4.3.3. Detailed Procedures
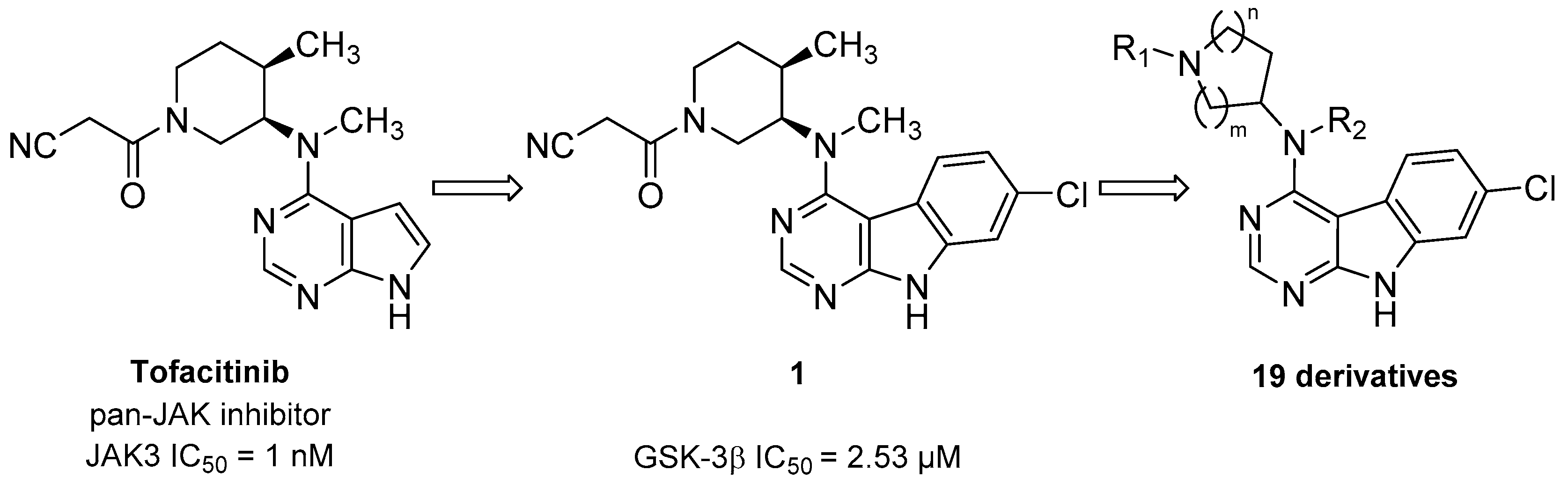
Preparation of 1
3-((3
R,4
R)-3-((7-Chloro-9
H-pyrimido[4,5-
b]indol-4-yl)(methyl)amino)-4-methylpiperidin-1-yl)-3-oxopropanenitrile (
1). 4,7-Dichloro-9
H-pyrimido[4,5-
b]indole (
9) (50.0 mg, 0.21 mmol) and 3-((3
R,4
R)-4-methyl-3-(methylamino)piperidin-1-yl)-3-oxopropanenitrile hydrochloride [
49] (48.7 mg, 0.21 mmol) were suspended in a mixture of dry dioxane (1 mL) and dry DMF (0.1 mL). DIPEA (67.9 mg, 0.53 mmol) was added and the mixture stirred under microwave irradiation (120 °C, 130W) in a sealed tube for 26 h. Volatiles were removed under reduced pressure. Purification of the residue by flash column chromatography (SiO
2, 1.EtOAc:
iPrOH 8:1, 2.DCM:MeOH 95:5) gave 24 mg of a light brown solid (29% yield); NMR shows a 3:1 mixture of amide bond rotamers,
1H-NMR (400 MHz, acetone-
d6) δ 11.30 (s, 1H), 8.55–8.38 (m, 1H), 7.81 (d,
J = 8.5 Hz, 1H), 7.62 (s, 1H), 7.33 (d,
J = 8.4 Hz, 1H), 4.85–4.74 (m, 0.75H), 4.27–4.17 (m, 0.25H), 4.11–3.64 (m, 6H), 3.19 (s, 2.25H), 3.05 (s, 0.75H), 2.51–2.31 (m, 1H), 2.01–1.79 (m, 2H), 1.24 (d,
J = 6.8 Hz, 0.75H), 1.15 (d,
J = 6.9 Hz, 2.25H);
13C-NMR (101 MHz, acetone-
d6) δ 164.5, 164.0, 161.0, 158.8, 155.3, 138.7, 131.4, 124.5, 122.0, 119.6, 116.0, 112.2, 100.0, 56.1, 53.3, 48.4, 46.6, 46.2, 34.1, 34.0, 32.9, 32.5, 32.2, 26.0, 25.7, 14.9; ESI-MS: (
m/z) 396.9 [M + H]
+, 418.9 [M + Na]
+, 394.9 [M − H]
-; HPLC: t
r = 7.201 min (100.0% purity).
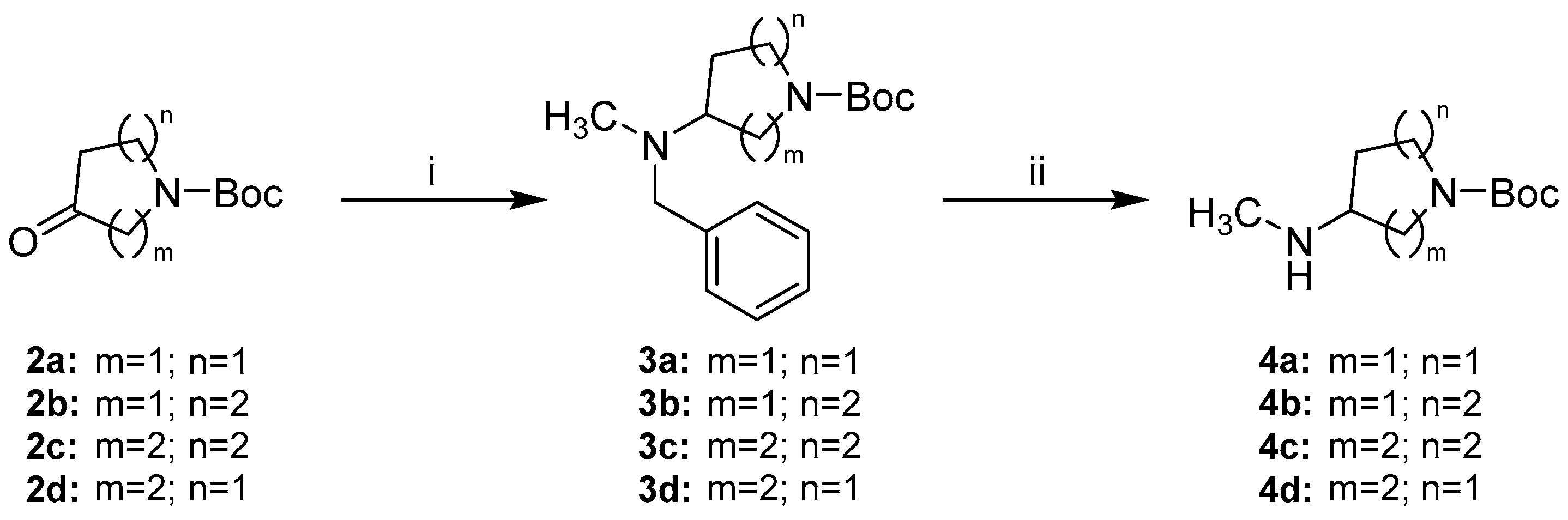
Detailed Procedures for the Preparation of Intermediates 3a–d
tert-Butyl-3-(benzyl(methyl)amino)pyrrolidine-1-carboxylate (3a). 3a was prepared from N-Boc-pyrrolidin-3-one (2a) (1.5 g, 8.10 mmol), N,N-benzylmethylamine (1.1 g, 8.91 mmol), glacial AcOH (534.9 mg, 8.91 mmol) and Na(OAc)3BH (2.6 g, 12.15 mmol) in dry DCM (17 mL) according to general procedure A. Purification by flash column chromatography (SiO2, n-hexane:EtOAc 3:1) gave 1.8 g of a yellow oil (76% yield); 1H-NMR (300 MHz, CDCl3) δ 7.38–7.22 (m, 5H, overlap with CHCl3 signal), 3.79–3.46 (m, 4H), 3.36–3.15 (m, 2H), 3.11–2.94 (m, 1H), 2.16 (s, 3H), 2.14–2.05 (m, 1H), 1.98–1.79 (m, 1H), 1.48 (s, 9H); 13C-NMR (75 MHz, CDCl3) δ 154.6, 138.7, 138.6, 129.2, 129.0, 128.4, 127.2, 79.3, 64.0, 63.2, 60.5, 50.0, 49.6, 45.2, 44.8, 39.8, 30.1, 29.1, 28.6; GC-MS method A: tr = 8.792 min, (m/z) 290 [M].
tert-Butyl-3-(benzyl(methyl)amino)piperidine-1-carboxylate (3b). 3b was prepared from N-Boc-piperidin-3-one (2b) (2.5 g, 12.55 mmol), N,N-benzylmethylamine (2.0 g, 16.31 mmol), glacial acetic acid (904.1 mg, 15.06 mmol) and Na(OAc)3BH (4.3 g, 20.08 mmol) in dry DCM (30 mL) according to general procedure A. After stirring overnight reaction control indicated incomplete conversion, therefore a second portion of N,N-benzylmethylamine (494.1 mg, 4.08 mmol) and Na(OAc)3BH (1.1 g, 5.02 mmol) was added and stirring continued for 1 h. Purification by flash column chromatography (SiO2, petroleum ether:EtOAc 3:1) gave 2.6 g of a yellow oil (69% yield); 1H-NMR (300 MHz, CDCl3) δ 7.32–7.07 (m, 5H, overlap with CHCl3 signal), 4.35–3.78 (m, 2H), 3.59 (d, J = 13.5 Hz, 1H), 3.51 (d, J = 13.4 Hz, 1H), 2.74–2.49 (m, 2H), 2.46–2.31 (m, 1H), 2.15 (s, 3H), 1.96– 1.82 (m, 1H), 1.70–1.57 (m, 1H), 1.48–1.29 (m, 11H); 13C-NMR (75 MHz, CDCl3) δ 155.0, 139.7, 128.8, 128.3, 126.9, 79.4, 59.4, 58.4, 46.2 (br), 44.4 (br), 38.0, 28.5, 27.6 (br), 24.7 (br).
tert-Butyl-4-(benzyl(methyl)amino)azepane-1-carboxylate (3c). 3c was prepared from N-Boc-hexahydro-1H-azepin-4-one (2c) (2.25 g, 10.55 mmol), N,N-benzylmethylamine (1.5 g, 12.66 mmol), glacial AcOH (696.9 mg, 11.61 mmol) and Na(OAc)3BH (3.4 g, 15.83 mmol) in dry DCM (25 mL) according to general procedure A. After stirring overnight reaction control indicated incomplete conversion, therefore a second portion of N,N-benzylmethylamine (639.2 mg, 5.27 mmol) and Na(OAc)3BH (1.1 g, 5.27 mmol) was added and stirring continued for 1 d. Purification by flash column chromatography (SiO2, n-hexane:EtOAc 3:1) gave 2.7 g of a yellow oil (81% yield); 1H-NMR (300 MHz, CDCl3) δ 7.36–7.20 (m, 5H, overlap with CHCl3 signal), 3.66–3.39 (m, 4H), 3.32–3.17 (m, 2H), 2.67–2.56 (m, 1H), 2.21–2.16 (m, 3H), 2.10–1.80 (m, 3H), 1.78–1.41 (m, 12H); 13C-NMR (75 MHz, CDCl3) δ 155.6, 140.0, 140.0, 128.7, 128.3, 126.9, 126.9, 79.2, 63.2, 63.1, 57.8, 57.7, 46.8, 46.4, 44.3, 44.0, 37.6, 30.3, 29.9, 29.8, 28.6, 25.9.
tert-Butyl-4-(benzyl(methyl)amino)piperidine-1-carboxylate (3d). 3d was prepared from N-Boc-piperidin-4-one (2d) (2.75 g, 13.80 mmol), N,N-benzylmethylamine (1.8 g, 15.18 mmol), glacial AcOH (911.7 mg, 15.18 mmol) and Na(OAc)3BH (4.4 g, 20.70 mmol) in dry DCM (30 mL) according to general procedure A. The reaction mixture was stirred over molecular sieves instead of Na2SO4, which was separated by filtration before stopping the reaction with saturated NaHCO3. Purification by flash column chromatography (SiO2, petroleum ether:EtOAc:3.5N NH3 in MeOH 25:73:2) gave 3.6 g of a white solid (86% yield) 1H-NMR (200 MHz, CDCl3) δ 7.37–7.17 (m, 5H, overlap with CHCl3 signal), 4.28–4.04 (m, 2H), 3.59 (s, 2H), 2.80–2.50 (m, 3H), 2.21 (s, 3H), 1.90–1.73 (m, 2H), 1.62–1.43 (m, 11H); 13C-NMR (50 MHz, CDCl3) δ 154.9, 139.5, 128.9, 128.4, 127.1, 79.6, 60.9, 58.1, 43.6, 37.7, 28.6, 28.0; GC-MS method A: tr = 9.663 min, (m/z) 304 [M].
