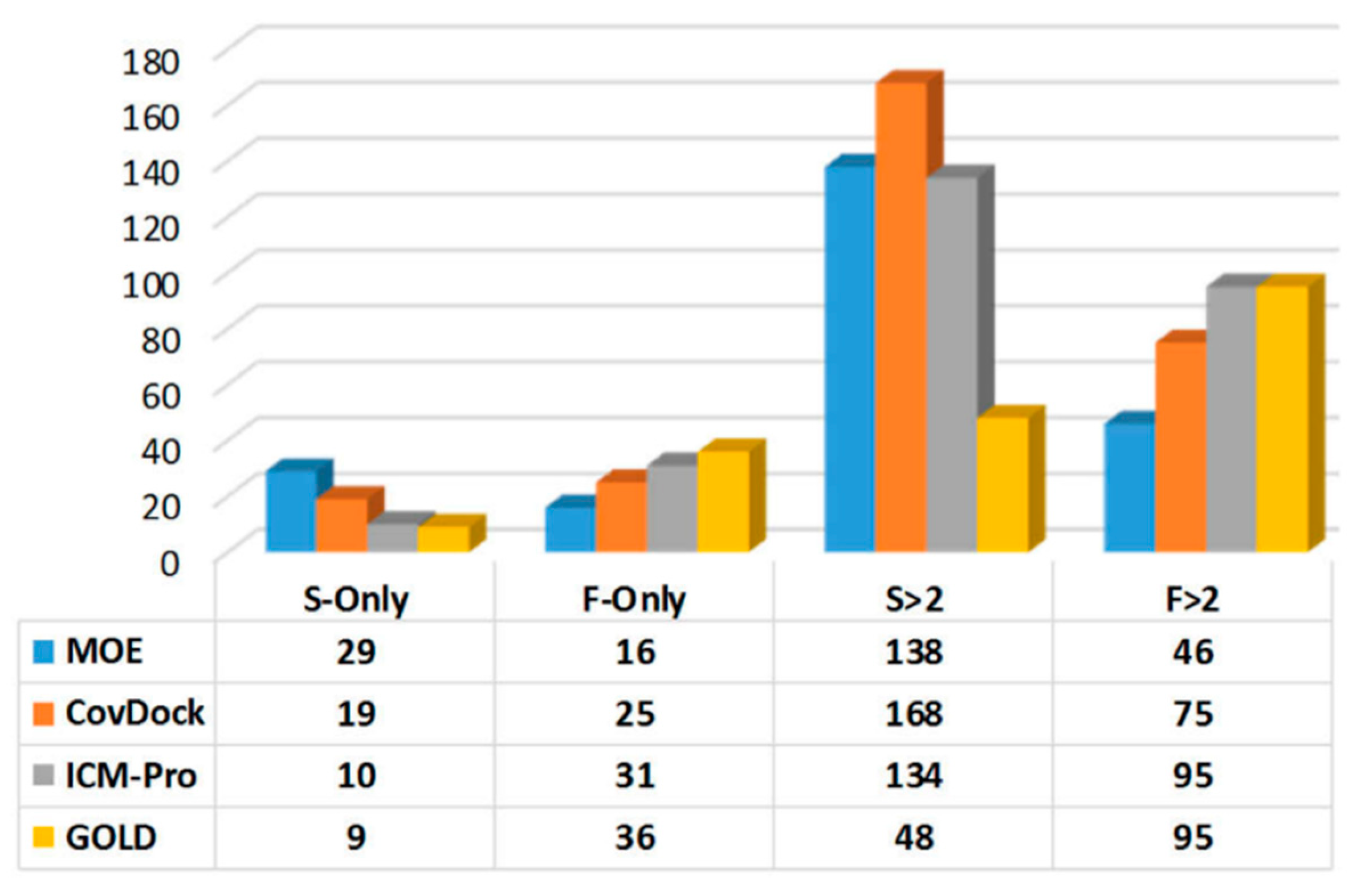
- Parameters with which to profile a docking tool. While analyzing the docking results, we revealed four parameters that can characterize a docking tool: (a) S-Only count—the number of cases in which only one docking tool predicted the successful poses, (b) F-Only count—the number of cases in which only one docking tool predicted the failing poses, (c) S > 2—the number of cases in which at least two docking tools predicted the successful poses, and (d) F > 2—the number of cases in which at least two docking tools predicted the failing poses. These parameters were derived from Table S13, and are listed in Table S15. The profiles for the four docking tools are depicted in Figure 6.
3. Discussion
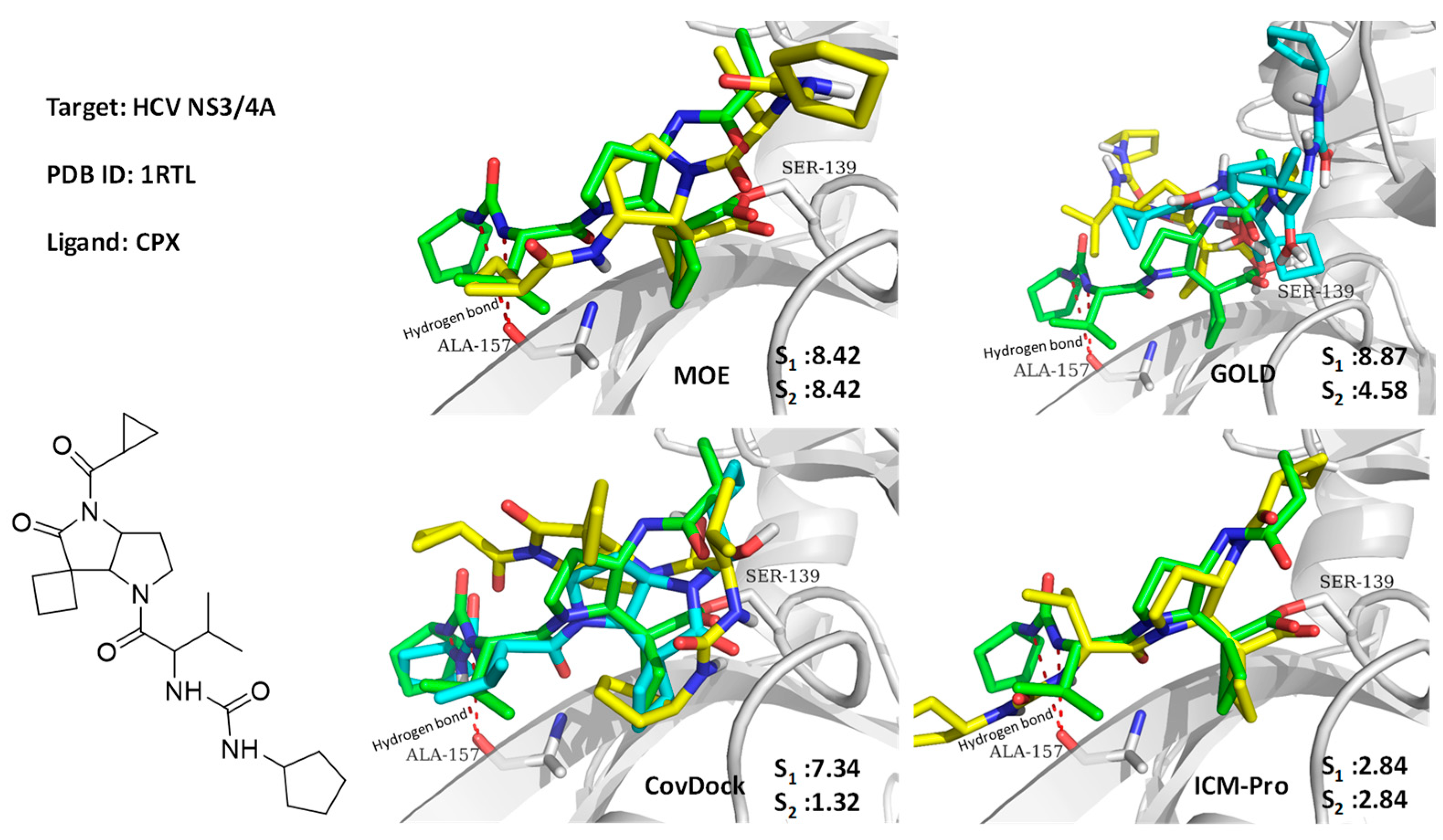
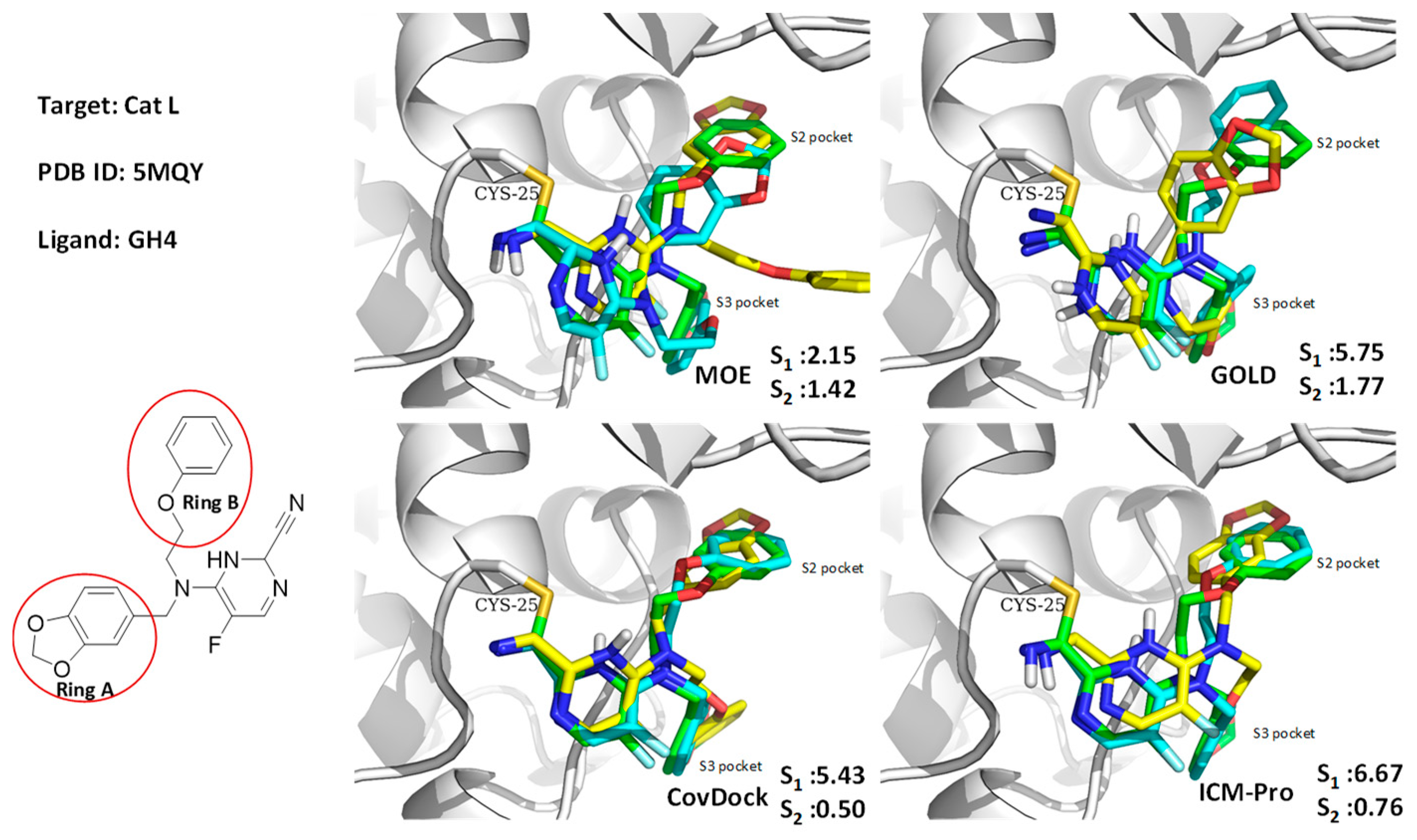
3.1. Case Studies of Abnormal Docking Results
3.1.1. Case Study 1
3.1.2. Case Study 2
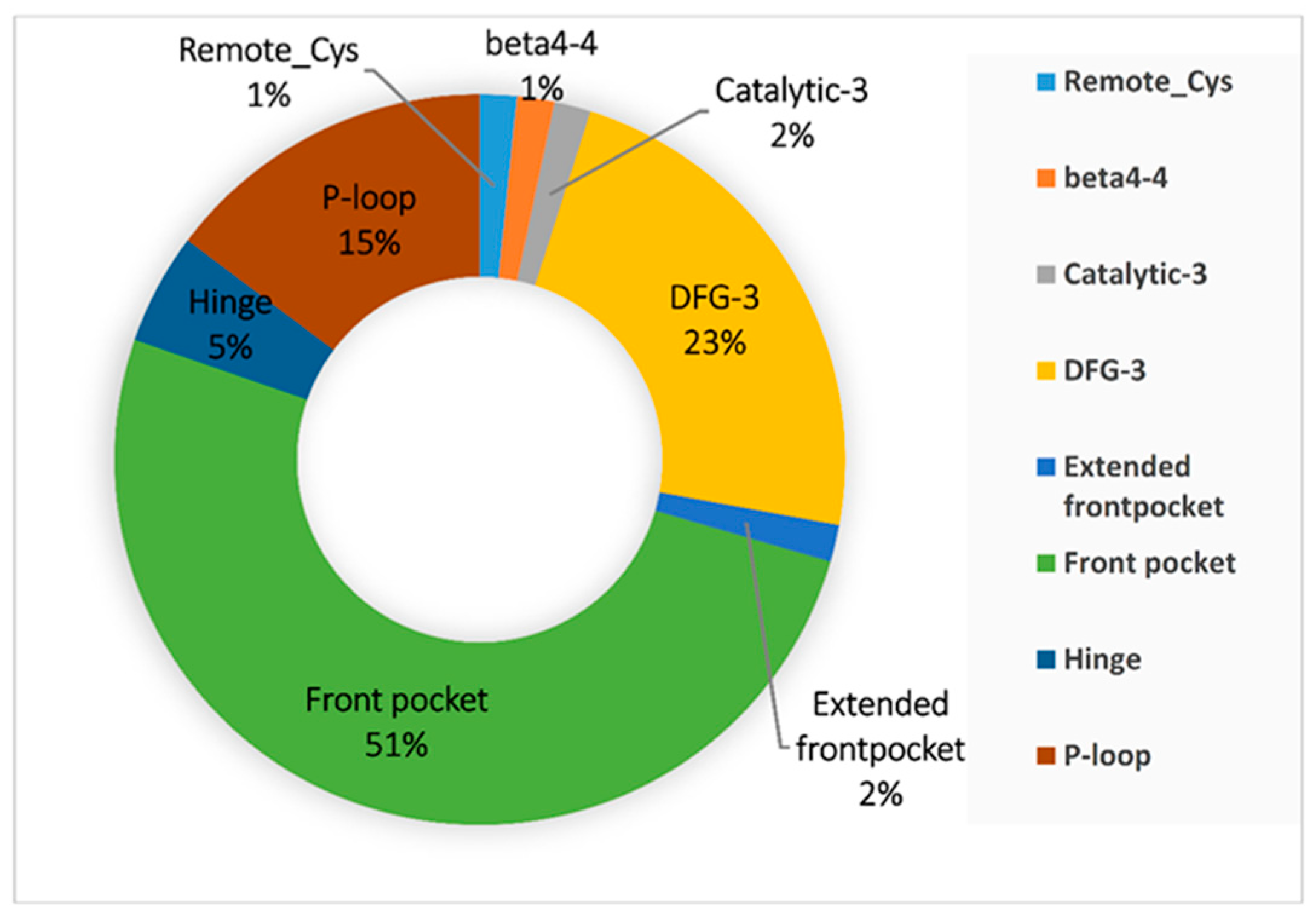
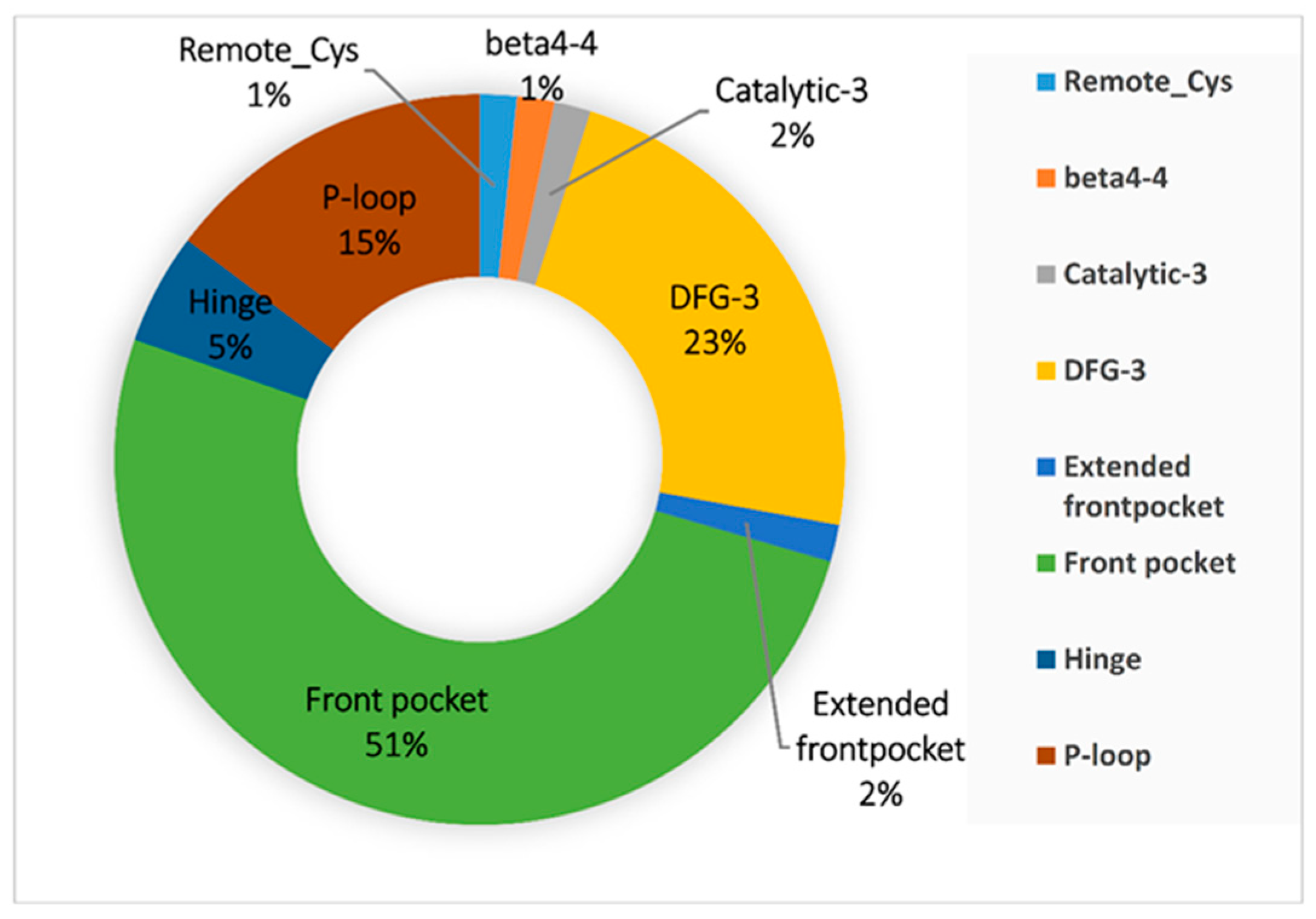
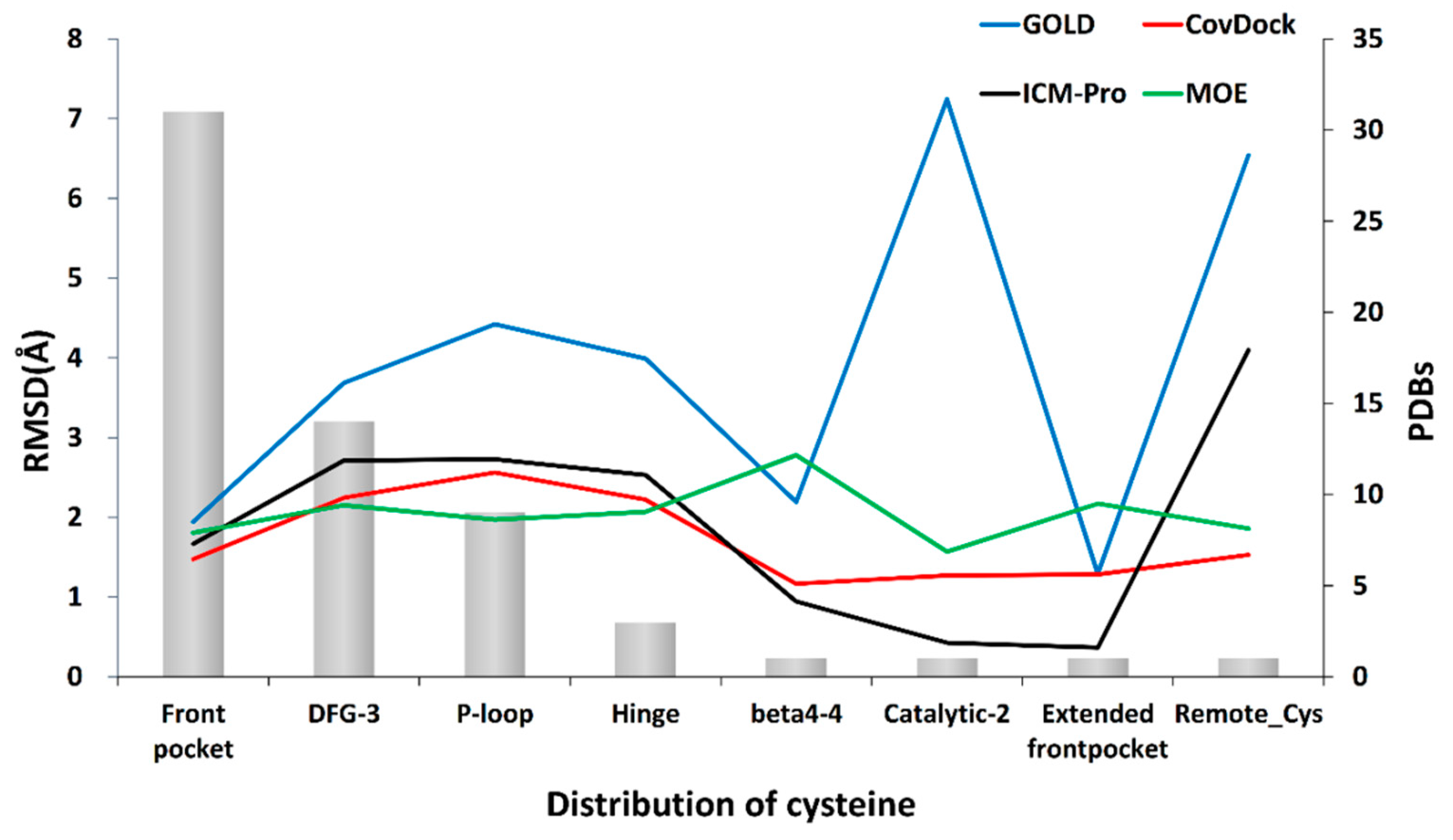
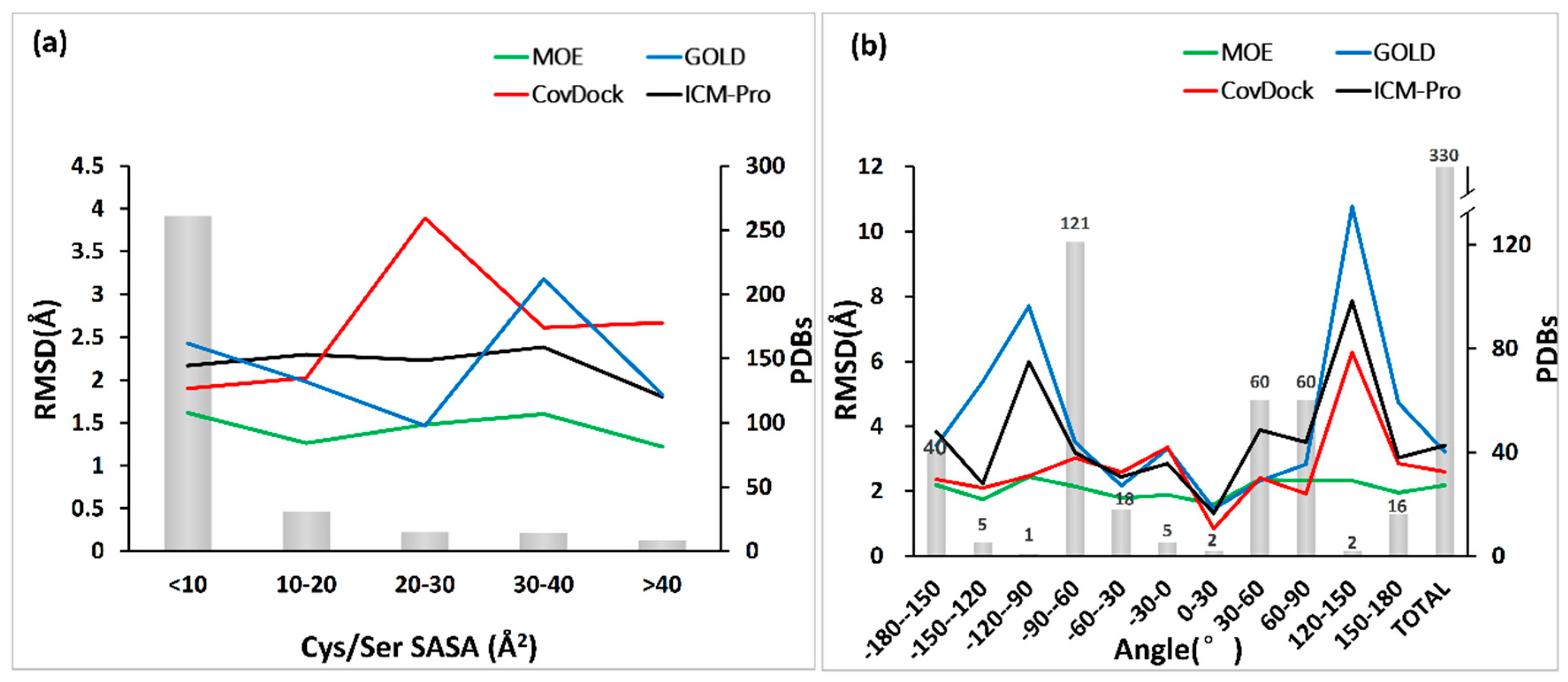
3.2. Binding Pocket Features and Docking Performance
4. Materials and Methods
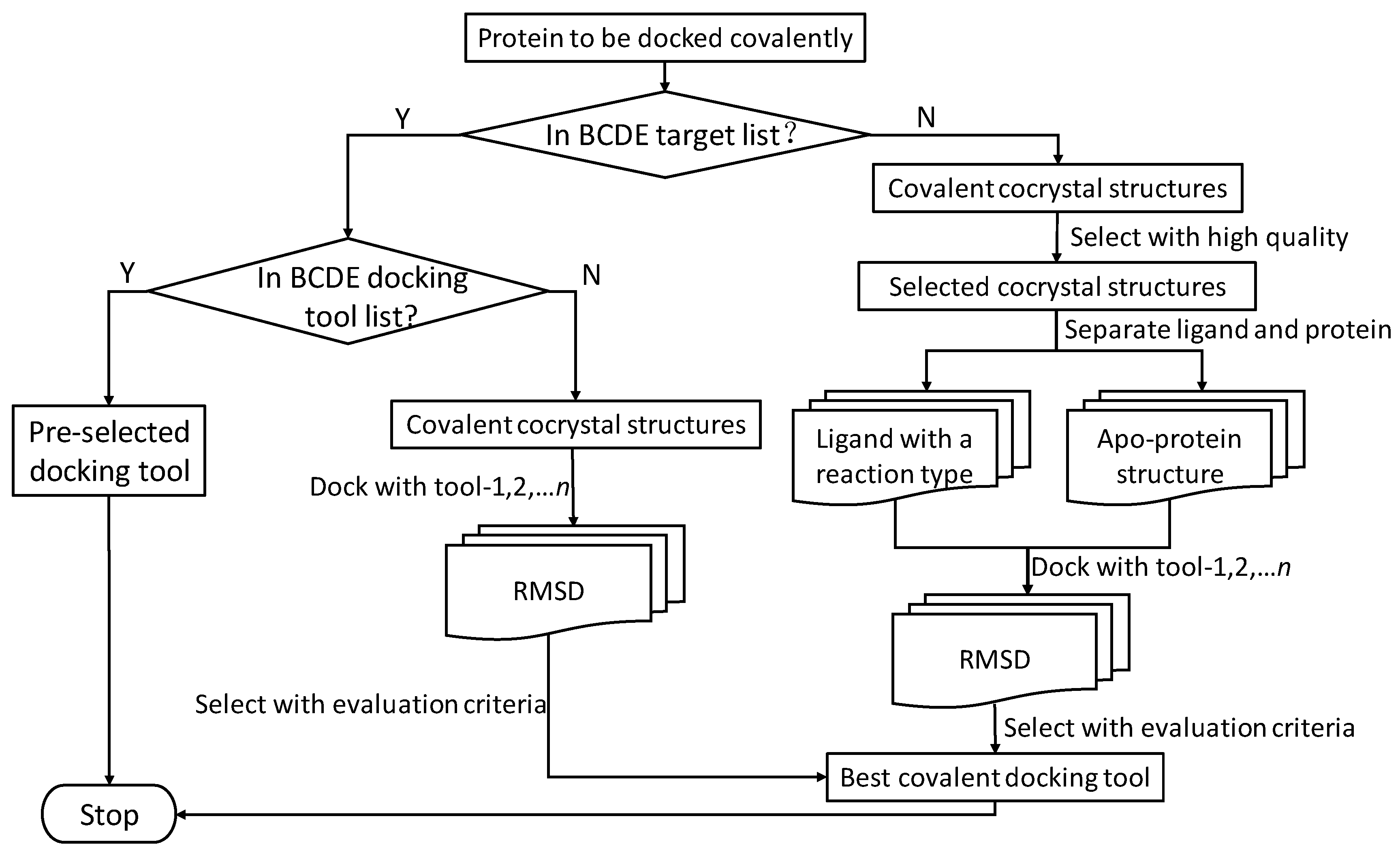
4.1. Benchmark Data for Covalent Docking Evaluation (BCDE)
- DNA complexes excluded
- proteins that have covalent bindings with ligands at cysteine and serine residues included
- ligand and receptor connected with a single bond included
- co-factors within 8 Å to the ligand excluded
- ligands with at least 5 non-hydrogen atoms and less than 30 rotatable bonds were included
4.2. Criteria for Evaluating Covalent Binding Tools
- Precision. This refers to the capacity of a covalent docking tool to reproduce the experimental binding mode, which is represented by the RMSD value of a calculated binding conformation and experimental data. The prediction accuracy was expressed in terms of the RMSD values of the ligand heavy atoms from the conformation co-crystallized in the PDB entry.
