
Discovery of Immunoproteasome Inhibitors Using Large-Scale Covalent Virtual Screening

 , ,
, , 
Abstract
1. Introduction
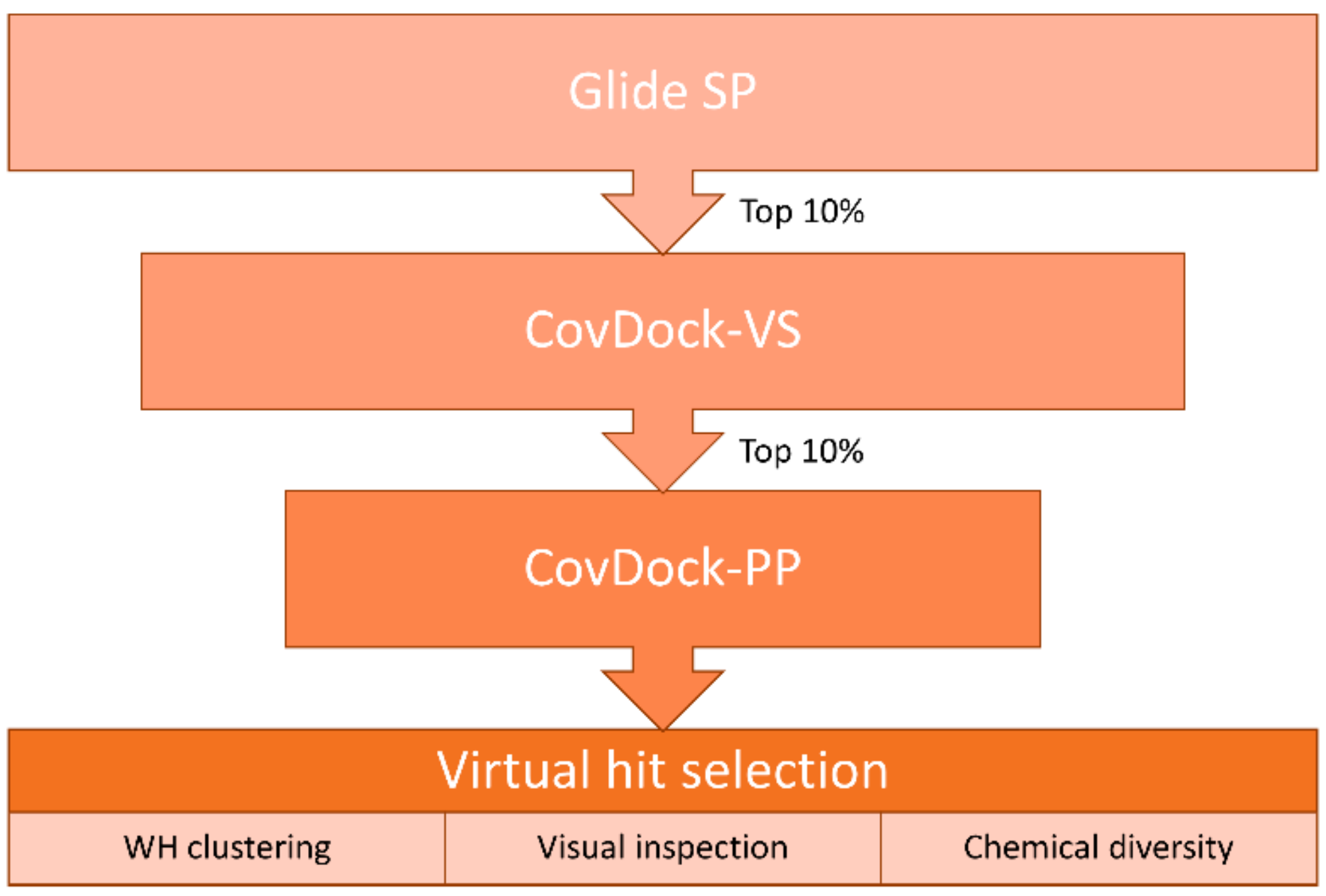
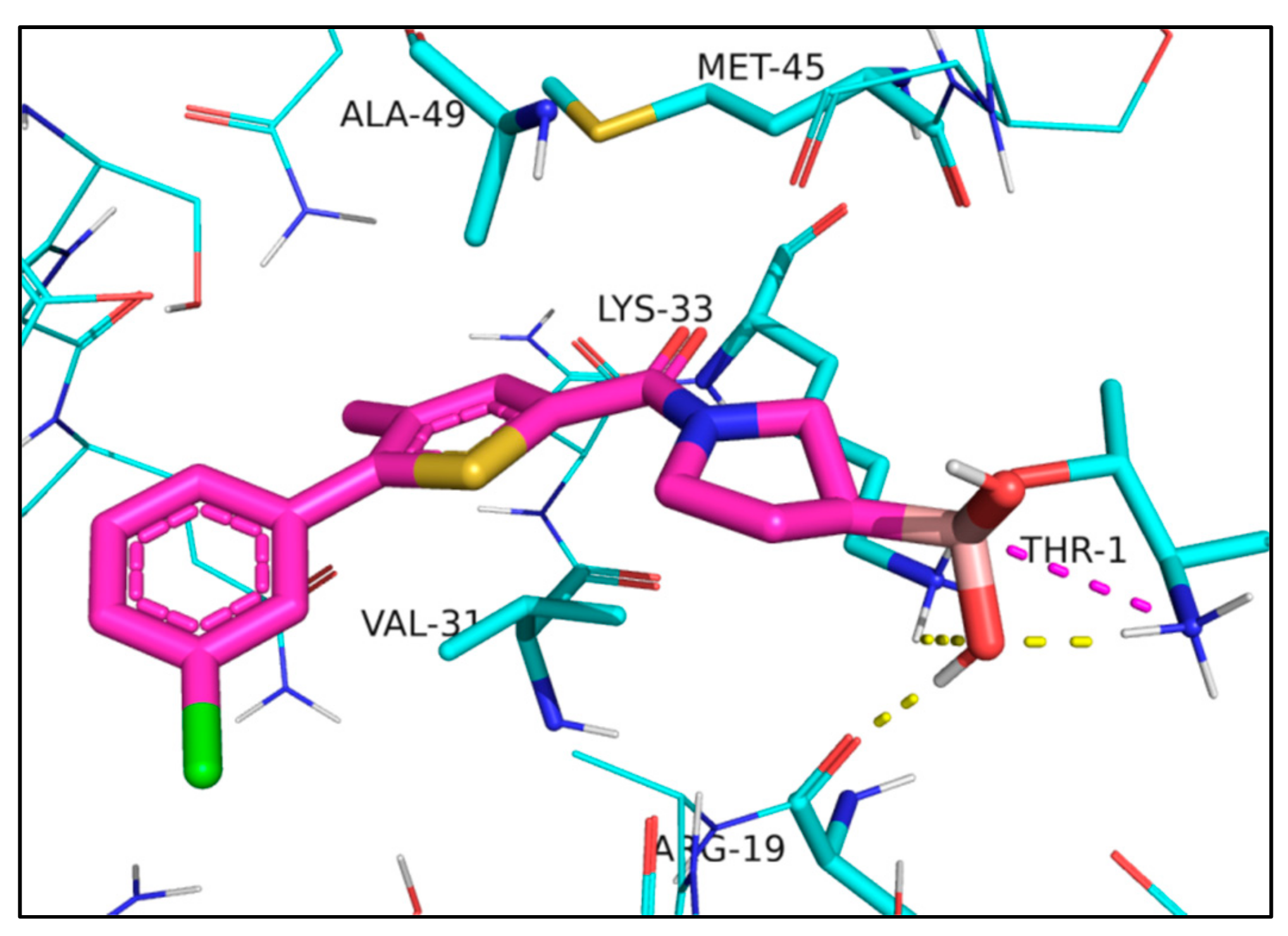
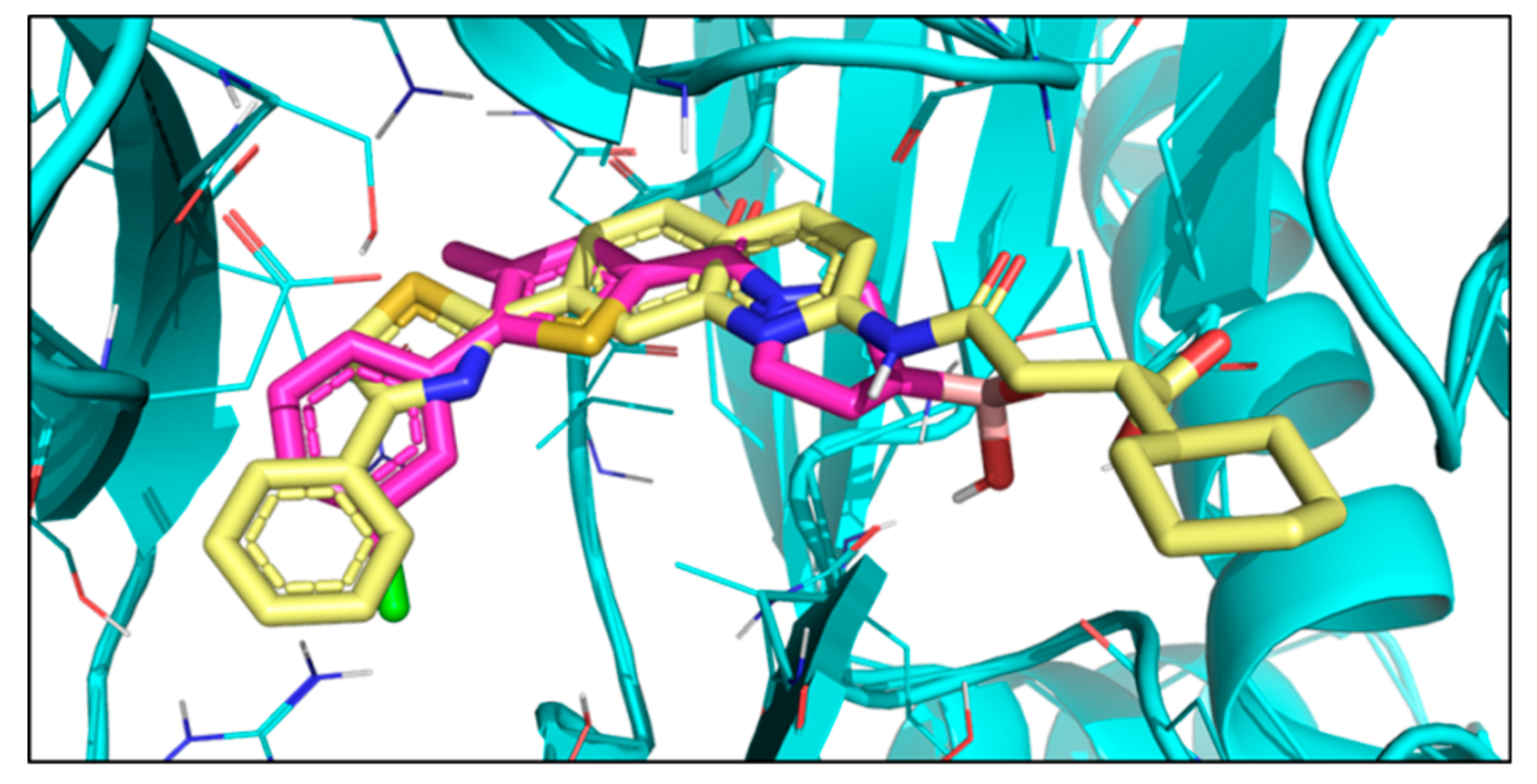
2. Results
3. Discussion
4. Materials and Methods
4.1. General Chemistry Methods
 |
 |
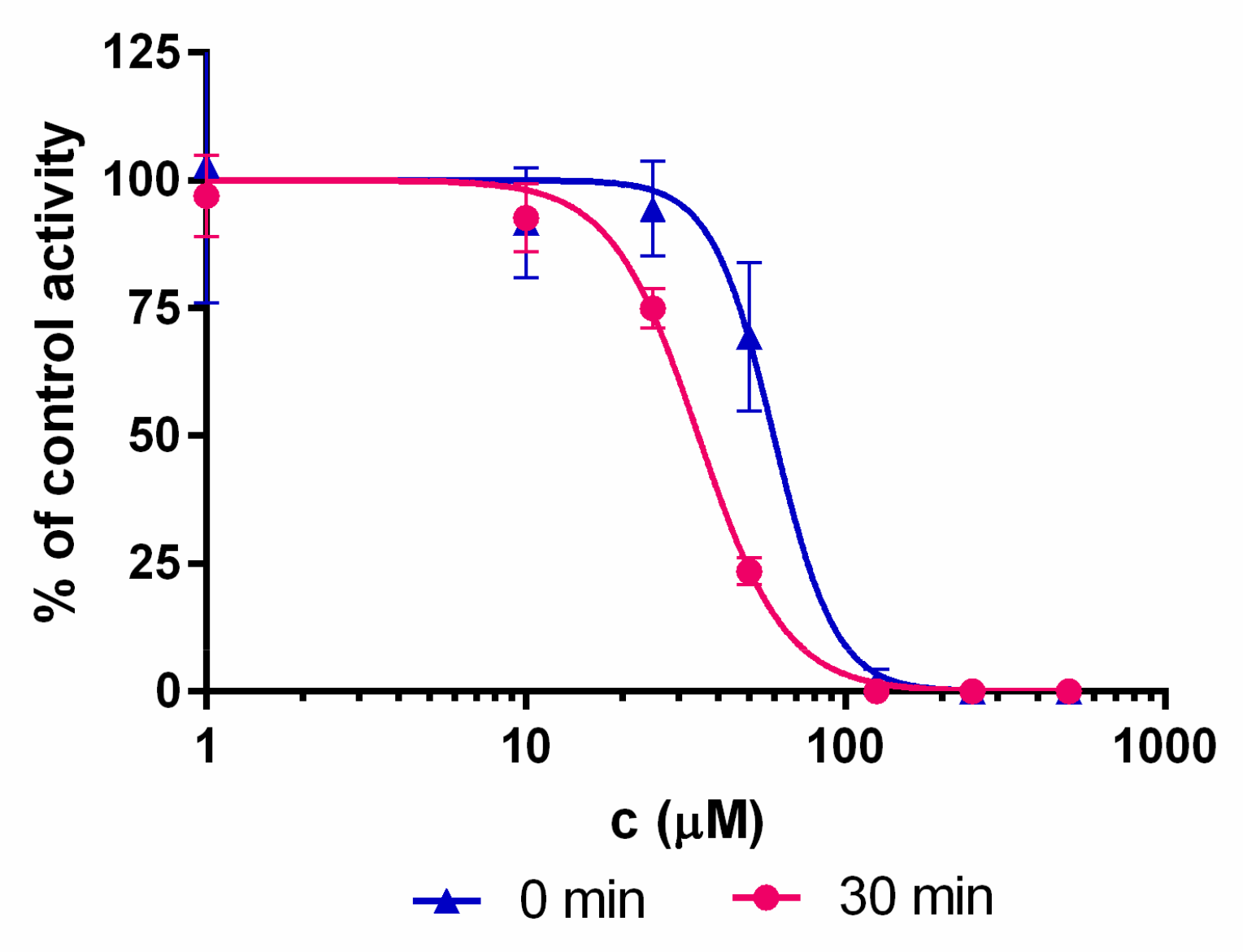
4.2. Kd Determination
4.3. Residual Activity Determination
4.4. IC50 Determination
4.5. Virtual Screening
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Cesco, S.; Kurian, J.; Dufresne, C.; Mittermaier, A.K.; Moitessier, N. European Journal of Medicinal Chemistry Covalent inhibitors design and discovery. Eur. J. Med. Chem. 2017, 138, 96–114. [Google Scholar] [CrossRef]
- Bauer, R.A. Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today 2015, 20, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-based virtual screening for drug discovery: principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef] [PubMed]
- Scarpino, A.; Ferenczy, G.G.; Keserü, G.M. Comparative Evaluation of Covalent Docking Tools. J. Chem. Inf. Model. 2018, 58, 1441–1458. [Google Scholar] [CrossRef] [PubMed]