Detailed Procedures for the Preparation of Intermediates 4a–d
tert-Butyl-3-(methylamino)pyrrolidine-1-carboxylate (4a). 3a (1.6 g, 5.51 mmol) was dissolved in HPLC grade MeOH (30 mL) and Pd/C 10% (m/m) (532.0 mg) was added. The suspension was stirred in a reactor charged with 5 bar of H2 pressure at rt for 4 h and then filtered over a pad of celite rinsing with fresh solvent. The filtrate was concentrated under reduced pressure to give 1.1 g of a green oil (96% crude yield), which was used in the next step without further purification; 1H-NMR (400 MHz, CDCl3) δ 3.48–3.19 (m, 3H), 3.17–2.93 (m, 2H), 2.33 (s, 3H), 2.00–1.88 (m, 1H), 1.67–1.54 (m, 1H), 1.38–1.33 (m, 9H), 1.30 (br s, 1H); 13C-NMR (101 MHz, CDCl3) δ 154.6, 79.0, 59.5, 58.7, 51.6, 51.1, 44.3, 44.0, 34.7, 31.7, 31.0, 28.5; GC-MS method A: tr = 3.039 min, (m/z) 200 [M].
tert-Butyl-3-(methylamino)piperidine-1-carboxylate (4b). 3b (2.2 g, 7.16 mmol) was dissolved in a solvent mixture of EtOAc (27 mL) and MeOH (18 mL). Pd/C 10% (m/m) (727.0 mg) was added and the mixture stirred in a reactor charged with 5 bar of H2 pressure at rt for 3 h. The mixture was filtered over a pad of celite rinsing with fresh solvent. The filtrate was concentrated under reduced pressure to give 1.5 g of a green oil (96% crude yield), which was used in the next step without further purification; 1H-NMR (400 MHz, CDCl3) δ 4.18–3.67 (m, 2H), 2.99–2.51 (m, 2H), 2.49–2.37 (m, 4H), 1.95–1.85 (m, 1H), 1.70–1.60 (m, 1H), 1.49–1.37 (m, 10H), 1.35–1.21 (m, 2H); 13C-NMR (101 MHz, CDCl3) δ 155.0, 79.5, 55.6, 48.8 (br), 44.3 (br), 33.9, 31.3, 28.6, 23.6 (br). GC-MS method B: tr = 11.729 min, (m/z) 214 [M].
tert-Butyl-4-(methylamino)azepane-1-carboxylate (4c). 3c (662.0 mg, 2.08 mmol) was dissolved in a solvent mixture of EtOAc (9 mL) and MeOH (6 mL). Pd/C 10% (m/m) (220.7 mg) was added and the mixture stirred in a reactor charged with 5 bar of H2 pressure at rt for 3 h. The mixture was filtered over a pad of celite rinsing with fresh solvent. The filtrate was concentrated under reduced pressure to give 463 mg of a green oil (98% crude yield), which was used in the next step without further purification; 1H-NMR (400 MHz, CDCl3) δ 3.54–3.36 (m, 2H), 3.36–3.09 (m, 2H), 2.51–2.41 (m, 1H), 2.36 (s, 3H), 1.97–1.87 (m, 1H), 1.87–1.72 (m, 2H), 1.59–1.31 (m, 13H); 13C-NMR (101 MHz, CDCl3) δ 155.6, 79.2, 60.2, 59.8, 46.6, 46.0, 43.3, 42.8, 34.8, 34.5, 34.2, 33.2, 32.5, 28.6, 24.8, 24.4. GC-MS method B: tr = 13.406 min, (m/z) 228 [M].
tert-Butyl-4-(methylamino)piperidine-1-carboxylate (4d). 3d (5.7 g, 18.72 mmol) was dissolved in MeOH (150 mL). Pd/C 10% (m/m) (600 mg) and Pd(OH)2/C 20% (m/m) (300 mg) were added and the mixture stirred in a reactor charged with 5 bar of H2 pressure at rt overnight. The mixture was filtered over a pad of celite rinsing with fresh solvent. The filtrate was concentrated under reduced pressure to give 3.9 g of a green oil (97% crude yield), which was used in the next step without further purification; 1H-NMR (200 MHz, CDCl3) δ 4.17–3.83 (m, 2H), 2.88–2.65 (m, 2H), 2.55–2.42 (m, 1H), 2.40 (s, 3H), 1.92–1.74 (m, 2H), 1.42 (s, 9H), 1.33–1.04 (m, 2H); 13C-NMR (50 MHz, CDCl3) δ 155.0, 79.5, 56.8, 42.6 (br), 33.5, 32.2, 28.5; GC-MS method A: tr = 3.499 min, (m/z) 214 [M].
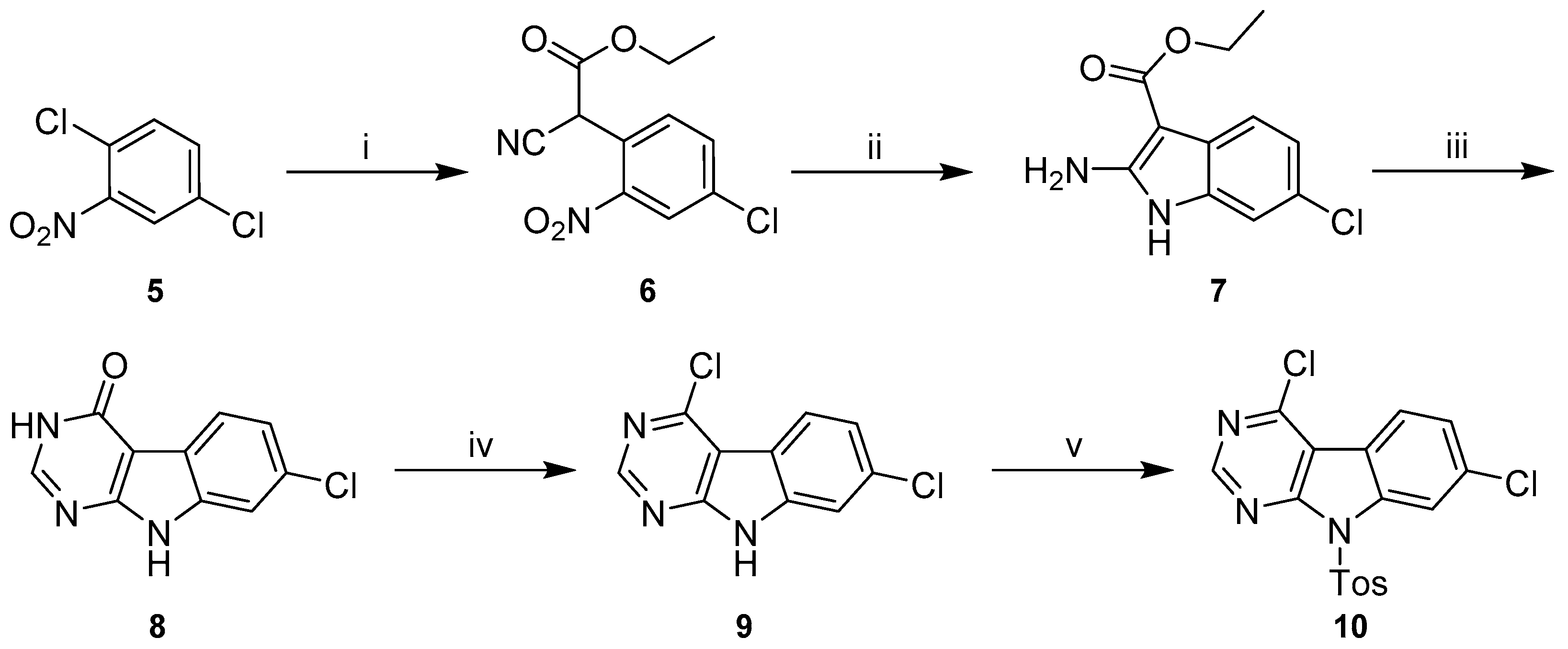
Detailed Procedures for the Preparation of 4,7-dichloro-9-tosyl-9H-pyrimido[4,5-b]indole (10)
Ethyl-2-(4-chloro-2-nitrophenyl)-2-cyanoacetate (6). A solution of ethyl-2-cyanoacetate (12.4 g, 109.38 mmol) in dry DMF (10 mL) was drop-added to a stirring, ice-cooled suspension of NaH (4.4 g of a 60% dispersion in mineral oil, 109.38 mmol) in dry DMF (20 mL). After complete addition, the dropping funnel was purged with additional dry DMF (5 mL) and ice-cooling was removed. After stirring at rt for 0.5 h, a solution of 1,4-dichloro-2-nitrobenzene (5) (10.0 g, 52.08 mmol) in dry DMF (10 mL) was drop-added. The stirring mixture was subsequently heated to 80 °C for 0.5 h, when reaction control via HPLC indicated complete conversion. The mixture was left to cool to rt and acidified with 10% HCl(aq) (50 mL). EtOAc (100 mL) was added, phases were separated and the aqueous layer extracted with additional EtOAc (3 × 30 mL). Combined organic layers were washed with saturated NaCl solution (5 × 50 mL) and dried over Na2SO4. The mixture was concentrated under reduced pressure and the liquid residue treated with ice-cold water and stirred with ice-cooling. The resulting yellow precipitate was triturated with the ice-cold water, filtered washing with ice-cold water and dried over P2O5 in vacuo. 14.5 g of a yellow solid (> 100% crude yield) that may contain traces of excessive ethyl-2-cyanoacetate, but was used in the next step without further purification; 1H-NMR (200 MHz, DMSO-d6) δ 8.35 (d, J = 2.0 Hz, 1H), 8.02 (dd, J = 8.2, 2.1 Hz, 1H), 7.78 (d, J = 8.2 Hz, 1H), 6.27 (s, 1H), 4.22 (q, J = 7.1 Hz, 2H), 1.19 (t, J = 7.1 Hz, 3H); 13C-NMR (50 MHz, DMSO-d6) δ 163.7, 147.4, 135.2, 134.9, 134.6, 126.0, 124.5, 115.0, 63.1, 40.7, 13.8; ESI-MS: (m/z) 266.9 [M − H]−; HPLC: tr = 6.655 min.
Ethyl-2-amino-6-chloro-1
H-indole-3-carboxylate (
7).
6 (7.0 g, 26.06 mmol) was dissolved in glacial AcOH (60 mL). The solution was stirred at 85 °C and Zinc dust (20.4 g, 312.72 mmol) was added in ten portions. The suspension was stirred at 85 °C for 75 min when reaction control via HPLC indicated complete consumption of the starting material. After cooling to rt, Zn dust was filtered off rinsing with AcOH (or EtOAc, alternatively) and the filtrate was concentrated under reduced pressure to leave a liquid residue. Careful addition of saturated NaHCO
3 solution neutralized residual AcOH resulting in a precipitate which was filtered off, washed with water and dried over P
2O
5 in vacuo. 5.8 g of a red-brown solid (93% crude yield), which was used in the next step without further purification. A small batch was purified by flash column chromatography (SiO
2, DCM/MeOH 97.5:2.5) for analytical purposes.
1H-NMR shows a mixture of products, which are assumed to be the title compound and the corresponding 1-hydroxyindole [
27]; HPLC: t
r = 8.042 min.
7-Chloro-3,9-dihydro-4H-pyrimido[4,5-b]indol-4-one (8). 7 (5.7 g, 23.88 mmol) and NH4HCO2 (1.7 g, 27.46 mmol) were suspended in formamide (50 mL) and stirred at 160 °C for 28 h with reflux cooling when reaction control via HPLC indicated nearly full consumption of the starting material. After cooling to rt the mixture was poured into ice-cold water resulting in a precipitate which was filtered, washed thoroughly with ice-cold water and dried over P2O5 in vacuo. 4.5 g of a green-brown solid (86% crude yield), which was used in the next step without further purification; 1H-NMR (200 MHz, DMSO-d6) δ 12.32 (br s, 2H), 8.15 (s, 1H), 7.95 (d, J = 8.5 Hz, 1H), 7.48 (d, J = 1.4 Hz, 1H), 7.25 (dd, J = 8.5, 1.5 Hz, 1H); 13C-NMR (50 MHz, DMSO-d6) δ 158.0, 154.3, 148.0, 136.0, 128.4, 121.8, 121.4, 120.9, 111.4, 100.0; ESI-MS: (m/z) 217.9 [M − H]−; HPLC: tr = 5.489 min.