- Generality. This refers to the capacity of a covalent docking tool to encompass different covalent binding systems. It is a measure of the overall precision across protein targets and reactions of all kinds. We marked the minimum RMSD among these tools for a single object then counted the number of marks in each tool. The goal was to find a covalent docking tool that is sufficiently general to be able to cope with most covalent systems.
- Robustness. This refers to the consistency of a covalent docking tool with the same docking process. Notwithstanding different structures of the same target or different targets of the same family or different families, there should be no unreasonable errors in the docking results. We calculated the average and deviation of the RMSD given by each docking tool in the whole data set.
4.3. Protocol for Selecting Tools
4.4. Docking Programs and Scoring Functions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Robertson, J.G. Mechanistic basis of enzyme-targeted drugs. Biochemistry 2005, 44, 5561–5571. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Boceprevir. In Drugs and Lactation Database (LactMed), Bethesda (MD). 2006. Available online: https://www.ncbi.nlm.nih.gov/books/NBK500610/ (accessed on 8 June 2019).
- Osimertinib. In Drugs and Lactation Database (LactMed), Bethesda (MD). 2018. Available online: https://www.ncbi.nlm.nih.gov/books/NBK500861/ (accessed on 8 June 2019).
- Damas, J.; Grek, V.; Remacle-Volon, G. Inhibition of the thrombocytopenic effect of exogenous and endogenous thrombin by PCR 4099 (d,1)methyl 2-(2-chlorophenyl)-2(4,5,6,7-tetrahydrothieno (3,2-c)pyridin-5-yl) acetate.hydrochloride.monohydrate. Thromb. Res. 1987, 48, 585–589. [Google Scholar] [CrossRef]
- Bradshaw, J.M.; McFarland, J.M.; Paavilainen, V.O.; Bisconte, A.; Tam, D.; Phan, V.T.; Romanov, S.; Finkle, D.; Shu, J.; Patel, V.; et al. Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat. Chem. Biol. 2015, 11, 525–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, R.; Peters, U.; Babbar, A.; Chen, Y.; Feng, J.; Janes, M.R.; Li, L.S.; Ren, P.; Liu, Y.; Zarrinkar, P.P. The reactivity-driven biochemical mechanism of covalent KRAS(G12C) inhibitors. Nat. Struct. Mol. Biol. 2018. [Google Scholar] [CrossRef]