- London, N.; Miller, R.M.; Krishnan, S.; Uchida, K.; Irwin, J.J.; Eidam, O.; Gibold, L.; Cimermančič, P.; Bonnet, R.; Shoichet, B.K.; et al. Covalent docking of large libraries for the discovery of chemical probes. Nat. Chem. Biol. 2014, 10, 1066–1072. [Google Scholar] [CrossRef] [PubMed]
- Katritch, V.; Byrd, C.M.; Tseitin, V.; Dai, D.; Raush, E.; Totrov, M.; Abagyan, R.; Jordan, R.; Hruby, D.E. Discovery of small molecule inhibitors of ubiquitin-like poxvirus proteinase I7L using homology modeling and covalent docking approaches. J. Comput. Aided. Mol. Des. 2007, 21, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Nnadi, C.I.; Jenkins, M.L.; Gentile, D.R.; Bateman, L.A.; Zaidman, D.; Balius, T.E.; Nomura, D.K.; Burke, J.E.; Shokat, K.M.; London, N. Novel K-Ras G12C Switch-II Covalent Binders Destabilize Ras and Accelerate Nucleotide Exchange. J. Chem. Inf. Model. 2018, 58, 464–471. [Google Scholar] [CrossRef]
- Marques, A.J.; Palanimurugan, R.; Matias, A.C.; Ramos, P.C.; Dohmen, R.J. Catalytic Mechanism and Assembly of the Proteasome. Chem. Rev. 2009, 109, 1509–1536. [Google Scholar] [CrossRef]