4,7-Dichloro-9H-pyrimido[4,5-b]indole (9). 8 (4.5 g, 20.44 mmol) was suspended in chlorobenzene (30 mL) and DIPEA (4.0 g, 30.66 mmol) was added. The mixture was stirred at rt and under N2 atmosphere when POCl3 (4.4 g, 28.62 mmol) was added carefully dropwise. After stirring at rt for 1 h, the mixture was heated to 80 °C for additional 4.5 h with reflux cooling when HPLC indicated complete consumption of the starting material. The mixture was left to cool down and dropped carefully into stirring water (300 mL) at rt resulting in a brown precipitate. Saturated NaHCO3 solution was added carefully and the suspension left to stir overnight for neutralization. The precipitate was filtered, washed with water and dried over P2O5 in vacuo. 4.2 g of a brown solid (87% crude yield), which was purified by the following recrystallization procedure: 2 g of crude 4,7-dichloro-9H-pyrimido[4,5-b]indole were suspended in boiling toluene (650 mL) and stirred for 0.5 h. The hot suspension was filtered rinsing the brown filter cake with fresh hot toluene. The filtrate was concentrated under reduced pressure resulting in precipitation of the product. The suspension was cooled and subsequently the precipitate was filtered, washed with cold toluene and dried under reduced pressure giving 1.2 g of a yellow solid (59% recrystallization yield, 51% total yield); 1H-NMR (200 MHz, DMSO-d6) δ 12.85 (s, 1H), 8.76 (s, 1H), 8.14 (d, J = 8.5 Hz, 1H), 7.57 (d, J = 1.9 Hz, 1H), 7.37 (dd, J = 8.5, 1.9 Hz, 1H); 13C-NMR (50 MHz, DMSO-d6) δ 156.3, 154.2, 151.3, 139.1, 132.7, 123.6, 121.9, 116.5, 112.0, 110.7; ESI-MS: (m/z) 235.7 [M − H]−; HPLC: tr = 8.590 min.
4,7-Dichloro-9-tosyl-9H-pyrimido[4,5-b]indole (10). NaH (151.2 mg of a 60% dispersion in mineral oil, 3.78 mmol) was added in three portions to a stirring suspension of 4,7-dichloro-9H-pyrimido[4,5-b]indole (9) (600.0 mg, 2.52 mmol) in dry THF (20 mL). p-Toluenesulfonyl chloride (576.6 mg, 3.02 mmol) was added after 20 min and stirring continued for another 0.5 h at rt and under N2 atmosphere when TLC indicated complete consumption of the starting material. The mixture was poured into ice-cold water and saturated NH4Cl solution (60 mL) was added. The precipitate was filtered, washed with cold water and dried over P2O5 in vacuo. 982 mg of a yellow solid (99% crude yield), which was directly used in the next step without further purification; ESI-MS: (m/z) 390.0 [M − H]−.
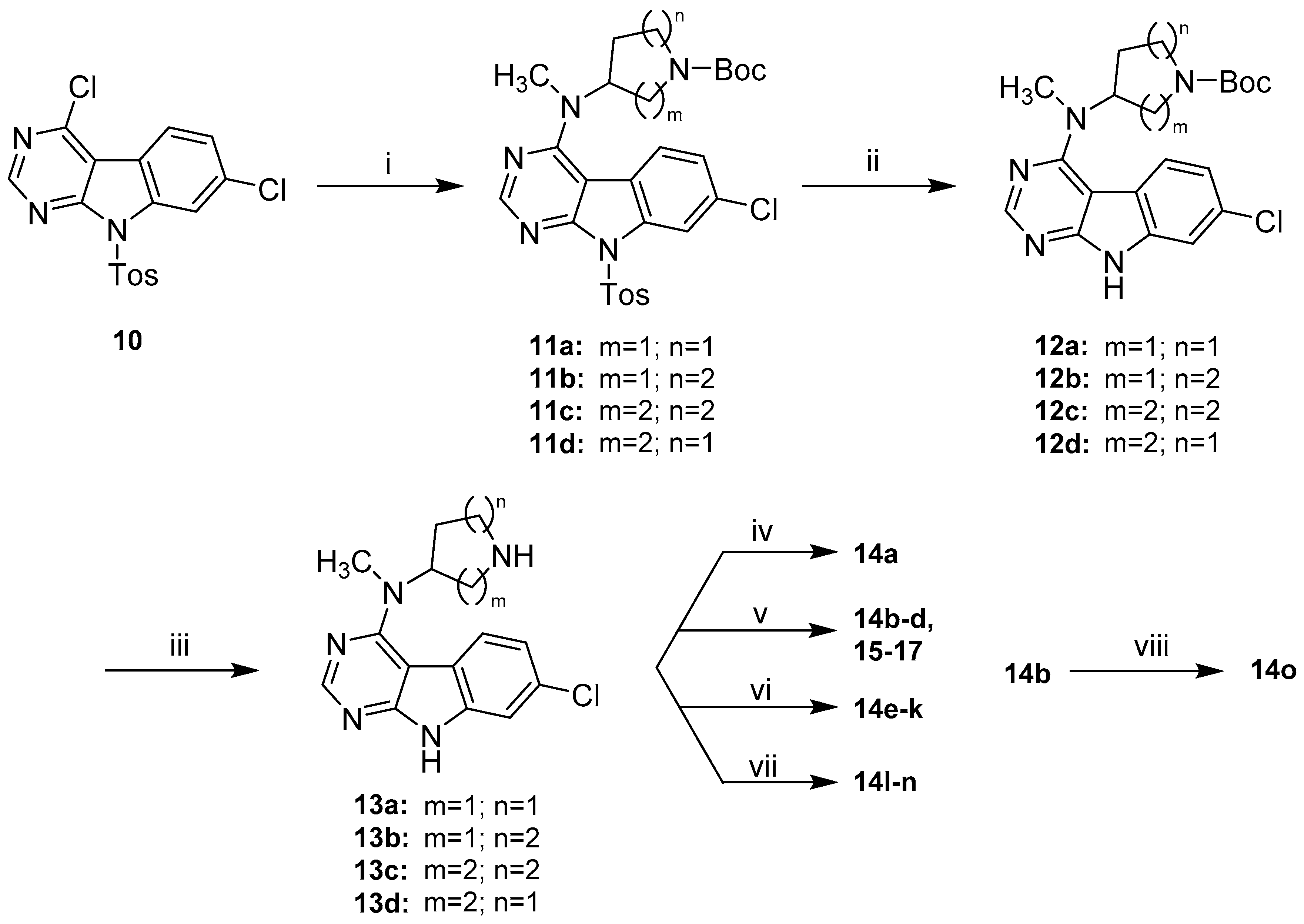
Detailed Procedures for the Preparation of Intermediates 11a–d
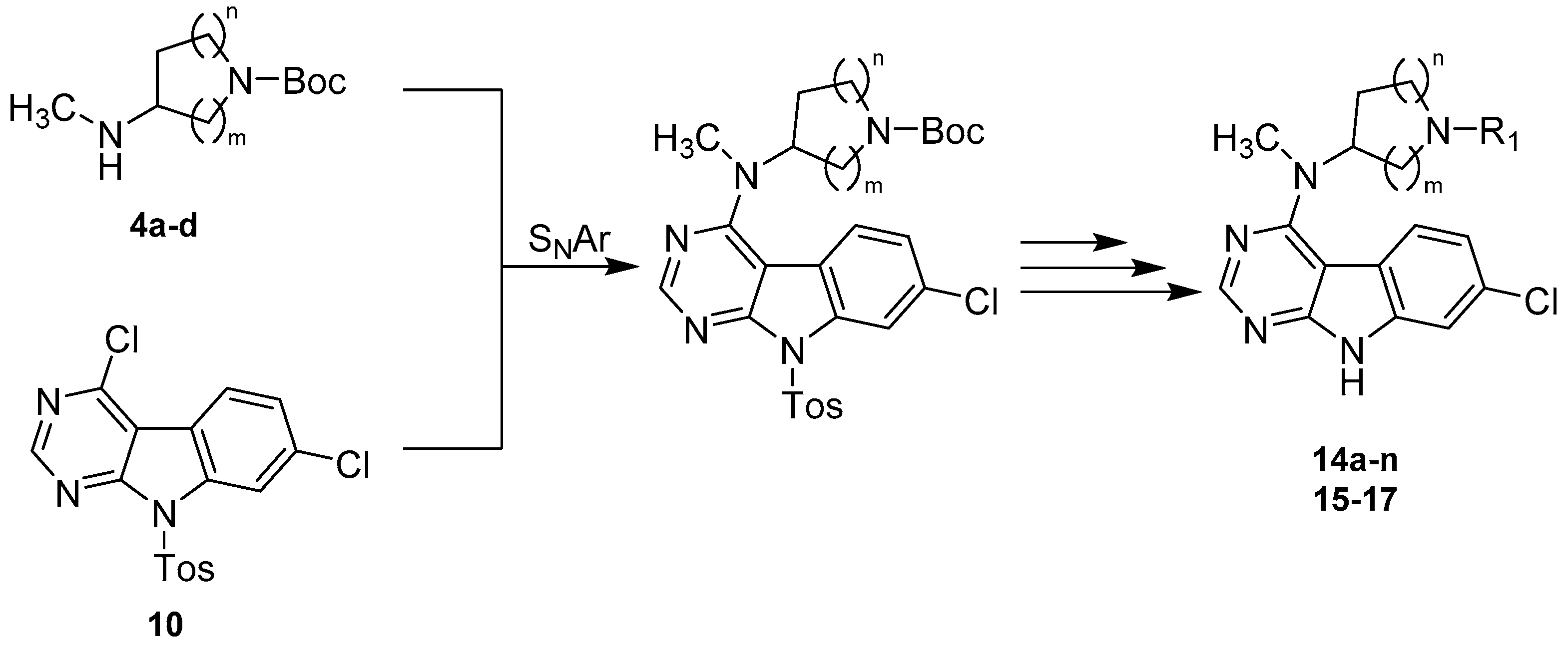
tert-Butyl-3-((7-chloro-9-tosyl-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)pyrrolidine-1-carboxyla-te (11a). 11a was prepared from 10 (1.0 g, 2.55 mmol), 4a (638.4 mg, 3.19 mmol) and DIPEA (988.8 mg, 7.65 mmol) in dry DMF (25 mL) according to general procedure B. 1.4 g of a beige solid (>100% crude yield), which was used in the next step without further purification. A small batch was purified by flash column chromatography (SiO2, DCM:MeOH 97.5:2.5) for analytical purposes; 1H-NMR (300 MHz, CDCl3) δ 8.64 (s, 1H), 8.54 (d, J = 1.8 Hz, 1H), 8.10 (d, J = 8.1 Hz, 2H), 7.63 (d, J = 8.5 Hz, 1H), 7.40 (dd, J = 8.5, 1.9 Hz, 1H), 7.27 (d, 2H, overlap with CHCl3 signal), 4.93–4.74 (m, 1H), 3.94–3.75 (m, 1H), 3.71–3.53 (m, 1H), 3.48–3.29 (m, 2H), 3.10 (s, 3H), 2.37 (s, 3H), 2.33–2.06 (m, 2H), 1.46 (s, 9H); 13C-NMR (50 MHz, CDCl3) δ 161.2, 157.2, 154.5, 154.3, 145.8, 136.2, 135.3, 132.9, 129.8, 128.1, 124.5, 123.3, 119.9, 114.7, 102.0, 79.6, 58.0, 57.4, 47.8, 44.7, 44.3, 35.2, 28.7, 28.6, 28.0, 21.7; ESI-MS: (m/z) 578.1 [M + Na]+, 554.3 [M − H]−; HPLC: tr = 11.123 min.
tert-Butyl-3-((7-chloro-9-tosyl-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)piperidine-1-carboxylate (11b). 11b was prepared from 10 (430.0 mg, 1.10 mmol), 4b (328.9 mg, 1.54 mmol) and DIPEA (425.1 mg, 3.29 mmol) in dry DMF (12 mL) according to general procedure B. 580 mg of a beige solid (93% crude yield), which was used in the next step without further purification. A small batch was purified by flash column chromatography (SiO2, DCM:MeOH 97.5:2.5) for analytical purposes; 1H-NMR (300 MHz, CDCl3) δ 8.60 (s, 1H), 8.52 (d, J = 1.9 Hz, 1H), 8.09 (d, J = 8.4 Hz, 2H), 7.60 (d, J = 8.1 Hz, 1H), 7.38 (dd, J = 8.5, 1.9 Hz, 1H), 7.25 (d, J = 8.1 Hz, 2H, overlap with CHCl3 signal), 4.49–3.96 (m, 3H), 3.10 (s, 3H), 3.08–2.98 (m, 1H), 2.76–2.61 (m, 1H), 2.35 (s, 3H), 2.00–1.71 (m, 3H), 1.63–1.50 (m, 1H), 1.39 (s, 9H); ESI-MS: (m/z) 569.9 [M + H]+, 591.9 [M + Na]+, 568.0 [M − H]-; HPLC: tr = 11.456 min.
tert-Butyl-4-((7-chloro-9-tosyl-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)azepane-1-carboxylate (11c). 11c was prepared from 10 (465.0 mg, 1.19 mmol), 4c (338.4 mg, 1.48 mmol) and DIPEA (459.7 mg, 3.56 mmol) in dry DMF (14 mL) according to general procedure B. 682 mg of a beige solid (98% crude yield), which was used in the next step without further purification. A small batch was purified by flash column chromatography (SiO2, DCM:MeOH 97.5:2.5) for analytical purposes; 1H-NMR (400 MHz, DMSO-d6) δ 8.49 (s, 1H), 8.36 (d, J = 1.9 Hz, 1H), 8.07–8.01 (m, 2H), 7.79–7.73 (m, 1H), 7.52 (dd, J = 8.6, 1.6 Hz, 1H), 7.40 (d, J = 8.2 Hz, 2H), 4.28–4.18 (m, 1H), 3.49–3.35 (m, 2H), 3.29–3.12 (m, 2H), 3.04 (s, 3H), 2.33 (s, 3H), 1.96–1.76 (m, 5H), 1.67–1.53 (m, 1H), 1.36–1.28 (m, 9H); ESI-MS: (m/z) 606.0 [M + Na]+, 582.1 [M − H]−; HPLC: tr = 11.827 min.
tert-Butyl-4-((7-chloro-9-tosyl-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)piperidine-1-carboxylate (11d). 11d was prepared from 10 (200.0 mg, 0.51 mmol), 4d (142.1 mg, 0.66 mmol) and DIPEA (197.7 mg, 1.53 mmol) in dry DMF (5.5 mL) according to general procedure B. Beige solid (>100% crude yield), which was used in the next step without further purification. A small batch was purified by flash column chromatography (SiO2, petroleum ether:EtOAc gradient elution from 9:1 to 4:6) for analytical purposes; 1H-NMR (200 MHz, CDCl3) δ 8.59 (s, 1H), 8.52 (d, J = 1.8 Hz, 1H), 8.09 (d, J = 8.3 Hz, 2H), 7.54 (d, J = 8.6 Hz, 1H), 7.37 (dd, J = 8.5, 1.9 Hz, 1H), 7.26 (d, J = 8.2 Hz, 2H, overlap with CHCl3 signal), 4.49–4.33 (m, 1H), 4.33–4.16 (m, 2H), 3.07 (s, 3H), 2.94–2.68 (m, 2H), 2.36 (s, 3H), 1.92–1.72 (m, 4H), 1.47 (s, 9H); 13C-NMR (50 MHz, CDCl3) δ 160.1, 157.4, 154.8, 154.1, 145.8, 136.2, 135.5, 132.7, 129.8, 128.2, 124.4, 123.1, 120.5, 114.8, 100.8, 80.0, 56.6, 43.4 (br), 33.4, 29.0, 28.6, 21.8. ESI-MS: (m/z) 592.0 [M + Na]+, 568.0 [M − H]-; HPLC: tr = 11.988 min.