- Goedken, E.R.; Argiriadi, M.A.; Banach, D.L.; Fiamengo, B.A.; Foley, S.E.; Frank, K.E.; George, J.S.; Harris, C.M.; Hobson, A.D.; Ihle, D.C.; et al. Tricyclic covalent inhibitors selectively target Jak3 through an active site thiol. J. Biol. Chem. 2015, 290, 4573–4589. [Google Scholar] [CrossRef]
- Sadowski, P.; Fooshee, D.; Subrahmanya, N.; Baldi, P. Synergies Between Quantum Mechanics and Machine Learning in Reaction Prediction. J. Chem. Inf. Model. 2016, 56, 2125–2128. [Google Scholar] [CrossRef]
- Khamis, M.A.; Gomaa, W.; Ahmed, W.F. Machine learning in computational docking. Artif. Intell. Med. 2015, 63, 135–152. [Google Scholar] [CrossRef]
- Miller, R.M.; Paavilainen, V.O.; Krishnan, S.; Serafimova, I.M.; Taunton, J. Electrophilic fragment-based design of reversible covalent kinase inhibitors. J. Am. Chem. Soc. 2013, 135, 5298–5301. [Google Scholar] [CrossRef]
- Adeniyi, A.A.; Muthusamy, R.; Soliman, M.E. New drug design with covalent modifiers. Expert Opin. Drug Discov. 2016, 11, 79–90. [Google Scholar] [CrossRef]
- Lonsdale, R.; Burgess, J.; Colclough, N.; Davies, N.L.; Lenz, E.M.; Orton, A.L.; Ward, R.A. Expanding the Armory: Predicting and Tuning Covalent Warhead Reactivity. J. Chem. Inf. Model. 2017, 57, 3124–3137. [Google Scholar] [CrossRef] [PubMed]
- Kathman, S.G.; Xu, Z.; Statsyuk, A.V. A fragment-based method to discover irreversible covalent inhibitors of cysteine proteases. J. Med. Chem. 2014, 57, 4969–4974. [Google Scholar] [CrossRef] [PubMed]
- London, N.; Farelli, J.D.; Brown, S.D.; Liu, C.; Huang, H.; Korczynska, M.; Al-Obaidi, N.F.; Babbitt, P.C.; Almo, S.C.; Allen, K.N.; et al. Covalent docking predicts substrates for haloalkanoate dehalogenase superfamily phosphatases. Biochemistry 2015, 54, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.Q.; Calhoun, S.; Fan, H.; Kalyanaraman, C.; Branch, M.C.; Mashiyama, S.T.; London, N.; Jacobson, M.P.; Babbitt, P.C.; Shoichet, B.K.; et al. Prediction of substrates for glutathione transferases by covalent docking. J. Chem. Inf. Model. 2014, 54, 1687–1699. [Google Scholar] [CrossRef] [PubMed]