- Thibaudeau, T.A.; Smith, D.M. A Practical Review of Proteasome Pharmacology. Pharmacol. Rev. 2019, 71, 170–197. [Google Scholar] [CrossRef]
- Genin, E.; Reboud-Ravaux, M.; Vidal, J. Proteasome inhibitors: recent advances and new perspectives in medicinal chemistry. Curr. Top. Med. Chem. 2010, 10, 232–256. [Google Scholar] [CrossRef]
- Demo, S.D.; Kirk, C.J.; Aujay, M.A.; Buchholz, T.J.; Dajee, M.; Ho, M.N.; Jiang, J.; Laidig, G.J.; Lewis, E.R.; Parlati, F.; et al. Antitumor Activity of PR-171, a Novel Irreversible Inhibitor of the Proteasome. Cancer Res. 2007, 67, 6383–6391. [Google Scholar] [CrossRef] [PubMed]
- Huber, E.M.; Groll, M. Inhibitors for the immuno- and constitutive proteasome: Current and future trends in drug development. Angew. Chemie Int. Ed. 2012, 51, 8708–8720. [Google Scholar] [CrossRef] [PubMed]
- Ettari, R.; Zappalà, M.; Grasso, S.; Musolino, C.; Innao, V.; Allegra, A. Immunoproteasome-selective and non-selective inhibitors: A promising approach for the treatment of multiple myeloma. Pharmacol. Ther. 2018, 182, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Sosič, I.; Gobec, M.; Brus, B.; Knez, D.; Živec, M.; Konc, J.; Lešnik, S.; Ogrizek, M.; Obreza, A.; Žigon, D.; et al. Nonpeptidic Selective Inhibitors of the Chymotrypsin-Like (β5 i) Subunit of the Immunoproteasome. Angew. Chemie Int. Ed. 2016, 55, 5745–5748. [Google Scholar] [CrossRef] [PubMed]
- Kasam, V.; Lee, N.-R.; Kim, K.-B.; Zhan, C.-G. Selective immunoproteasome inhibitors with non-peptide scaffolds identified from structure-based virtual screening. Bioorg. Med. Chem. Lett. 2014, 24, 3614–3617. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Baur, R.; Le Chapelain, C.; Dubiella, C.; Heinemeyer, W.; Huber, E.M.; Groll, M. Structural Elucidation of a Nonpeptidic Inhibitor Specific for the Human Immunoproteasome. ChemBioChem 2017, 18, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Angelo, N.G.; Warren, J.D.; Nathan, C.F.; Lin, G. Oxathiazolones Selectively Inhibit the Human Immunoproteasome over the Constitutive Proteasome. ACS Med. Chem. Lett. 2014, 5, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Bosc, E.; Nastri, J.; Lefort, V.; Valli, M.; Contiguiba, F.; Pioli, R.; Furlan, M.; da