Detailed Procedures for the Preparation of Intermediates 12a–d
tert-Butyl-3-((7-chloro-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)pyrrolidine-1-carboxylate (12a). 12a was prepared from 11a (697.0 mg, 1.26 mmol) and KtBuO (986.1 mg, 8.79 mmol) in dry THF (40 mL) according to general procedure C in a reaction time of 1.5 h. Purification by flash column chromatography (SiO2, DCM:MeOH gradient elution from 97.5:2.5 to 93:7) gave 394 mg of a beige solid (78% yield); 1H-NMR (300 MHz, CDCl3) δ 11.68 (br s, 1H), 8.65–8.51 (m, 1H), 7.74 (d, J = 8.6 Hz, 1H), 7.53 (d, J = 1.8 Hz, 1H), 7.29 (dd, 1H, overlap with CHCl3 signal), 5.23–5.02 (m, 1H), 3.99–3.80 (m, 1H), 3.78–3.37 (m, 3H), 3.30 (s, 3H), 2.39–2.14 (m, 2H), 1.49 (s, 9H); 13C-NMR (50 MHz, DMSO-d6) δ 160.2, 157.4, 153.7, 153. 6, 137.5, 129.5, 124.0, 120.6, 118.3, 110.9, 98.1, 78.4, 56.8, 56.2, 47.0, 44.4, 44.1, 34.1, 28.2, 27.9, 27.0; ESI-MS: (m/z) 424.2 [M + Na]+, 400.2 [M − H]−; HPLC: tr = 9.653 min.
tert-Butyl-3-((7-chloro-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)piperidine-1-carboxylate (12b). 12b was prepared from 11b (580.0 mg, 1.02 mmol) and KtBuO (799.1 mg, 7.12 mmol) in dry THF (32 mL) according to general procedure C in a reaction time of 1 h. Purification by flash column chromatography (SiO2, DCM:MeOH gradient elution from 97.5:2.5 to 93:7) gave 320 mg of a beige solid (76% yield); 1H-NMR (200 MHz, CDCl3) δ 12.25 (br s, 1H), 8.57 (s, 1H), 7.70 (d, J = 8.6 Hz, 1H), 7.50 (d, J = 1.8 Hz, 1H), 7.24 (dd, 1H, overlap with CHCl3 signal), 4.59–3.95 (m, 3H), 3.26 (s, 3H), 3.17–2.96 (m, 1H), 2.84–2.56 (m, 1H), 2.19–1.55 (m, 4H), 1.43 (s, 9H); 13C-NMR (50 MHz, CDCl3) δ 160.2, 157.2, 155.0, 152.5, 137.5, 131.1, 123.7, 121.5, 118.8, 111.6, 98.7, 80.0, 55.2, 46.8, 44.1, 33.4, 28.5, 28.2, 25.0; ESI-MS: (m/z) 438.0 [M + Na]+, 413.9 [M − H]−; HPLC: tr = 9.001 min.
tert-Butyl-4-((7-chloro-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)azepane-1-carboxylate (12c). 12c was prepared from 11c (682.0 mg, 1.17 mmol) and KtBuO (917.0 mg, 8.17 mmol) in dry THF (40 mL) following general procedure C in a reaction time of 0.75 h. Purification by flash column chromatography (SiO2, DCM:MeOH gradient elution from 97.5:2.5 to 93:7) gave 392 mg of a beige solid (78% yield); 1H-NMR (400 MHz, CDCl3) δ 12.43 (br s, 1H), 8.55 (s, 1H), 7.69–7.62 (m, 1H), 7.50–7.45 (m, 1H), 7.25–7.20 (m, 1H), 4.64–4.52 (m, 1H), 3.88–3.78 (m, 0.5H), 3.72–3.63 (m, 0.5H), 3.57–3.38 (m, 2H), 3.28–3.15 (m, 4H), 2.11–1.67 (m, 6H), 1.47 (s, 9H); 13C-NMR (101 MHz, CDCl3) δ 159.9, 157.3, 155.7, 152.7, 137.5, 130.9, 123.5, 121.3, 119.0, 111.6, 98.4, 79.6, 58.9, 58.4, 46.9, 46.0, 43.6, 43.5, 33.1, 32.3, 31.82, 31.79, 31.5, 28.7, 25.43, 25.36; ESI-MS: (m/z) 430.0 [M + H]+, 452.0 [M + Na]+, 428.0 [M − H]−; HPLC: tr = 9.734 min.
tert-Butyl-4-((7-chloro-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)piperidine-1-carboxylate (12d). 12d was prepared from 11d (290.8 mg, 0.51 mmol) and KtBuO (400.7 mg, 3.57 mmol) in HPLC grade THF (32 mL) according to general procedure C in a reaction time of 2 h. Purification by flash column chromatography (SiO2, DCM:MeOH gradient elution from 97.5:2.5 to 93:7) gave 138 mg of an off-white solid (65% yield); 1H-NMR (400 MHz, CDCl3) δ 12.09 (s, 1H), 8.56 (s, 1H), 7.67 (d, J = 8.6 Hz, 1H), 7.48 (d, J = 1.9 Hz, 1H), 7.24 (dd, J = 8.6, 1.9 Hz, 1H), 4.70–4.58 (m, 1H), 4.46–4.11 (m, 2H), 3.23 (s, 3H), 2.99–2.75 (m, 2H), 1.96–1.80 (m, 4H), 1.50 (s, 9H); 13C-NMR (50 MHz, CDCl3) δ 160.0, 157.2, 154.9, 152.5, 137.5, 130.9, 123.4, 121.2, 118.9, 111.6, 98.3, 79.9, 55.9, 43.6, 33.2, 29.1, 28.6; ESI-MS: (m/z) 416.1 [M + H]+, 438.1 [M + Na]+, 414.1 [M − H]−; HPLC: tr = 10.236 min.
Detailed Procedures for the Preparation of Intermediates 13a–d
7-Chloro-N-methyl-N-(pyrrolidin-3-yl)-9H-pyrimido[4,5-b]indol-4-amine (13a). 13a was prepared from 12a (340.0 mg, 0.85 mmol) and TFA (1.8 mL) in dry DCM (9 mL) according to general procedure D. 220 mg of a beige solid (86% yield), which was used in the next step without further purification; 1H-NMR (300 MHz, DMSO-d6) δ 8.40 (s, 1H), 7.83 (d, J = 8.6 Hz, 1H), 7.48 (d, J = 2.0 Hz, 1H), 7.27 (dd, J = 8.6, 2.0 Hz, 1H), 4.97–4.85 (m, 1H), 3.17 (s, 3H), 3.14–3.07 (m, 1H), 3.00–2.75 (m, 3H), 2.12–1.98 (m, 1H), 1.94–1.80 (m, 1H). Both N-H not detected due to hydrogen bonding; 13C-NMR (75 MHz, DMSO-d6) δ 160.1, 157.4, 153.7, 137.4, 129.2, 123.8, 120.4, 118.6, 110.8, 97.6, 58.8, 49.2, 46.4, 33.4, 29.0; ESI-MS: (m/z) 301.9 [M + H]+, 299.9 [M − H]−, HPLC: tr = 3.556 min.
7-Chloro-N-methyl-N-(piperidin-3-yl)-9H-pyrimido[4,5-b]indol-4-amine (13b). 13b was prepared from 12b (314.0 mg, 0.76 mmol) and TFA (1.7 mL) in dry DCM (8.5 mL) according to general procedure D in a reaction time of 1 h. 236 mg of a beige solid (99% yield), which was used in the next step without further purification; 1H-NMR (300 MHz, MeOD) δ 8.34 (s, 1H), 7.75 (d, J = 8.6 Hz, 1H), 7.49 (d, J = 1.9 Hz, 1H), 7.26 (dd, J = 8.6, 2.0 Hz, 1H), 4.48 (tt, J = 11.2, 4.0 Hz, 1H), 3.25 (s, 3H), 3.17–3.08 (m, 1H), 3.05–2.96 (m, 1H), 2.96–2.86 (m, 1H), 2.63–2.49 (m, 1H), 2.16–1.85 (m, 3H), 1.78–1.60 (m, 1H); 13C-NMR (50 MHz, DMSO-d6) δ 159.6, 157.5, 153.7, 137.4, 129.1, 123.8, 120.4, 118.7, 110.8, 97.1, 55.7, 48.6, 45.5, 32.7, 28.2, 26.4; ESI-MS: (m/z) 316.0 [M + H]+, 338.0 [M + Na]+, 313.9 [M − H]-; HPLC: tr = 3.815 min.
N-(Azepan-4-yl)-7-chloro-N-methyl-9H-pyrimido[4,5-b]indol-4-amine (13c). 13c was prepared from 12c (125.0 mg, 0.29 mmol) and TFA (1 mL) in dry DCM (5 mL) according to general procedure D in a reaction time of 1.5 h. Purification by flash column chromatography (SiO2, DCM:2N NH3 in MeOH 9:1) gave 71 mg of a beige solid (74% yield); 1H-NMR (400 MHz, CDCl3) δ 8.53 (s, 1H), 7.66 (d, J = 8.6 Hz, 1H), 7.44 (d, J = 1.6 Hz, 1H), 7.20 (dd, J = 8.6, 1.8 Hz, 1H), 4.75–4.66 (m, 1H), 3.23 (s, 3H), 3.12–2.99 (m, 2H), 2.96–2.87 (m, 2H), 2.13–1.96 (m, 4H), 1.95–1.85 (m, 1H), 1.78–1.66 (m, 1H). Both N-H not detected due to hydrogen bonding; 13C-NMR (101 MHz, CDCl3) δ 160.1, 157.8, 153.1, 137.5, 130.7, 123.5, 121.1, 119.2, 111.5, 98.5, 58.6, 48.9, 46.1, 35.0, 32.7, 31.6, 27.6; ESI-MS: (m/z) 329.9 [M + H]+, 328.0 [M − H]-; HPLC: tr = 3.401 min.
7-Chloro-N-methyl-N-(piperidin-4-yl)-9H-pyrimido[4,5-b]indol-4-amine (13d). 13d was prepared from 12d (105.0 mg, 0.25 mmol) and TFA (0.6 mL) in dry DCM (3 mL) according to general procedure D in a reaction time of 1 h. 73 mg of a beige solid (92% yield) which was used in the next step without further purification; 1H-NMR (400 MHz, MeOD) δ 8.34 (s, 1H), 7.73 (d, J = 8.6 Hz, 1H), 7.50 (d, J = 1.9 Hz, 1H), 7.26 (dd, J = 8.6, 2.0 Hz, 1H), 4.55–4.46 (m, 1H), 3.23 (s, 3H), 3.21–3.13 (m, 2H), 2.76–2.66 (m, 2H), 2.02–1.90 (m, 2H), 1.90–1.83 (m, 2H); ESI-MS: (m/z) 316.0 [M + H]+, 314.0 [M − H]-; HPLC: tr = 3.485 min.