- Schroder, J.; Klinger, A.; Oellien, F.; Marhofer, R.J.; Duszenko, M.; Selzer, P.M. Docking-based virtual screening of covalently binding ligands: An orthogonal lead discovery approach. J. Med. Chem. 2013, 56, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.C.; Welker, A.; Rieger, M.; Sahu, P.K.; Sotriffer, C.A.; Schirmeister, T.; Engels, B. Protocol for rational design of covalently interacting inhibitors. Chemphyschem. 2014, 15, 3226–3235. [Google Scholar] [CrossRef] [PubMed]
- Scarpino, A.; Ferenczy, G.G.; Keseru, G.M. Comparative Evaluation of Covalent Docking Tools. J. Chem. Inf. Model. 2018, 58, 1441–1458. [Google Scholar] [CrossRef] [PubMed]
- Mjos, K.D.; Orvig, C. Metallodrugs in medicinal inorganic chemistry. Chem Rev 2014, 114, 4540–4563. [Google Scholar] [CrossRef] [PubMed]
- Robles, V.M.; Ortega-Carrasco, E.; Alonso-Cotchico, L.; Rodriguez-Guerra, J.; Lledos, A.; Marechal, J.D. Toward the computational design of artificial metalloenzymes: from protein-ligand docking to multiscale approaches. ACS Catal. 2015, 5, 2469–2480. [Google Scholar] [CrossRef]
- Sciortino, G.; Garribba, E.; Marechal, J.D. Validation and applications of protein-ligand docking approaches improved for metalloligands with multiple vacant sites. Inorg. Chem. 2019, 58, 294–306. [Google Scholar] [CrossRef]
- Sciortino, G.; Rodriguez-Guerra Pedregal, J.; Lledos, A.; Garribba, E.; Marechal, J.D. Prediction of the interaction of metallic moieties with proteins: An update for protein-ligand docking techniques. J. Comput. Chem. 2018, 39, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.; Knorr, S.; Hamacher, K.; Schmidt, B. DOCKTITE-a highly versatile step-by-step workflow for covalent docking and virtual screening in the molecular operating environment. J. Chem. Inf. Model. 2015, 55, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Bianco, G.; Forli, S.; Goodsell, D.S.; Olson, A.J. Covalent docking using autodock: Two-point attractor and flexible side chain methods. Protein Sci. 2016, 25, 295–301. [Google Scholar] [CrossRef] [PubMed]