Bolzani, V.S.; El Amri, C.; Reboud-Ravaux, M. Piperlongumine and some of its analogs inhibit selectively the human immunoproteasome over the constitutive proteasome. Biochem. Biophys. Res. Commun. 2018, 496, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, A.F.; Groettrup, M. Subunit specific inhibitors of proteasomes and their potential for immunomodulation. Curr. Opin. Chem. Biol. 2014, 23, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Basler, M.; Lindstrom, M.M.; LaStant, J.J.; Bradshaw, J.M.; Owens, T.D.; Schmidt, C.; Maurits, E.; Tsu, C.; Overkleeft, H.S.; Kirk, C.J.; et al. Co-inhibition of immunoproteasome subunits LMP2 and LMP7 is required to block autoimmunity. EMBO Rep. 2018, 19, e46512. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.W.B.; Lowe, E.; Anderl, J.L.; Fan, A.; Muchamuel, T.; Bowers, S.; Moebius, D.C.; Kirk, C.; McMinn, D.L. Required Immunoproteasome Subunit Inhibition Profile for Anti-Inflammatory Efficacy and Clinical Candidate KZR-616 ((2S,3R)-N-((S)-3-(Cyclopent-1-en-1-yl)-1-((R)-2-methyloxiran-2-yl)-1-oxopropan-2-yl)-3-hydroxy-3-(4-methoxyphenyl)-2-((S)-2-(2-morpholinoacetamido)propanamido)propenamide). J. Med. Chem. 2018, 61, 11127–11143. [Google Scholar] [PubMed]