Detailed Procedures for the Preparation of Compounds 14a–o
3-(3-((7-Chloro-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)piperidin-1-yl)-3-oxopropanenitrile (14a). Cyanoacetic acid (23.1 mg, 0.27 mmol) and PyBOP (141.4 mg, 0.27 mmol) were dissolved in dry DCM (5 mL) and stirred at rt and under N2 atmosphere for 20 min. A suspension of 13b (71.5 mg, 0.23 mmol) and DIPEA (87.8 mg, 0.68 mmol) in dry DCM (5 mL) was added and the mixture stirred at rt and under N2 atmosphere for 1.5 h. The mixture was then diluted with DCM, washed with saturated NaHCO3 solution (4 × 15 mL), dried over Na2SO4 and concentrated under reduced pressure. Purification of the residue by flash column chromatography (SiO2, 1. DCM:MeOH gradient elution from 95:5 to 92.5:7.5, 2. DCM:EtOH gradient elution from 96.5:3.5 to 93:7) gave 51 mg of a white solid (59% yield); NMR shows a 7:3 mixture of amide bond rotamers, 1H (400 MHz, CDCl3) δ 12.59 (br s, 1H), 8.51–8.30 (m, 1H), 7.67–7.48 (m, 1H), 7.41–7.30 (m, 1H), 7.21–7.09 (m, 1H), 4.90–4.78 (m, 0.3H), 4.70–4.58 (m, 0.7H), 4.45–4.23 (m, 1H), 4.22–4.11 (m, 0.7H), 4.04–3.79 (m, 1.4H), 3.78–3.69 (m, 0.3H), 3.66–3.54 (m, 0.6H), 3.30–3.12 (m, 4H), 3.05–2.94 (m, 0.3H), 2.70–2.56 (m, 0.7H), 2.26–1.88 (m, 3H), 1.86–1.64 (m, 1H); 13C-NMR (101 MHz, CDCl3) δ 161.2, 160.6, 159.8, 157.2, 156.7, 152.5, 152.0, 137.5, 137.4, 131.24, 131.17, 123.6, 123.5, 121.5, 121.4, 118.41, 118.35, 114.7, 114.1, 111.7, 111.6, 98.5, 77.4, 55.5, 54.6, 48.2, 46.9, 45.4, 43.5, 34.9, 34.0, 28.7, 27.6, 25.40, 25.37, 25.2, 24.8; ESI-MS: (m/z) 383.1 [M + H]+, 405.1[M + Na]+, 380.9 [M − H]-; HPLC: tr = 6.202 min (100.0% purity).
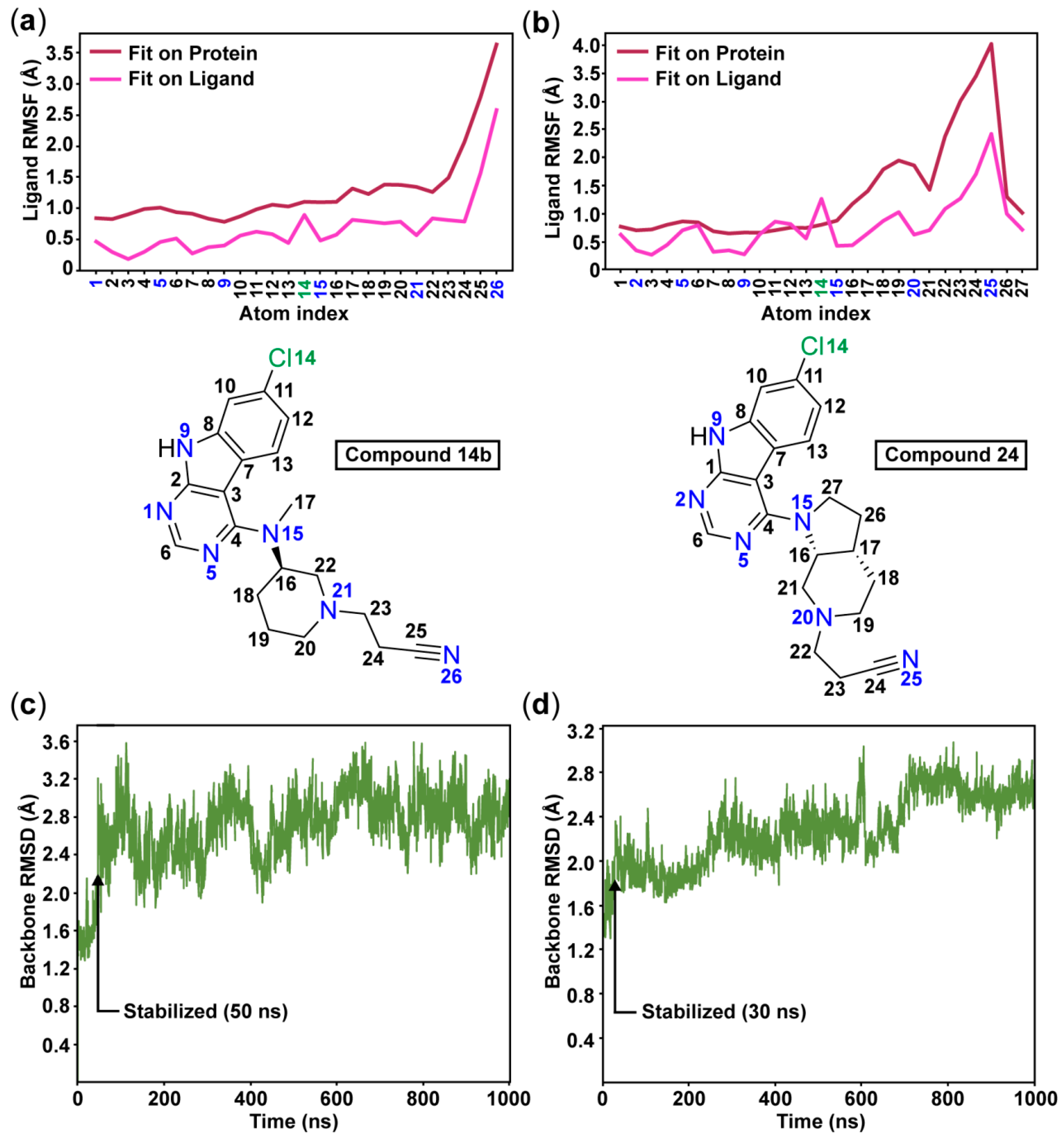
3-(3-((7-Chloro-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)piperidin-1-yl)propanenitrile (14b). 13b (125.0 mg, 0.40 mmol) and acrylonitrile (46.2 mg, 0.87 mmol) were stirred in dry MeOH (35 mL) at rt and under N2 atmosphere overnight. Volatiles were removed under reduced pressure. Purification of the residue by flash column chromatography (SiO2, DCM:MeOH gradient elution from 95.5:4.5 to 93.5:6.5) gave 92 mg of a white solid (63% yield); 1H-NMR (400 MHz, DMSO-d6) δ 12.22 (s, 1H), 8.40 (s, 1H), 7.78 (d, J = 8.6 Hz, 1H), 7.48 (s, 1H), 7.34 (d, J = 8.4 Hz, 1H), 4.56–4.33 (m, 1H), 3.14 (s, 3H), 3.11–3.03 (m, 1H), 2.92–2.81 (m, 1H), 2.78–2.68 (m, 2H), 2.68–2.58 (m, 2H), 2.44–2.32 (m, 1H), 2.02–1.88 (m, 1H), 1.84–1.63 (m, 3H), 1.59–1.41 (m, 1H); 13C-NMR (101 MHz, DMSO-d6) δ 159.4, 157.4, 153.7, 137.3, 129.1, 123.8, 120.6, 120.0, 118.5, 110.8, 97.0, 55.7, 54.7, 53.0, 52.2, 32.5, 27.2, 24.3, 15.0. ESI-MS: (m/z) 369.1 [M + H]+, 391.0 [M + Na]+, 366.9 [M − H]-; HPLC: tr = 3.695 min (100.0% purity).
3-(3-((7-Chloro-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)piperidin-1-yl)propanamide (14c). 13b (65.0 mg, 0.21 mmol) and acrylamide (16.1 mg, 0.23 mmol) were stirred in dry MeOH (11 mL) at rt and under N2 atmosphere overnight. Additional acrylamide (16.1 mg, 0.23 mmol) was added and stirring at rt continued for 5 days. Volatiles were removed under reduced pressure. Purification of the residue by flash column chromatography (SiO2, DCM:2N NH3 in MeOH gradient elution from 92:8 to 9:1) gave 65 mg of a white solid (82% yield); 1H-NMR (400 MHz, DMSO-d6) δ 12.21 (s, 1H), 8.39 (s, 1H), 7.76 (d, J = 8.7 Hz, 1H), 7.48 (d, J = 2.0 Hz, 1H), 7.34 (br s, 1H), 7.30 (dd, J = 8.6, 2.0 Hz, 1H), 6.78 (br s, 1H), 4.50–4.34 (m, 1H), 3.15 (s, 3H), 3.05–2.96 (m, 1H), 2.87–2.77 (m, 1H), 2.65–2.52 (m, 2H), 2.35–2.18 (m, 3H), 1.96–1.85 (m, 1H), 1.84–1.65 (m, 3H), 1.58–1.42 (m, 1H); 13C-NMR (101 MHz, DMSO-d6) δ 173.2, 159.4, 157.4, 153.7, 137.3, 129.1, 123.8, 120.5, 118.5, 110.8, 97.0, 55.9, 54.8, 54.2, 52.7, 33.0, 32.6, 27.4, 24.4; ESI-MS: (m/z) 387.4 [M + H]+, 409.4 [M + Na]+, 385.3 [M − H]−; HPLC: tr = 3.872 min (100.0% purity).
Methyl-3-(3-((7-chloro-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)piperidin-1-yl)propanoate (14d). 13b (65.0 mg, 0.21 mmol) and methyl acrylate (19.5 mg, 0.23 mmol) were stirred in dry MeOH (11 mL) at rt and under N2 atmosphere for 3 h. Additional methyl acrylate (4.4 mg, 0.05 mmol) was added and stirring at rt continued for 1 h. Volatiles were removed under reduced pressure. Purification of the residue by flash column chromatography (SiO2, DCM:MeOH gradient elution from 96:4 to 92:8) gave 74 mg of a beige solid (89% yield); 1H-NMR (400 MHz, DMSO-d6) δ 12.21 (s, 1H), 8.39 (s, 1H), 7.75 (d, J = 8.7 Hz, 1H), 7.47 (d, J = 2.0 Hz, 1H), 7.28 (dd, J = 8.6, 2.0 Hz, 1H), 4.45–4.34 (m, 1H), 3.55 (s, 3H), 3.14 (s, 3H), 3.00–2.92 (m, 1H), 2.84–2.75 (m, 1H), 2.62 (t, J = 6.9 Hz, 2H), 2.53–2.46 (m, 2H, overlap with DMSO-d5 signal), 2.36–2.26 (m, 1H), 1.95–1.85 (m, 1H), 1.83–1.65 (m, 3H), 1.55–1.41 (m, 1H); 13C-NMR (101 MHz, DMSO-d6) δ 172.4, 159.4, 157.4, 153.7, 137.3, 129.1, 123.8, 120.4, 118.5, 110.8, 97.0, 55.9, 54.8, 53.3, 52.5, 51.1, 32.6, 31.6, 27.3, 24.4; ESI-MS: (m/z) 402.5 [M + H]+, 424.6 [M + Na]+, 400.3 [M − H]-; HPLC: tr = 4.363 min (100.0% purity).
7-Chloro-N-methyl-N-(1-propylpiperidin-3-yl)-9H-pyrimido[4,5-b]indol-4-amine (14e). 14e was prepared from 13b (65.0 mg, 0.21 mmol), propionaldehyde (17.9 mg, 0.31 mmol), glacial AcOH (24.7 mg, 0.41 mmol) and Na(OAc)3BH (87.2 mg, 0.41 mmol) in dry DCM (10 mL) according to general procedure E in a reaction time of 2 h. Purification by flash column chromatography (SiO2, 1. DCM:2N NH3 in MeOH gradient elution from 95:5 to 92:8, 2. DCM:2N NH3 in MeOH gradient elution from 94:6 to 91.5:8.5) gave 46 mg of a white solid (62% yield); 1H-NMR (400 MHz, DMSO-d6) δ 2.21 (s, 1H), 8.39 (s, 1H), 7.78 (d, J = 8.7 Hz, 1H), 7.47 (d, J = 2.0 Hz, 1H), 7.23 (dd, J = 8.6, 2.0 Hz, 1H), 4.48–4.37 (m, 1H), 3.14 (s, 3H), 3.00–2.91 (m, 1H), 2.84–2.76 (m, 1H), 2.33–2.17 (m, 3H), 1.87–1.66 (m, 4H), 1.58–1.39 (m, 3H), 0.84 (t, J = 7.3 Hz, 3H); 13C-NMR (101 MHz, DMSO-d6) δ 159.4, 157.4, 153.7, 137.3, 129.1, 123.8, 120.3, 118.6, 110.8, 97.0, 60.0, 56.3, 54.9, 52.9, 32.6, 27.6, 24.5, 19.6, 11.7; ESI-MS: (m/z) 358.2 [M + H]+, 380.1 [M + Na]+, 356.2 [M − H]-; HPLC: tr = 4.200 min (99.3% purity).