- London, N.; Miller, R.M.; Krishnan, S.; Uchida, K.; Irwin, J.J.; Eidam, O.; Gibold, L.; Cimermancic, P.; Bonnet, R.; Shoichet, B.K.; et al. Covalent docking of large libraries for the discovery of chemical probes. Nat. Chem. Biol. 2014, 10, 1066–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, X.; Zhou, S.; Ge, Z.; Li, R.; Kwoh, C.K. CovalentDock Cloud: A web server for automated covalent docking. Nucleic Acids Res. 2013, 41, W329–W332. [Google Scholar] [CrossRef] [PubMed]
- Bohari, M.H.; Sastry, G.N. FDA approved drugs complexed to their targets: Evaluating pose prediction accuracy of docking protocols. J. Mol. Model. 2012, 18, 4263–4274. [Google Scholar] [CrossRef]
- Hauser, A.S.; Windshugel, B. LEADS-PEP: A benchmark data set for assessment of peptide docking performance. J. Chem. Inf. Model. 2016, 56, 188–200. [Google Scholar] [CrossRef]
- Vogel, S.M.; Bauer, M.R.; Boeckler, F.M. DEKOIS: Demanding evaluation kits for objective in silico screening—a versatile tool for benchmarking docking programs and scoring functions. J. Chem. Inf. Model. 2011, 51, 2650–2665. [Google Scholar] [CrossRef]
- Kumalo, H.M.; Bhakat, S.; Soliman, M.E. Theory and applications of covalent docking in drug discovery: Merits and pitfalls. Molecules 2015, 20, 1984–2000. [Google Scholar] [CrossRef]
- Sotriffer, C. Docking of covalent ligands: Challenges and approaches. Mol. Inform. 2018. [Google Scholar] [CrossRef]
- Chen, M.; Zeng, G.M.; Lai, C.; Li, J.; Xu, P.; Wu, H.P. Molecular basis of laccase bound to lignin: Insight from comparative studies on the interaction of Trametes versicolor laccase with various lignin model compounds. RSC Adv. 2015, 5, 52307–52313. [Google Scholar] [CrossRef]
