
Identification of Azoxystrobin Glutathione Conjugate Metabolites in Maize Roots by LC-MS



Abstract
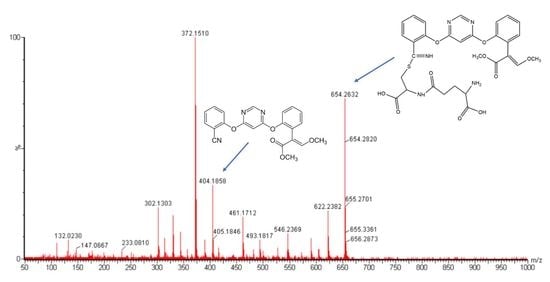
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Maize Root Culture Growth
4.3. Azoxystrobin Uptake
4.4. Sample Preparation and Extraction
4.5. LC-MS/MS Analysis
4.5.1. Q-TOF Analysis
4.5.2. QTRAP Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MSe | Waters® acquisition method synonymous of “all ions fragmentation” and data independent acquisition (DIA) |
| GSH | reduced glutathione |
| LC-MS | Liquid Chromatography followed by Mass Spectroscopy |
| Q-TOF | Quadrupole-Time of Flight |
| Q-TRAP | Triple Quadrupole (QQQ) Linear Ion Traps |
| MRM-IDA | Multiple Reaction Monitoring using Information Dependent Acquisition (IDA) |
| MRM-EPI | Multiple Reaction Monitoring (MRM) with Enhanced Product Ion (EPI) |
References
- Joseph, R.S. Metabolism of Azoxystrobin in Plants and Animals. Special Publication-Royal Soc. Chem. 1999, 233, 265–278. [Google Scholar] [CrossRef]
- Gautam, M.; Fomsgaard, I.S. Liquid chromatography-tandem mass spectrometry method for simultaneous quantification of azoxystrobin and its metabolites, azoxystrobin free acid and 2-hydroxybenzonitrile, in greenhouse-grown lettuce. Food Addit. Contam. A 2017, 34, 2173–2180. [Google Scholar] [CrossRef] [PubMed]
- Gautam, M.; Elhiti, M.; Fomsgaard, I.S. Maize root culture as a model system for studying azoxystrobin biotransformation in plants. Chemosphere 2018, 195, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Gautam, M.; Etzerodt, T.; Fomsgaard, I.S. Quantification of azoxystrobin and identification of two novel metabolites in lettuce via liquid chromatography–quadrupole-linear ion trap (QTRAP) mass spectrometry. Int. J. Environm. Anal. Chem. 2017, 97, 419–430. [Google Scholar] [CrossRef]
- Bryant, D. Glutathione conjugation of herbicides and fungicides in plants and fungi: Functional characterization of glutathione transferases from phytopathogens. Ph.D. Thesis, Durham University, Durham, UK, 2004. [Google Scholar]
- Laird, W.J.D.; Gledhill, A.J.; Lappin, G.J. Metabolism of methyl-(E)-2-{2-[6-(2-cyanophenoxy)pyrimidin-4-yloxy]phenyl}-3-methoxyacrylate (azoxystrobin) in rat. Xenobiotica 2003, 33, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Ohkama-Ohtsu, N.; Zhao, P.; Xiang, C.; Oliver, D.J. Glutathione conjugates in the vacuole are degraded by gamma-glutamyl transpeptidase GGT3 in Arabidopsis. Plant. J. 2007, 49, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Zhong, D.; Chen, X. A fragmentation-based method for the differentiation of glutathione conjugates by high-resolution mass spectrometry with electrospray ionization. Anal. Chim. Acta 2013, 788, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Marrs, K.A. THE FUNCTIONS AND REGULATION OF GLUTATHIONE S-TRANSFERASES IN PLANTS. Annual Rev. Plant Biol. 1996, 47, 127–158. [Google Scholar] [CrossRef]
- Edwards, R.; Dixon, D.P.; Walbot, V. Plant glutathione S-transferases: Enzymes with multiple functions in sickness and in health. Trends Plant. Sci. 2000, 5, 193–198. [Google Scholar] [CrossRef]
- Dixon, D.P.; Lapthorn, A.; Edwards, R. Plant glutathione transferases. Genome Biol. 2002, 3, reviews3004. [Google Scholar] [CrossRef][Green Version]