7-Chloro-N-(1-isopropylpiperidin-3-yl)-N-methyl-9H-pyrimido[4,5-b]indol-4-amine (14f). 14f was prepared from 13b (62.0 mg, 0.20 mmol), acetone (228.1 mg, 3.93 mmol), glacial AcOH (23.6 mg, 0.39 mmol) and Na(OAc)3BH (83.2 mg, 0.39 mmol) in dry DCM (10 mL) according to general procedure E in a reaction time of 5 h. A second portion of acetone (228.1 mg, 3.93 mmol) was added after 3 h. Purification by flash column chromatography (SiO2, DCM:2N NH3 in MeOH 95:5 to 92.5:7.5) gave 30 mg of a beige solid (43% yield); 1H-NMR (400 MHz, DMSO-d6) δ 12.20 (s, 1H), 8.39 (s, 1H), 7.79 (d, J = 8.7 Hz, 1H), 7.47 (d, J = 2.0 Hz, 1H), 7.24 (dd, J = 8.6, 2.0 Hz, 1H), 4.46–4.35 (m, 1H), 3.16 (s, 3H), 2.96–2.86 (m, 1H), 2.79–2.66 (m, 2H), 2.48–2.41 (m, 1H), 2.16–2.03 (m, 1H), 1.86–1.67 (m, 3H), 1.55–1.41 (m, 1H), 1.05–0.92 (m, 6H); 13C-NMR (50 MHz, DMSO-d6) δ 159.4, 157.4, 153.8, 137.3, 129.1, 123.9, 120.3, 118.6, 110.8, 97.0, 55.5, 54.0, 51.3, 48.1, 32.7, 28.0, 24.8, 18.1, 17.7; ESI-MS: (m/z) 357.8 [M + H]+, 355.8 [M − H]-; HPLC: tr = 4.291 min (99.0% purity).
7-Chloro-N-(1-cyclopentylpiperidin-3-yl)-N-methyl-9H-pyrimido[4,5-b]indol-4-amine (14g). 14g was prepared from 13b (60.0 mg, 0.19 mmol), cyclopentanone (63.9 mg, 0.76 mmol), glacial AcOH (22.8 mg, 0.38 mmol) and Na(OAc)3BH (80.5 mg, 0.38 mmol) in dry DCM (10 mL) according to general procedure E in a reaction time of 3 h. Purification by flash column chromatography (SiO2, DCM:2N NH3 MeOH gradient elution from 96:4 to 93:7) gave 54 mg of a white solid (74% yield); 1H-NMR (400 MHz, DMSO-d6) δ 12.21 (s, 1H), 8.38 (s, 1H), 7.79 (d, J = 8.7 Hz, 1H), 7.47 (d, J = 1.9 Hz, 1H), 7.24 (dd, J = 8.6, 2.0 Hz, 1H), 4.47–4.35 (m, 1H), 3.14 (s, 3H), 3.09–3.01 (m, 1H), 2.94–2.84 (m, 1H), 2.58–2.52 (m, 1H, overlap with DMSO-d5), 2.30–2.17 (m, 1H), 1.93–1.67 (m, 6H), 1.64–1.39 (m, 5H), 1.38–1.25 (m, 2H); 13C-NMR (101 MHz, DMSO-d6) δ 159.4, 157.4, 153.7, 137.3, 129.1, 123.8, 120.2, 118.6, 110.8, 96.9, 66.7, 55.0, 54.8, 51.5, 32.6, 29.8, 29.6, 27.6, 24.5, 23.6; ESI-MS: (m/z) 383.8 [M + H]+, 381.8 [M − H]-; HPLC: tr = 4.839 min (100.0% purity).
7-Chloro-N-(1-(furan-2-ylmethyl)piperidin-3-yl)-N-methyl-9H-pyrimido[4,5-b]indol-4-amine (14h). 14h was prepared from 13b (80.0 mg, 0.25 mmol), furan-2-carbaldehyde (30.4 mg, 0.32 mmol), glacial AcOH (30.4 mg, 0.51 mmol) and Na(OAc)3BH (80.5 mg, 0.38 mmol) in dry DCM (10 mL) according to general procedure E in a reaction time of 6.5 h. Purification by flash column chromatography (SiO2, 1.DCM:MeOH gradient elution from 96:4 to 92.5:7.5, 2.DCM:MeOH gradient elution from 96:4 to 92:8) gave 55 mg of an off-white solid (55% yield); 1H-NMR (300 MHz, CDCl3) δ 11.76 (br s, 1H), 8.54 (s, 1H), 7.63 (d, J = 8.6 Hz, 1H), 7.49–7.37 (m, 2H), 7.21 (dd, J = 8.6, 1.8 Hz, 1H), 6.39–6.31 (m, 1H), 6.30–6.22 (m, 1H), 4.69–4.50 (m, 1H), 3.71 (d, J = 14.2 Hz, 1H), 3.64 (d, J = 14.2 Hz, 1H), 3.28–3.14 (m, 4H), 3.03–2.92 (m, 1H), 2.50–2.37 (m, 1H), 2.14–2.02 (m, 1H), 2.00–1.90 (m, 1H), 1.88–1.71 (m, 3H); 13C-NMR (50 MHz, CDCl3) δ 160.3, 157.6, 153.1, 151.4, 142.5, 137.3, 130.8, 123.7, 121.3, 119.0, 111.4, 110.4, 109.4, 98.7, 56.1, 55.7, 54.9, 53.2, 33.2, 28.0, 24.7.; ESI-MS: (m/z) 396.1 [M + H]+, 418.1 [M + Na]+, 393.9 [M − H]-; HPLC: tr = 4.781 min (99.7% purity).
7-Chloro-N-methyl-N-(1-(pyridin-4-ylmethyl)piperidin-3-yl)-9H-pyrimido[4,5-b]indol-4-amine (14i). 14i was prepared from 13b (75.0 mg, 0.24 mmol), pyridin-4-carbaldehyde (38.2 mg, 0.36 mmol), glacial AcOH (28.5 mg, 0.48 mmol) and Na(OAc)3BH (100.7 mg, 0.48 mmol) in dry DCM (10 mL) according to general procedure E in a reaction time of 2.5 h. Purification by flash column chromatography (SiO2, DCM:MeOH gradient elution from 95:5 to 90.5:9.5) gave 71 mg of a white solid (73% yield); 1H-NMR (400 MHz, DMSO-d6) δ 12.22 (s, 1H), 8.49 (d, J = 5.7 Hz, 2H), 8.37 (s, 1H), 7.69 (d, J = 8.6 Hz, 1H), 7.48 (d, J = 1.8 Hz, 1H), 7.32 (d, J = 5.6 Hz, 2H), 7.22 (dd, J = 8.6, 1.8 Hz, 1H), 4.54–4.41 (m, 1H), 3.59–3.48 (m, 2H), 3.13 (s, 3H), 2.98–2.88 (m, 1H), 2.80–2.70 (m, 1H), 2.37–2.27 (m, 1H), 2.00–1.89 (m, 1H), 1.86–1.65 (m, 3H), 1.62–1.48 (m, 1H); 13C-NMR (50 MHz, DMSO-d6) δ 159.4, 157.5, 153.7, 149.5, 147.5, 137.4, 129.2, 123.8, 123.7, 120.3, 118.5, 110.9, 97.0, 60.8, 55.9, 54.8, 52.8, 32.8, 27.2, 24.4; ESI-MS: (m/z) 407.4 [M + H]+, 405.2 [M − H]−; HPLC: tr = 6.121 min (99.5% purity).
3-((3-((7-Chloro-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)piperidin-1-yl)methyl)benzonitrile (14j). 14j was prepared from 13b (25.0 mg, 0.08 mmol), 3-formylbenzonitrile (31.1 mg, 0.24 mmol), glacial AcOH (9.5 mg, 0.16 mmol) and Na(OAc)3BH (33.6 mg, 0.16 mmol) in dry DCM (4 mL) according to general procedure E in a reaction time of 4 h. Purification by flash column chromatography (SiO2, DCM:MeOH gradient elution from 96:4 to 93:7) gave 20 mg of a white solid (59% yield); 1H-NMR (400 MHz, DMSO-d6) δ 12.20 (s, 1H), 8.37 (s, 1H), 7.77 (br s, 1H), 7.75–7.64 (m, 3H), 7.54 (t, J = 7.7 Hz, 1H), 7.47 (d, J = 2.0 Hz, 1H), 7.21 (dd, J = 8.6, 2.0 Hz, 1H), 4.54–4.41 (m, 1H), 3.61 (d, J = 13.7 Hz, 1H), 3.54 (d, J = 13.7 Hz, 1H), 3.14 (s, 3H), 3.01–2.91 (m, 1H), 2.82–2.72 (m, 1H), 2.37–2.26 (m, 1H), 2.02–1.90 (m, 1H), 1.84–1.67 (m, 3H), 1.61–1.48 (m, 1H); 13C-NMR (50 MHz, DMSO-d6) δ 159.4, 157.4, 153.7, 140.2, 137.4, 133.7, 132.1, 130.9, 129.4, 129.2, 123.8, 120.3, 118.9, 118.5, 111.2, 110.9, 97.0, 61.0, 55.6, 54.8, 52.8, 32.8, 27.2, 24.4; ESI-MS: (m/z) 431.2 [M + H]+, 453.2 [M + Na]+, 429.2 [M − H]-; HPLC: tr = 4.879 min (100.0% purity).
7-Chloro-N-methyl-N-(1-(3,3,3-trifluoropropyl)piperidin-3-yl)-9H-pyrimido[4,5-b]indol-4-amine (14k). 14k was prepared from 13b (55.0 mg, 0.17 mmol), trifluoropropanal (97.6 mg, 0.87 mmol), glacial AcOH (20.9 mg, 0.35 mmol) and Na(OAc)3BH (73.8 mg, 0.35 mmol) in dry DCM (10 mL) according to general procedure E in a reaction time of 3 h. Purification by flash column chromatography (SiO2, DCM:MeOH gradient elution from 97.5:2.5 to 92.5:7.5) gave 38 mg of a white solid (53% yield); 1H-NMR (400 MHz, DMSO-d6) δ 12.20 (s, 1H), 8.39 (s, 1H), 7.76 (d, J = 8.7 Hz, 1H), 7.48 (d, J = 2.0 Hz, 1H), 7.26 (dd, J = 8.6, 2.0 Hz, 1H), 4.49–4.37 (m, 1H), 3.15 (s, 3H), 3.03–2.96 (m, 1H), 2.88–2.79 (m, 1H), 2.61–2.53 (m, 2H), 2.51–2.41 (m, 2H, overlapping with DMSO-d5), 2.37–2.26 (m, 1H), 1.96–1.87 (m, 1H), 1.86–1.67 (m, 3H), 1.59–1.45 (m, 1H); 13C-NMR (101 MHz, DMSO-d6) δ 159.4, 157.4, 153.7, 137.3, 129.1, 127.2 (q, J = 276.9 Hz), 123.7, 120.3, 118.5, 110.8, 97.0, 55.8, 54.7, 52.4, 50.4 (q), 32.6, 30.4 (q, J = 26.3 Hz), 27.2, 24.3; ESI-MS: (m/z) 412.3 [M + H]+, 434.4 [M + Na]+, 410.3 [M − H]-; HPLC: tr = 7.463 min (97.2% purity).
2-(3-((7-Chloro-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)piperidin-1-yl)acetonitrile (14l). 13b (40.0 mg, 0.13 mmol) was dissolved in dry DMF (2 mL). 2-Bromoacetonitrile (16.7 mg, 0.14 mmol) and Et3N (38.5 mg, 0.38 mmol) were added and the mixture stirred at rt for 1.5 h. Saturated NaHCO3 solution (5 mL) was added and the mixture extracted with EtOAc (6 × 2 mL). Combined organic layers were diluted with additional EtOAc, washed with saturated NaHCO3 solution (3 × 20 mL), dried over Na2SO4 and concentrated under reduced pressure. Purification of the residue by flash column chromatography (SiO2, DCM:MeOH 96:4) gave 41 mg of a beige solid (91% yield); 1H-NMR (400 MHz, DMSO-d6) δ 12.22 (s, 1H), 8.41 (s, 1H), 7.76 (d, J = 8.6 Hz, 1H), 7.48 (d, J = 1.9 Hz, 1H), 7.27 (dd, J = 8.6, 2.0 Hz, 1H), 4.52–4.41 (m, 1H), 3.75 (s, 2H), 3.18 (s, 3H), 2.95–2.87 (m, 1H), 2.81–2.72 (m, 1H), 2.48–2.42 (m, 1H), 2.20–2.09 (m, 1H), 1.95–1.72 (m, 3H), 1.66–1.53 (m, 1H); 13C-NMR (101 MHz, DMSO-d6) δ 159.5, 157.4, 153.7, 137.4, 129.2, 123.8, 120.4, 118.4, 115.8, 110.8, 97.2, 54.4, 54.1, 51.3, 45.3, 32.8, 26.6, 24.1; ESI-MS: (m/z) 355.0 [M + H]+, 376.9 [M + Na]+, 352.9 [M − H]-; HPLC: tr = 8.358 min (99.4% purity).