- Dietzen, M.; Kalinina, O.V.; Taskova, K.; Kneissl, B.; Hildebrandt, A.K.; Jaenicke, E.; Decker, H.; Lengauer, T.; Hildebrandt, A. Large oligomeric complex structures can be computationally assembled by efficiently combining docked interfaces. Proteins 2015, 83, 1887–1899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slater, M.J.; Amphlett, E.M.; Andrews, D.M.; Bamborough, P.; Carey, S.J.; Johnson, M.R.; Jones, P.S.; Mills, G.; Parry, N.R.; Somers, D.O.; et al. Pyrrolidine-5,5-trans-lactams. 4. Incorporation of a P3/P4 urea leads to potent intracellular inhibitors of hepatitis C virus NS3/4A protease. Org. Lett. 2003, 5, 4627–4630. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, B.; Tichy, M.; Wang, L.; Robinson, S.; Martin, R.E.; Kuglstatter, A.; Benz, J.; Giroud, M.; Schirmeister, T.; Abel, R.; et al. Prospective evaluation of free energy calculations for the prioritization of cathepsin L inhibitors. J. Med. Chem. 2017, 60, 2485–2497. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Liu, Q.; Bliven, S.; Xie, L.; Bourne, P.E. Determining cysteines available for covalent inhibition across the human kinome. J. Med. Chem. 2017, 60, 2879–2889. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Bourne, P.E. Progress with covalent small-molecule kinase inhibitors. Drug Discov. Today 2018, 23, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.Y.; Doerksen, R.J. Protein kinase-inhibitor database: Structural variability of and inhibitor interactions with the protein kinase P-loop. J. Proteome Res. 2010, 9, 4433–4442. [Google Scholar] [CrossRef]
- Fraczkiewicz, R.; Braun, W. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J. Comput. Chem. 1998, 19, 319–333. [Google Scholar] [CrossRef]
- Grant, B.J.; Rodrigues, A.P.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef]
- Du, J.; Yan, X.; Liu, Z.; Cui, L.; Ding, P.; Tan, X.; Li, X.; Zhou, H.; Gu, Q.; Xu, J. cBinderDB: A covalent binding agent database. Bioinformatics 2017, 33, 1258–1260. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE) 2013.08; Chemical Computing Group ULC: Montreal, QC, Canada, 2018.