- Caccuri, A.M.; Lo Bello, M.; Nuccetelli, M.; Nicotra, M.; Rossi, P.; Antonini, G.; Federici, G.; Ricci, G. Proton release upon glutathione binding to glutathione transferase P1-1: Kinetic analysis of a multistep glutathione binding process. Biochemistry 1998, 37, 3028–3034. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, D.; Meade, G.; Foley, V.M.; Dowd, C.A. Structure, function and evolution of glutathione transferases: Implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem. J. 2001, 360, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Caccuri, A.M.; Antonini, G.; Board, P.G.; Parker, M.W.; Nicotra, M.; Lo Bello, M.; Federici, G.; Ricci, G. Proton release on binding of glutathione to alpha, Mu and Delta class glutathione transferases. Biochem. J. 1999, 344, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, D.W.; Clough, J.M.; Godwin, J.R.; Hall, A.A.; Hamer, M.; Parr-Dobrzanski, B. The strobilurin fungicides. Pest. Manage. Sci. 2002, 58, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.E.; Dietz, K.J.; Schroder, P. Degradation of glutathione S-conjugates by a carboxypeptidase in the plant vacuole. FEBS Lett. 1996, 384, 31–34. [Google Scholar] [CrossRef]
- Beck, A.; Lendzian, K.; Oven, M.; Christmann, A.; Grill, E. Phytochelatin synthase catalyzes key step in turnover of glutathione conjugates. Phytochemistry 2003, 62, 423–431. [Google Scholar] [CrossRef]
- Martin, M.N.; Slovin, J.P. Purified gamma-glutamyl transpeptidases from tomato exhibit high affinity for glutathione and glutathione S-conjugates. Plant. Physiol. 2000, 122, 1417–1426. [Google Scholar] [CrossRef][Green Version]
- Huber, C.; Bartha, B.; Harpaintner, R.; Schroder, P. Metabolism of acetaminophen (paracetamol) in plants--two independent pathways result in the formation of a glutathione and a glucose conjugate. Environ. Sci. Pollut. Res. Int. 2009, 16, 206–213. [Google Scholar] [CrossRef]
- Murphy, C.M.; Fenselau, C.; Gutierrez, P.L. Fragmentation characteristic of glutathione conjugates activated by high-energy collisions. J. Am. Soc. Mass Spectr. 1992, 3, 815–822. [Google Scholar] [CrossRef]
- Jian, W.; Liu, H.-F.; Zhao, W.; Jones, E.; Zhu, M. Simultaneous Screening of Glutathione and Cyanide Adducts Using Precursor Ion and Neutral Loss Scans-Dependent Product Ion Spectral Acquisition and Data Mining Tools. J. Am. Soc. Mass Spectr. 2012, 23, 964–976. [Google Scholar] [CrossRef][Green Version]
- Kassahun, K.; Davis, M.; Hu, P.; Martin, B.; Baillie, T. Biotransformation of the Naturally Occurring Isothiocyanate Sulforaphane in the Rat: Identification of Phase I Metabolites and Glutathione Conjugates. Chem. Res. Toxicol. 1997, 10, 1228–1233. [Google Scholar] [CrossRef] [PubMed]
- Mali’n, T.J.; Lindberg, S.; Åstot, C. Novel glutathione conjugates of phenyl isocyanate identified by ultra-performance liquid chromatography/electrospray ionization mass spectrometry and nuclear magnetic resonance. J. Mass Spectr. 2014, 49, 68–79. [Google Scholar] [CrossRef]
- Mueller, L.N.; Brusniak, M.-Y.; Mani, D.R.; Aebersold, R. An Assessment of Software Solutions for the Analysis of Mass Spectrometry Based Quantitative Proteomics Data. J. Proteome Res. 2008, 7, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Marcotte, E.M. Label-Free Protein Quantitation Using Weighted Spectral Counting. In Quantitative Methods in Proteomics; Marcus, K., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 321–341. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound (RT_m/z) | m/z | z | RT | PW | Anova (p) | q Value | FC | HM | LM | Max Abundance |
|---|---|---|---|---|---|---|---|---|---|---|
| 18.18_711.2189 m/z | 711.2189 | 1 | 18.18 | 0.61 | <1.1 × 10−16 | <1.1 × 10−16 | 18.7 | AZO_96h | CTRL_24h | 139.9 |
| 19.33_552.1740 m/z | 552.1740 | 1 | 19.33 | 0.69 | 1.11 × 10−16 | 4.58 × 10−15 | 27.8 | AZO_96h | CTRL_48h | 50.7 |
| 18.47_654.1935 m/z | 654.1935 | 1 | 18.47 | 0.61 | 2.22 × 10−16 | 8.10 × 10−15 | 110 | AZO_96h | CTRL_3h | 162.0 |