7-Chloro-N-(1-(cyclopropylmethyl)piperidin-3-yl)-N-methyl-9H-pyrimido[4,5-b]indol-4-amine (14m). (Bromomethyl)cyclopropane (34.4 mg, 0.25 mmol) and Et3N (33.6 mg, 0.33 mmol) were added to a suspension of 13b (70.0 mg, 0.22 mmol) in HPLC grade acetonitrile (10 mL). The mixture was stirred at 60 °C for 2 days and then concentrated under reduced pressure. Purification of the residue by flash column chromatography (SiO2, DCM:2N NH3 in MeOH gradient elution from 98:2 to 91.5:8.5) gave 64 mg of a white solid (78% yield); 1H-NMR (400 MHz, DMSO-d6) δ 12.22 (s, 1H), 8.39 (s, 1H), 7.80 (d, J = 8.7 Hz, 1H), 7.47 (d, J = 2.0 Hz, 1H), 7.25 (dd, J = 8.6, 2.0 Hz, 1H), 4.49–4.38 (m, 1H), 3.14 (s, 3H), 3.12–3.06 (m, 1H), 2.94–2.87 (m, 1H), 2.30–2.12 (m, 3H), 1.92–1.65 (m, 4H), 1.60–1.45 (m, 1H), 0.91–0.78 (m, 1H), 0.51–0.36 (m, 2H), 0.13–0.00 (m, 2H); 13C-NMR (101 MHz, DMSO-d6) δ 159.4, 157.4, 153.7, 137.3, 129.1, 123.8, 120.3, 118.6, 110.8, 97.0, 62.9, 56.1, 54.9, 52.8, 32.5, 27.6, 24.4, 8.3, 3.8, 3.6; ESI-MS: (m/z) 370.1 [M + H]+, 392.1 [M + Na]+, 368.2 [M − H]-; HPLC: tr = 4.318 min (100.0% purity).
7-Chloro-N-(1-(3-(dimethylamino)propyl)piperidin-3-yl)-N-methyl-9H-pyrimido[4,5-b]indol-4-amine (14n). 13b (100.0 mg, 0.32 mmol) and 3-chloro-N,N-dimethylpropan-1-amine hydrochloride (65.1 mg, 0.41 mmol) were suspended in HPLC grade acetonitrile (15 mL). Et3N (96.1 mg, 0.95 mmol) was added. The mixture was stirred at 90 °C for 2–3 days and then concentrated under reduced pressure. Purification of the residue by flash column chromatography (SiO2, DCM:2N NH3 in MeOH 9:1) gave 50 mg of an off-white solid (39% yield); 1H-NMR (300 MHz, CDCl3) δ 12.07 (br s, 1H), 8.54 (s, 1H), 7.69 (d, J = 8.6 Hz, 1H), 7.44 (d, J = 1.0 Hz, 1H), 7.21 (dd, J = 8.6, 1.2 Hz, 1H), 4.66–4.51 (m, 1H), 3.23 (s, 3H), 3.18–3.10 (m, 1H), 2.99–2.90 (m, 1H), 2.53–2.20 (m, 11H), 2.02–1.66 (m, 7H); 13C-NMR (101 MHz, CDCl3) δ 160.3, 157.8, 153.2, 137.5, 130.8, 123.7, 121.2, 119.2, 111.4, 98.6, 58.0, 57.0, 56.8, 55.6, 53.8, 45.3, 33.1, 28.5, 25.0, 24.9; ESI-MS: (m/z) 401.4 [M + H]+, 399.4 [M − H]-; HPLC: tr = 4.318 min.
3-(3-((7-Chloro-9-methyl-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)piperidin-1-yl)propanenitrile (14o). NaH (8.1 mg of a 60% dispersion in mineral oil, 0.20 mmol) was added to a stirring suspension of 14b (50.0 mg, 0.14 mmol) in dry THF (10 mL). The mixture was stirred at rt and under N2 atmosphere for 0.5 h. Methyl iodide (0.5 mL of a freshly prepared 0.4M solution in THF, 0.20 mmol) was drop-added and the mixture stirred for 20 h at rt and under N2 atmosphere. Dimethylamine (0.075 mL of a 2M solution in THF, 0.15 mmol) was added to quench excessive methyl iodide and stirring continued for 1 h. Saturated NH4Cl solution (15 mL) was added to stop the reaction and the mixture then basified with saturated NaHCO3 solution. EtOAc was added and phases were separated. The organic layer was washed with saturated NaHCO3 solution (2 × 20 mL), dried over Na2SO4 and concentrated under reduced pressure. Purification of the residue by flash column chromatography (SiO2, DCM:MeOH 96.7:3.3 to 93.5:6.5) gave 31 mg of a light beige solid (60% yield); 1H-NMR (400 MHz, DMSO-d6) δ 8.46 (s, 1H), 7.83–7.74 (m, 2H), 7.38 (dd, J = 8.6, 1.3 Hz, 1H), 4.49–4.39 (m, 1H), 3.83 (s, 3H), 3.14 (s, 3H), 3.11–3.03 (m, 1H), 2.90–2.82 (m, 1H), 2.76–2.59 (m, 4H), 2.43–2.34 (m, 1H), 2.00–1.89 (m, 1H), 1.81–1.66 (m, 3H), 1.55–1.42 (m, 1H); 13C-NMR (101 MHz, DMSO-d6) δ 159.3, 156.8, 153.5, 138.5, 129.5, 123.7, 120.8, 120.0, 118.0, 109.7, 96.7, 55.6, 54.9, 53.0, 52.2, 32.6, 27.9, 27.2, 24.3, 15.0; ESI-MS: (m/z) 383.0 [M + H]+, 405.0 [M + Na]+; HPLC: tr = 4.740 min (100.0% purity).
Detailed Procedures for the Preparation of Compounds 15–17
3-(3-((7-Chloro-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)pyrrolidin-1-yl)propanenitrile (15). 13a (60.0 mg, 0.20 mmol) and acrylonitrile (23.2 mg, 0.44 mmol) were stirred in dry MeOH (17 mL) at rt overnight. Volatiles were removed under reduced pressure. Purification of the residue by flash column chromatography (SiO2, DCM:MeOH 96:4 to 93:7) gave 51 mg of an off-white solid (72% yield); 1H-NMR (400 MHz, DMSO-d6) δ 12.22 (s, 1H), 8.41 (s, 1H), 7.84 (d, J = 8.6 Hz, 1H), 7.48 (d, J = 1.9 Hz, 1H), 7.29 (dd, J = 8.6, 1.9 Hz, 1H), 5.11–5.01 (m, 1H), 3.24 (s, 3H), 2.98–2.86 (m, 2H), 2.78–2.57 (m, 5H), 2.43–2.33 (m, 1H), 2.27–2.16 (m, 1H), 2.05–1.94 (m, 1H); 13C-NMR (101 MHz, DMSO-d6) δ 160.0, 157.3, 153.7, 137.4, 129.2, 123.8, 120.4, 119.8, 118.5, 110.8, 97.6, 56.8, 56.2, 52.9, 50.3, 33.3, 27.8, 16.5; ESI-MS: (m/z) 355.0 [M + H]+, 377.0 [M + Na]+, 353.0 [M − H]-; HPLC: tr = 3.414 min (100.0% purity).
3-(4-((7-Chloro-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)azepan-1-yl)propanenitrile (16). 13c (65.0 mg, 0.20 mmol) and acrylonitrile (23.0 mg, 0.43 mmol) were stirred in HPLC grade MeOH (20 mL) at rt and under N2 overnight. Additional acrylonitrile was added repeatedly, but conversion seized at ~ 80% as calculated by HPLC. Volatiles were removed under reduced pressure. Purification of the residue by flash column chromatography (SiO2, DCM:MeOH 95:5) gave 50 mg of a beige solid (66% yield); 1H-NMR (400 MHz, CDCl3) δ 12.59 (br s, 1H), 8.53 (s, 1H), 7.65 (d, J = 8.5 Hz, 1H), 7.44 (s, 1H), 7.21 (d, J = 8.5 Hz, 1H), 4.74–4.62 (m, 1H), 3.22 (s, 3H), 2.91 (t, J = 6.8 Hz, 2H), 2.87–2.71 (m, 4H), 2.52 (t, J = 6.6 Hz, 2H), 2.13–1.87 (m, 5H), 1.80–1.67 (m, 1H); 13C-NMR (101 MHz, CDCl3) δ 160.0, 157.7, 153.0, 137.4, 130.8, 123.5, 121.1, 119.1, 119.0, 111.5, 98.4, 58.4, 54.8, 54.0, 51.7, 32.9, 32.5, 30.9, 25.7, 16.8; ESI-MS: (m/z) 405.1 [M + Na]+, 381.1 [M − H]-; HPLC: tr = 3.102 min (98.4% purity).
3-(4-((7-Chloro-9H-pyrimido[4,5-b]indol-4-yl)(methyl)amino)piperidin-1-yl)propanenitrile (17). 13d (34.0 mg, 0.11 mmol) and acrylonitrile (8.6 mg, 0.16 mmol) were stirred in dry MeOH (10 mL) at rt and under N2 atmosphere overnight. Volatiles were removed under reduced pressure. Purification of the residue by flash column chromatography (SiO2, DCM:MeOH 97:3 to 95:5) gave 31 mg of a pale yellow solid (78% yield); 1H-NMR (200 MHz, DMSO-d6) δ 12.19 (s, 1H), 8.39 (s, 1H), 7.74 (d, J = 8.7 Hz, 1H), 7.48 (d, J = 1.9 Hz, 1H), 7.26 (dd, J = 8.6, 2.0 Hz, 1H), 4.41–4.19 (m, 1H), 3.14 (s, 3H), 3.07–2.91 (m, 2H), 2.73–2.53 (m, 4H), 2.19–1.65 (m, 6H); 13C-NMR (50 MHz, DMSO-d6) δ 159.6, 157.5, 153.8, 137.4, 129.2, 123.7, 120.4, 120.1, 118.6, 110.9, 97.2, 55.5, 52.7, 52.2, 32.5, 28.3, 15.2; ESI-MS: (m/z) 369.0 [M + H]-; 391.0 [M + Na]+, 367.0 [M − H]-; HPLC: tr = 3.474 min (100.0% purity).
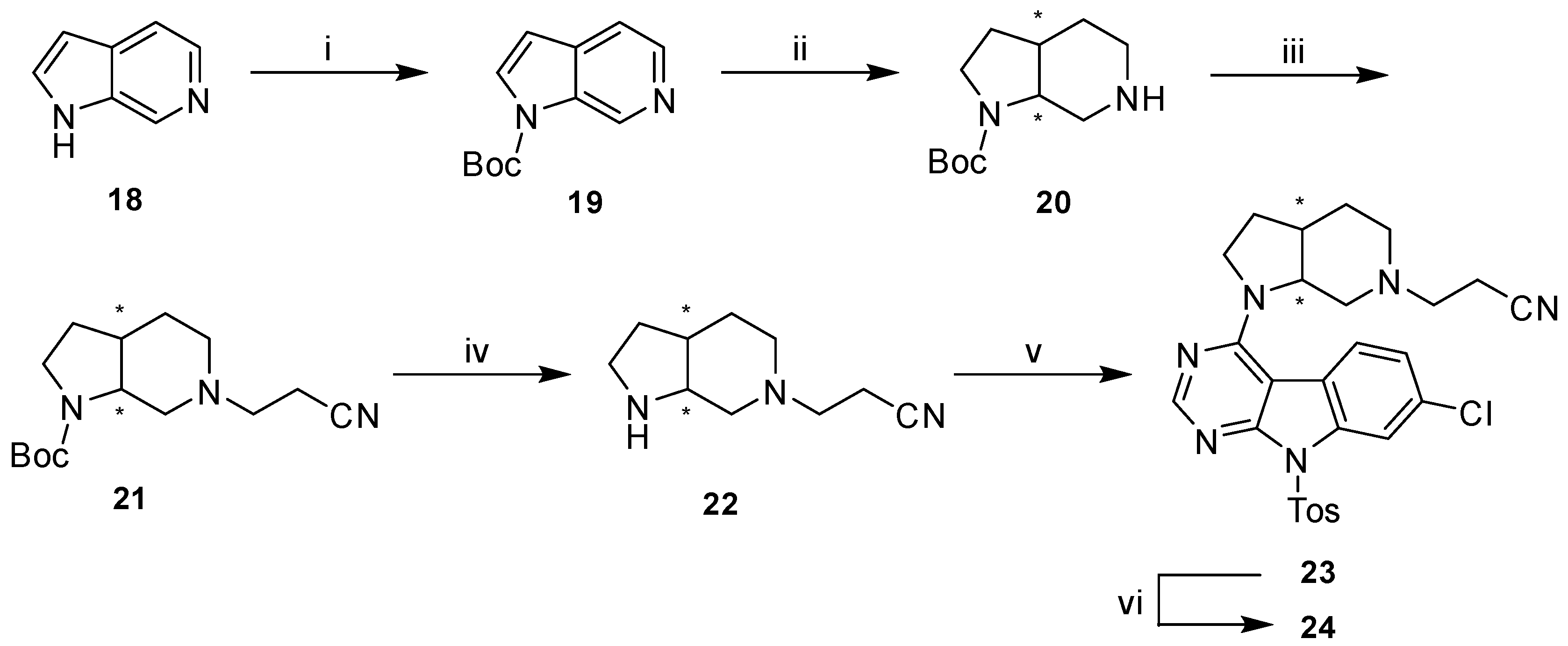
Detailed Procedures for the Preparation of Intermediates 19–23 and Compound 24
tert-Butyl-1H-pyrrolo[2,3-c]pyridine-1-carboxylate (19). A solution of 1H-pyrrolo[2,3-c]pyridine (18) (500.0 mg, 4.23 mmol) in dry THF (5 mL) was stirred under N2 atmosphere and with ice-cooling. Boc anhydride (1108.4 mg, 5.08 mmol) was added dropwise. The mixture was left to warm to rt and stirred overnight under N2 atmosphere. After diluting with EtOAc the mixture was washed with saturated NaHCO3 solution (2 × 20 mL) and saturated NaCl solution (20 mL), dried over Na2SO4 and concentrated under reduced pressure. Purification of the residue by flash column chromatography (SiO2, DCM:MeOH 97.5:2.5) gave 846 mg of a yellow oil (92% yield); 1H-NMR (300 MHz, DMSO-d6) δ 9.25 (s, 1H), 8.34 (d, J = 5.3 Hz, 1H), 7.86 (d, J = 3.6 Hz, 1H), 7.63 (dd, J = 5.3, 1.1 Hz, 1H), 6.77 (dd, J = 3.6, 0.6 Hz, 1H), 1.64 (s, 9H); 13C-NMR (75 MHz, DMSO-d6) δ 148.4, 141.7, 136.5, 135.5, 131.5, 129.5, 115.7, 106.5, 84.8, 27.6; HPLC: tr = 2.259 min.