- MarvinSketch, Marvin 17.21.0: ChemAxon. Available online: https://chemaxon.com (accessed on 8 June 2019).
- Labute, P. The generalized Born/volume integral implicit solvent model: Estimation of the free energy of hydration using London dispersion instead of atomic surface area. J. Comput. Chem. 2008, 29, 1693–1698. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 1995, 245, 43–53. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Dev. and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.; Cramer, R.D., III; Van Opdenbosch, N. Validation of the general purpose tripos 5.2 force field. J. Comput. Chem. 1989, 10, 982–1012. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins 2011, 79, 2794–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toledo Warshaviak, D.; Golan, G.; Borrelli, K.W.; Zhu, K.; Kalid, O. Structure-based virtual screening approach for discovery of covalently bound ligands. J. Chem. Inf. Model. 2014, 54, 1941–1950. [Google Scholar] [CrossRef]
- Ruben, A.; Maxim, T.; Dmitry, K. ICM—A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar]
- Abagyan, R.; Totrov, M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 1994, 235, 983–1002. [Google Scholar] [CrossRef]
- Maxim, T.; Ruben, A. Derivation of sensitive discrimination potential for virtual ligand screening. In Proceedings of the RECOMB ’99 Proceedings of the third annual international conference on Computational molecular biology, Lyon, France, 11–14 April 1999; pp. 312–320. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Class | Reaction Type | Warhead Class | Warhead Core | Example | Frequency * |
|---|---|---|---|---|---|
| Nucleophilic Addition | Nucleophilic Addition by Cys | Nitrile(cys) |  | 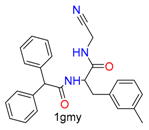 | 42 |
| Alkene(cys) | 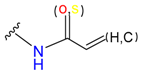 | 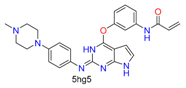 | 84 | ||
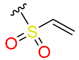 | 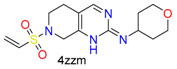 | 18 | |||
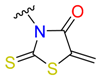 | 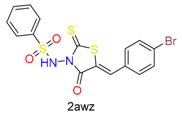 | 15 | |||
| Carbonyl(cys) |  | 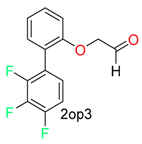 | 13 | ||
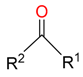 | 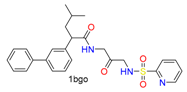 | 9 | |||
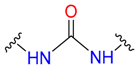 |  | 3 | |||
| Alkyne | 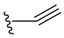 |  | 1 | ||
| Guanyl | 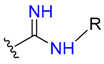 | 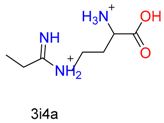 | 2 | ||
| Nucleophilic Addition by Ser | Nitrile (ser) | 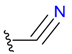 |  | 3 | |
| Carbonyl (ser) | 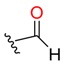 | 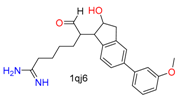 | 3 | ||
 | 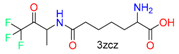 | 26 | |||
| Boric acid | 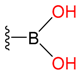 | 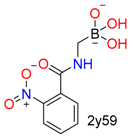 | 8 | ||
| Nucleophilic Substitution | Nucleophilic Substitution (cys) | Halide |  | 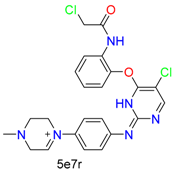 | 13 |
 | 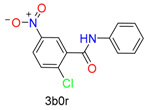 | 2 | |||
| Others | 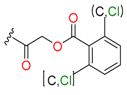 | 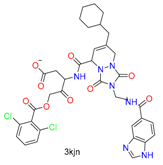 | 13 | ||
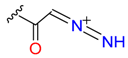 |  | 3 | |||
| Nucleophilic Substitution by Ser | Phosphonyl | 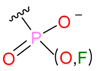 | 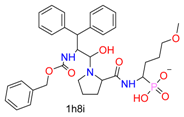 | 3 | |
| Ring Opening | Ring Opening by Cys | Heterocyclic |  | 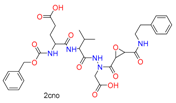 | 11 |
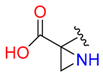 | 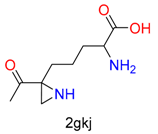 | 6 | |||
| Ring Opening by Ser | Lactam | 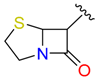 |  | 39 | |
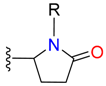 |  | 1 | |||
| Lactone | 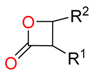 |  | 2 | ||