| 28.70_403.0995 m/z | 404.1068 | 1 | 28.70 | 3.12 | 3.33 × 10−16 | 1.13 × 10−14 | 2.98 × 10−3 | AZO_3h | CTRL_48h | 8260.1 |
| 28.70_372.0798 m/z | 372.0798 | 1 | 28.70 | 2.94 | 5.33× 10−16 | 1.28 × 10−13 | 8.05 × 103 | AZO_3h | CTRL_48h | 6041.6 |
| 16.10_534.1777 m/z | 534.1777 | 1 | 16.10 | 0.64 | 2.12 × 10−10 | 1.60 × 10−9 | 3.91 | AZO_96h | CTRL_3h | 57.2 |
| 23.22_557.1621 m/z | 557.1621 | 1 | 23.22 | 1.25 | 4.47 × 10−9 | 2.41 × 10−8 | 6.76 | CTRL_3h | AZO_96h | 3340.4 |
| 13.32_743.2417 m/z | 743.2417 | 1 | 13.32 | 0.59 | 5.68 × 10−9 | 3.01 × 10−8 | 3.46 | AZO_96h | CTRL_3h | 63.9 |
| 11.19_549.1369 m/z | 549.1369 | 1 | 11.19 | 0.96 | 4.20 × 10−6 | 1.27 × 10−5 | 4.68 | AZO_96h | CTRl_3h | 229.8 |
| 21.12_557.1727 m/z | 557.1727 | 1 | 21.12 | 1.17 | 7.97 × 10−6 | 2.30 × 10−5 | 2.39 | AZO_3h | CTRl_72h | 104.5 |
| 34.25_404.1264 m/z | 404.1264 | 1 | 34.25 | 0.80 | 1.02 × 10−5 | 2.89 × 10−5 | Infinity | AZO_3h | CTRL_3h | 12.9 |
| 3.90_1103.2994 m/z | 1103.2994 | 1 | 3.90 | 0.45 | 2.50 × 10−5 | 6.57 × 10−5 | 4.87 | CTRL_3h | CTRL_48h | 20.4 |
| 21.25_663.2184 m/z | 663.2184 | 1 | 21.25 | 0.59 | 9.41 × 10−5 | 0.00022 | 2.78 | AZO_3h | AZO_72h | 26.5 |
| 13.21_426.1160 m/z | 426.1160 | 1 | 13.21 | 0.85 | 0.000114 | 0.000263 | 1.74 | CTRL_72h | CTRL_3h | 94.7 |
| 13.37_549.1715 m/z | 549.1715 | 1 | 13.37 | 2.59 | 0.0258 | 4 | 1.42 | AZO_3h | CTRL_24h | 330.3 |
| 9.32_367.0946 m/z | 367.0946 | 2 | 9.32 | 0.51 | 0.0296 | 456 | 2.84 | AZO_3h | CTRL_3h | 57.5 |
| 1.98_404.1227 m/z | 404.1227 | 1 | 1.98 | 0.24 | 0.143 | 0.197 | Infinity | AZO_24h | CTRL_24h | 14.2 |
| 3.82_370.0885 m/z | 370.0885 | 2 | 3.82 | 0.51 | 0.208 | 0.278 | 2.36 | CTRL_96h | AZO_96h | 5.3 |
| 1.52_404.1226 m/z | 404.1226 | 1 | 1.52 | 0.35 | 0.31 | 0.4 | Infinity | CTRL_3h | CTRL_24h | 3.6 |
| 9.32_355.0933 m/z | 355.0933 | 2 | 9.32 | 0.51 | 0.336 | 0.432 | 4.08 | AZO_3h | CTRL_3h | 7.5 |
| 38.31_404.1245 m/z | 404.1245 | 1 | 38.31 | 0.45 | 0.508 | 0.629 | Infinity | AZO_48h | CTRL_3h | 13.4 |
| 37.53_404.1205 m/z | 404.1205 | 1 | 37.53 | 0.56 | 0.607 | 0.74 | Infinity | AZO_72h | CTRL_3h | 16.6 |
| 24.29_743.2418 m/z | 743.2418 | 1 | 24.29 | 0.61 | 0.704 | 0.838 | 1.38 | AZO_3h | AZO_96h | 11.4 |
| 3.71_380.1017 m/z | 380.1017 | 2 | 3.71 | 0.32 | 0.978 | >0.9995 | 4.22 | CTRL_96h | CTRL_24h | 18.3 |
| Scan type | Q1 | Q3 | DP (V) | EP (V) | CE (V) | CXP (V) | CUR (psi) | CAD | IS (V) | TEM (°C) | GS 1 (psi) | GS 2 (psi) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MRM | 711 | 404 | 60 | 10 | 30 | 13 | 35 | High | 4300 | 450 | 90 | 50 |
| 711 | 372 | 60 | 10 | 30 | 13 | 35 | High | 4300 | 450 | 90 | 50 | |
| 711 | 582 | 60 | 10 | 30 | 13 | 35 | High | 4300 | 450 | 90 | 50 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dionisio, G.; Gautam, M.; Fomsgaard, I.S. Identification of Azoxystrobin Glutathione Conjugate Metabolites in Maize Roots by LC-MS. Molecules 2019, 24, 2473. https://doi.org/10.3390/molecules24132473
Dionisio G, Gautam M, Fomsgaard IS. Identification of Azoxystrobin Glutathione Conjugate Metabolites in Maize Roots by LC-MS. Molecules. 2019; 24(13):2473. https://doi.org/10.3390/molecules24132473
Chicago/Turabian StyleDionisio, Giuseppe, Maheswor Gautam, and Inge Sindbjerg Fomsgaard. 2019. "Identification of Azoxystrobin Glutathione Conjugate Metabolites in Maize Roots by LC-MS" Molecules 24, no. 13: 2473. https://doi.org/10.3390/molecules24132473
APA StyleDionisio, G., Gautam, M., & Fomsgaard, I. S. (2019). Identification of Azoxystrobin Glutathione Conjugate Metabolites in Maize Roots by LC-MS. Molecules, 24(13), 2473. https://doi.org/10.3390/molecules24132473