tert-Butyl-octahydro-1H-pyrrolo[2,3-c]pyridine-1-carboxylate (20). 19 (746.0 mg, 3.42 mmol) was dissolved in glacial AcOH (50 mL) and PtO2 (150.0 mg, 20% (m/m)) was added. The mixture was stirred in a reactor charged with 5 bar of H2 pressure at rt for 35 h, then diluted with EtOAc and filtered over a pad of celite rinsing with EtOAc. The filtrate was concentrated under reduced pressure, redissolved in DCM and washed with saturated NaHCO3 solution (2 × 20 mL) adding saturated NaCl solution to improve phase separation. Combined aqueous layers were re-extracted with DCM (25 mL). Combined organic layers were dried over Na2SO4 and concentrated under reduced pressure to give 680 mg of a yellow oil (88% crude yield), which was used in the next step without further purification; GC-MS method A: tr = 4.974 min, (m/z) 226 [M].
tert-Butyl-6-(2-cyanoethyl)octahydro-1H-pyrrolo[2,3-c]pyridine-1-carboxylate (21). 20 (340.0 mg, 1.50 mmol) was dissolved in HPLC grade MeOH (50 mL) and acrylonitrile (175.4 mg, 3.31 mmol) was added. The mixture was stirred at rt and under N2 atmosphere overnight. Volatiles were removed under reduced pressure. Purification of the residue by flash column chromatography (SiO2, petroleum ether:EtOAc 35:65) gave 350 mg of a yellow oil (83% yield); 1H-NMR (300 MHz, CDCl3) δ 3.91–3.73 (m, 1H), 3.43–3.18 (m, 2H), 2.99–2.86 (m, 1H), 2.65 (t, J = 7.2 Hz, 2H), 2.60–2.48 (m, 1H), 2.44 (t, J = 6.8 Hz, 2H), 2.33–1.54 (m, 7H), 1.40 (s, 9H); 13C-NMR (75 MHz, CDCl3) δ 154.5, 154.3, 118.9, 118.8, 79.3, 79.1, 55.0, 53.8, 53.4, 53.1, 49.2, 48.6, 45.4, 44.9, 35.1, 34.5, 28.5, 26.7, 25.8, 25.7, 25.5, 15.8; GC-MS method A: tr = 8.701 min, (m/z) 279 [M].
3-(Octahydro-6H-pyrrolo[2,3-c]pyridin-6-yl)propanenitrile (22). 21 (300.0 mg, 1.07 mmol) was dissolved in dry DCM (6 mL) under Ar atmosphere. 4N HCl in dioxane (2.7 mL) was added to the stirring solution resulting in a waxy precipitate. The reaction progress was monitored by a ninhydrin stained TLC. After full consumption of 21, demineralized water was added to dissolve the waxy precipitate. The pH of the aqueous layer was adjusted to 14 with 50% NaOH(aq). Phases were separated and the aqueous layer was extracted with DCM (15 × 15 mL). Combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. 160 mg of an orange oil (83% yield), which was used in the next step without further purification. A partial hydrolysis of the nitrile group to an amide group was observed in the NMR spectra of the crude product. A small batch was purified by flash column chromatography for analytical purposes (SiO2, DCM:2N NH3 in MeOH 9:1); 1H-NMR (400 MHz, CDCl3) δ 3.82–3.52 (m, 1H), 3.22–3.13 (m, 1H), 3.12–3.05 (m, 1H), 3.04–2.95 (m, 1H), 2.94–2.88 (m, 1H), 2.72–2.60 (m, 3H), 2.49 (t, J = 6.9 Hz, 2H), 2.45–2.38 (m, 1H), 2.16–2.07 (m, 1H), 2.01–1.93 (m, 1H), 1.91–1.82 (m, 1H), 1.64–1.54 (m, 2H), 1.49–1.40 (m, 1H); 13C-NMR (75 MHz, CDCl3) δ 118.9, 57.8, 53.7, 53.3, 51.9, 44.2, 35.8, 31.2, 27.1, 16.0; GC-MS method B: tr = 14.130 min, (m/z) 179 [M].
3-(1-(7-Chloro-9-tosyl-9H-pyrimido[4,5-b]indol-4-yl)octahydro-6H-pyrrolo[2,3-c]pyridin-6-yl)propa-nenitrile (23). 10 (200.0 mg, 0.51 mmol), 22 (109.7 mg, 0.61 mmol) and DIPEA (210.9 mg, 1.63 mmol) were mixed with dry DMF (6 mL). The mixture was stirred at 80 °C for 1 h and was then left to cool to rt. Saturated NaCl solution (15 mL) was added and the mixture extracted with EtOAc (3 × 25 mL). Combined organic layers were washed with saturated NaCl solution (3 × 20 mL), dried over Na2SO4 and concentrated under reduced pressure. Purification of the residue by flash column chromatography (SiO2, petroleum ether:(EtOAc + MeOH 95 + 5) 4:6) gave 175 mg of a beige solid (64% yield); 1H-NMR (300 MHz, CDCl3) δ 8.56–8.48 (m, 2H), 8.10 (d, J = 8.4 Hz, 2H), 7.77 (br s, 1H), 7.36 (dd, J = 8.5, 1.2 Hz, 1H), 7.27 (d, 2H, overlapping with CHCl3 signal), 4.65–4.48 (m, 1H), 4.24–4.06 (m, 1H), 3.53–3.41 (m, 1H), 3.17–3.01 (m, 1H), 2.82–2.52 (m, 4H), 2.49–2.27 (m, 7H), 1.98–1.69 (m, 4H); 13C-NMR (75 MHz, CDCl3) δ 158.0, 156.9, 154.2, 145.7, 136.0, 135.6, 132.2, 129.8, 128.2, 124.2, 123.5, 120.7, 118.8, 114.5, 100.3, 57.5, 53.5, 53.4, 50.8, 34.6, 29.8, 26.8, 21.8, 15.8. Signal overlap assumed at 50.8; ESI-MS: (m/z) 534.9 [M + H]+, 556.8 [M + Na]+, 532.9 [M − H]-; HPLC: tr = 7.197 min.
3-(3a
RS,7a
SR)-(1-(7-Chloro-9
H-pyrimido[4,5-
b]indol-4-yl)octahydro-6
H-pyrrolo[2,3-c]pyridin-6-yl)-propanenitrile (
24).
23 (150.0 mg, 0.28 mmol) was dissolved in dry THF (10 mL) and K
tBuO (220.2 mg, 1.96 mmol) was added. The mixture was stirred at rt and under N
2 atmosphere for 75 min. Saturated NaCl solution (25 mL) was added and the mixture extracted with EtOAc (3 × 25 mL). Combined organic layers were dried over Na
2SO
4 and concentrated under reduced pressure. Purification of the residue by flash column chromatography (SiO
2, DCM:MeOH 95:5) gave 59 mg of a light beige solid (55% yield);
1H-NMR (300 MHz, pyridine-
d5) δ 13.68 (br s, 1H), 8.84 (s, 1H), 8.23 (d,
J = 8.8 Hz, 1H), 7.81 (d,
J = 2.0 Hz, 1H), 7.52 (dd,
J = 8.7, 2.0 Hz, 1H), 4.95–4.86 (m, 1H), 4.42–4.22 (m, 1H), 4.03–3.86 (m, 1H), 3.18–3.02 (m, 1H), 2.74–2.53 (m, 5H), 2.50–2.25 (m, 3H), 1.98–1.62 (m, 4H);
13C-NMR (101 MHz, DMSO-
d6) δ 156.9, 156.6, 153.8, 137.3, 128.7, 123.8, 120.3, 119.9, 118.5, 110.7, 96.0, 56.6, 53.2, 52.9, 49.1, 48.4, 34.1, 26.7, 25.5, 14.9; ESI-MS: (
m/z) 381.2 [M + H]
+, 403.2 [M + Na]
+, 379.2 [M − H]
−; HPLC: t
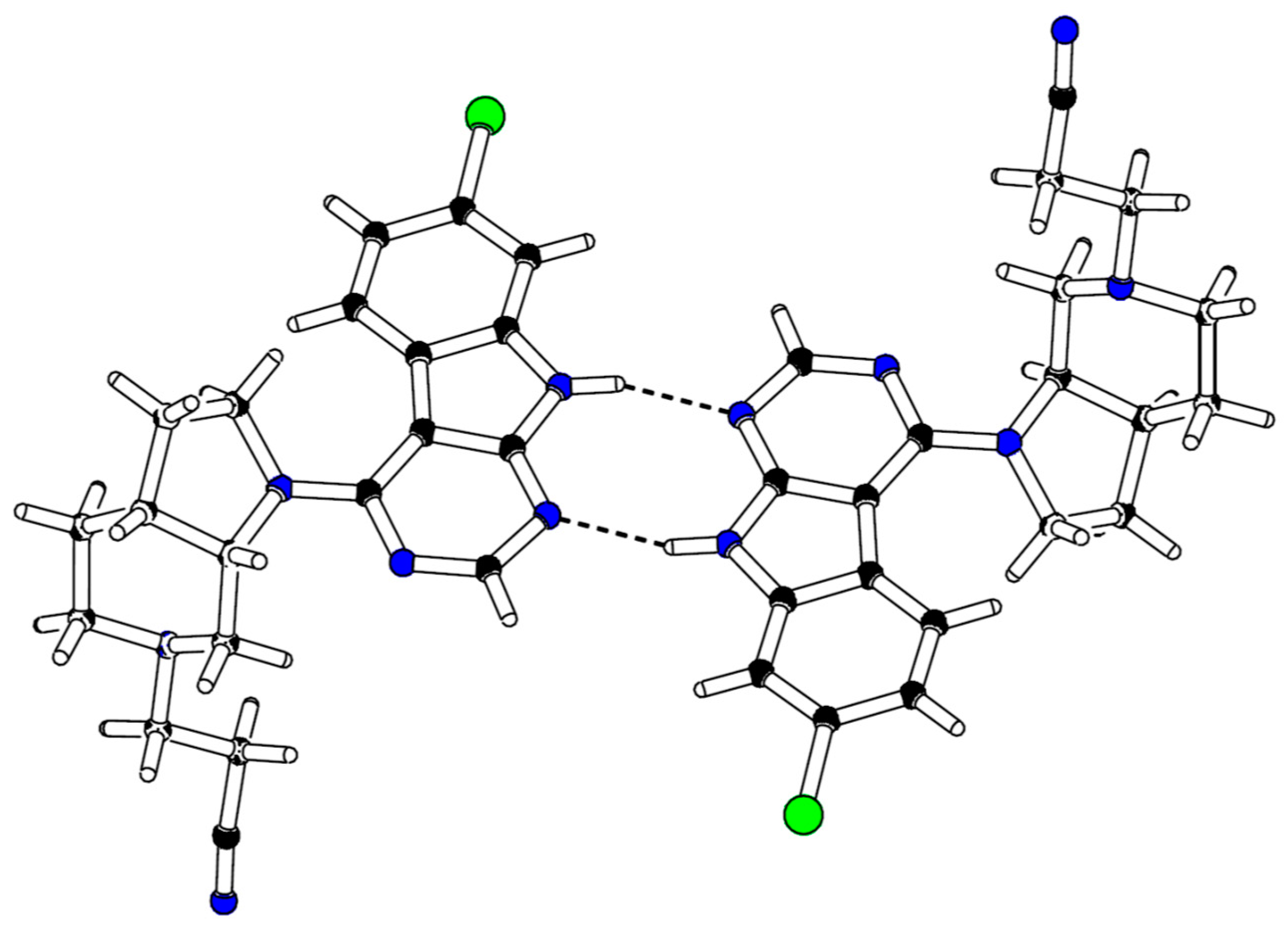
r = 4.362 min (99.9% purity). Crystals suitable for X-ray determination were obtained by slow evaporation of a solution of
24 in methanol and chloroform at 298 K under atmospheric pressure. CCDC 1,917,242 contains the
supplementary crystallographic data. These data can be obtained free of charge via
http://www.ccdc.cam.ac.uk/conts/retrieving.html.


 and
and 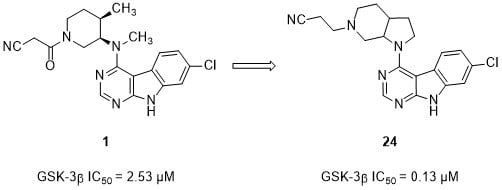











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





