| Disulfide Formation | Disulfide Formation | Sulfydryl |  |  | 10 |
| Reaction Class | Warhead | n | Best Scored Pose (RMSD) | Best Sampled Pose (RMSD) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MOE | GOLD | CovDock | ICM-Pro | MOE | GOLD | CovDock | ICM-Pro | ||||
| Nucleophilic Addition by Cys | Nitrile | 42 | 2.26 | 1.1 | 1.27 | 3.74 | 1.6 | 0.91 | 1.24 | 1.56 | 5 Å 1 Å |
| Alkene | 117 | 1.99 | 2.47 | 1.47 | 1.68 | 1.4 | 1.59 | 1.18 | 1.41 | ||
| Carbonyl | 25 | 2.27 | 2.12 | 2.05 | 3.72 | 1.5 | 1.77 | 1.33 | 1.52 | ||
| Alkyne | 1 | 2.01 | 0.67 | 0.42 | 0.39 | 1.9 | 0.67 | 0.35 | 0.39 | ||
| Guanyl | 2 | 1.78 | 1.46 | 0.81 | 1.37 | 1.4 | 0.98 | 0.81 | 1.37 | ||
| Nucleophilic Substitution by Cys | Halide | 15 | 1.67 | 2.31 | 4.37 | 1.06 | 1.3 | 2.17 | 4.37 | 0.88 | |
| Others | 16 | 2.1 | 2.19 | 2.12 | 2.09 | 1.6 | 1.47 | 1.3 | 1.77 | ||
| Ring Opening by Cys | Heterocyclic | 17 | 1.71 | 1.74 | 1.87 | 4.88 | 1.4 | 1.48 | 1.33 | 4.72 | |
| Disulfide Formation | Sulfydryl | 10 | 1.94 | 2.94 | 2.52 | 2.92 | 1.3 | 2.47 | 2.52 | 1.95 | |
| Nucleophilic Addition by Ser | Nitrile | 3 | 1.38 | 2.54 | 0.73 | 2.66 | 1.3 | 1.77 | 0.52 | 2.66 | |
| Carbonyl | 29 | 1.61 | 3.54 | 3.1 | 2.03 | 1.3 | 1.58 | 1.4 | 1.36 | ||
| Boronic Acid | 8 | 1.94 | 2.57 | 2.94 | 2.44 | 1.5 | 1.88 | 2.74 | 2.44 | ||
| Nucleophilic Substitution by Ser | Phosphonyl | 3 | 2.32 | 2.33 | 5.81 | 1.68 | 1.5 | 2.08 | 3.25 | 1.68 | |
| Ring Opening by Ser | Lactam | 40 | 1.73 | 2.95 | 1.7 | 2.53 | 1.5 | 1.62 | 1.7 | 1.48 | |
| Lactone | 2 | 2.09 | 2.56 | 2.52 | 5.66 | 1.5 | 1.6 | 1.6 | 4.98 | ||
| P(i,x) | 1.94 | 2.33 | 2.05 | 2.53 | 1.5 | 1.6 | 1.33 | 1.56 | |||
| Receptor Type | n | Best Scored Pose (RMSD) | Best Sampled Pose (RMSD) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MOE | GOLD | CovDock | ICM-Pro | MOE | GOLD | CovDock | ICM-Pro | |||
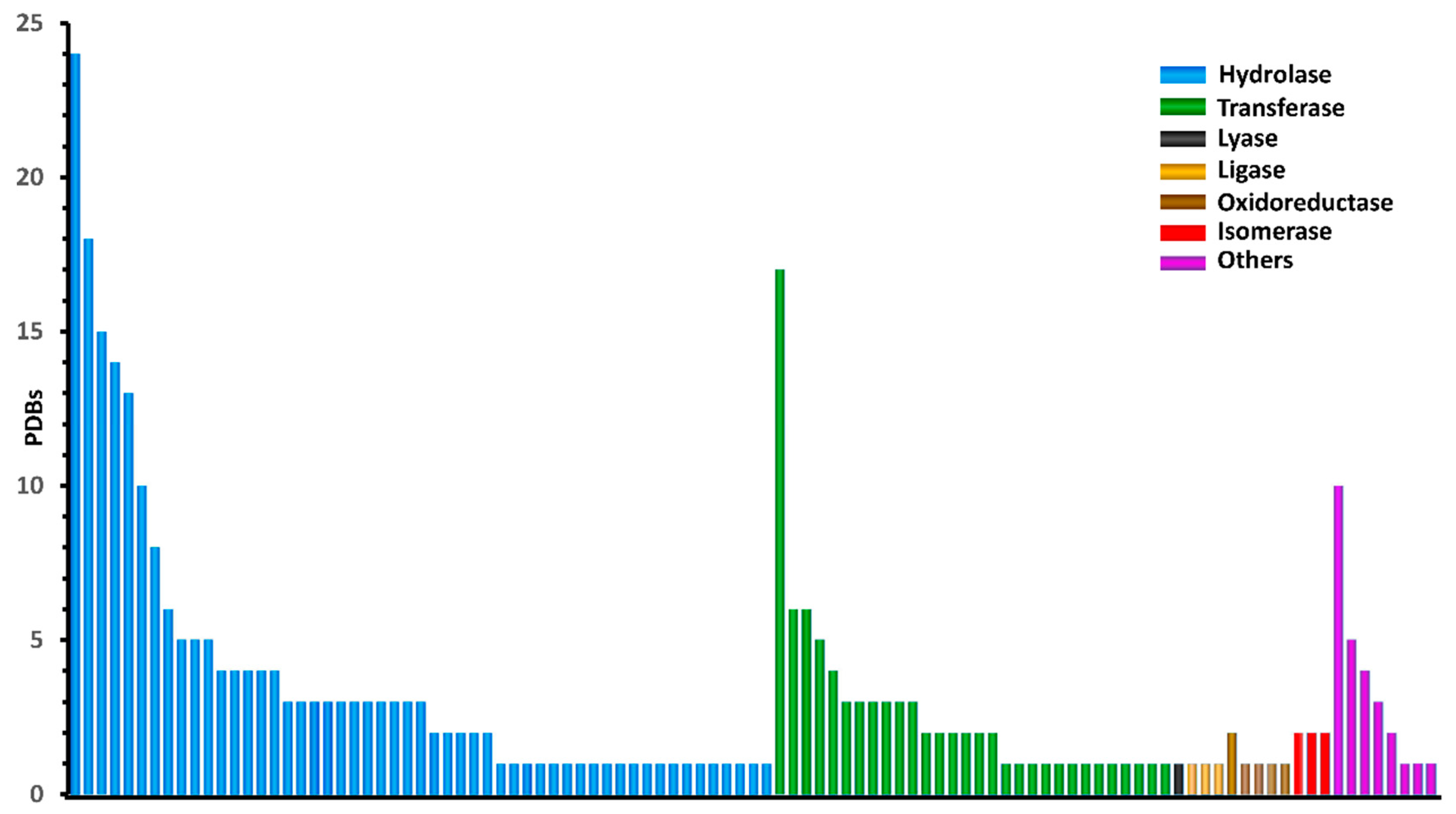
| Hydrolase | 204 | 2.07 | 2.33 | 1.71 | 2.76 | 1.54 | 1.51 | 1.39 | 1.61 | 5 Å 1 Å |
| Transferase | 83 | 1.8 | 2.24 | 1.3 | 1.34 | 1.34 | 1.46 | 1.06 | 1.24 | |
| Ligase | 3 | 2.9 | 2.63 | 1.08 | 1.41 | 1.25 | 1.04 | 0.84 | 1.41 | |
| Lyase | 1 | 0.96 | 1.23 | 1.24 | 1.34 | 0.9 | 1.17 | 1.19 | 1.34 | |
| Oxidoreductase | 6 | 1.7 | 1.44 | 2.37 | 1.13 | 1.18 | 1.06 | 2.37 | 1.13 | |
| Isomerase | 6 | 1.49 | 1.2 | 1.02 | 2.25 | 1.33 | 0.78 | 1.01 | 2.25 | |
| Transcription | 18 | 2.38 | 6.15 | 2.15 | 3.42 | 1.6 | 3.03 | 1.71 | 2.1 | |
| Viral Protein | 4 | 3.91 | 0.81 | 4.29 | 4.93 | 2.97 | 0.81 | 1.3 | 4.93 | |
| Metal binding protein | 5 | 1.72 | 5.54 | 5.43 | 2.37 | 1.04 | 4.54 | 3 | 1.67 | |
| P(i,x) | 1.8 | 2.24 | 1.71 | 2.25 | 1.33 | 1.17 | 1.3 | 1.61 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, C.; Yan, X.; Gu, Q.; Du, J.; Wu, D.; Lu, Y.; Zhou, H.; Xu, J. Systematic Studies on the Protocol and Criteria for Selecting a Covalent Docking Tool. Molecules 2019, 24, 2183. https://doi.org/10.3390/molecules24112183
Wen C, Yan X, Gu Q, Du J, Wu D, Lu Y, Zhou H, Xu J. Systematic Studies on the Protocol and Criteria for Selecting a Covalent Docking Tool. Molecules. 2019; 24(11):2183. https://doi.org/10.3390/molecules24112183
Chicago/Turabian StyleWen, Chang, Xin Yan, Qiong Gu, Jiewen Du, Di Wu, Yutong Lu, Huihao Zhou, and Jun Xu. 2019. "Systematic Studies on the Protocol and Criteria for Selecting a Covalent Docking Tool" Molecules 24, no. 11: 2183. https://doi.org/10.3390/molecules24112183
APA StyleWen, C., Yan, X., Gu, Q., Du, J., Wu, D., Lu, Y., Zhou, H., & Xu, J. (2019). Systematic Studies on the Protocol and Criteria for Selecting a Covalent Docking Tool. Molecules, 24(11), 2183. https://doi.org/10.3390/molecules24112183