- Fu, H.; Fang, H.; Sun, J.; Wang, H.; Liu, A.; Sun, J.; Wu, Z. Boronic acid-based enzyme inhibitors: a review of recent progress. Curr. Med. Chem. 2014, 21, 3271–3280. [Google Scholar] [CrossRef] [PubMed]
- Li, A.C.; Yu, E.; Ring, S.C.; Chovan, J.P. Boronic Acid-Containing Proteasome Inhibitors: Alert to Potential Pharmaceutical Bioactivation. Chem. Res. Toxicol. 2013, 26, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- eMolecules. Available online: https://www.emolecules.com/ (accessed on 4 October 2018).
- Zhu, K.; Borrelli, K.W.; Greenwood, J.R.; Day, T.; Abel, R.; Farid, R.S.; Harder, E. Docking Covalent Inhibitors: A Parameter Free Approach To Pose Prediction and Scoring. J. Chem. Inf. Model. 2014, 54, 1932–1940. [Google Scholar] [CrossRef]
- Toledo Warshaviak, D.; Golan, G.; Borrelli, K.W.; Zhu, K.; Kalid, O. Structure-Based Virtual Screening Approach for Discovery of Covalently Bound Ligands. J. Chem. Inf. Model. 2014, 54, 1941–1950. [Google Scholar] [CrossRef] [PubMed]
- Brogi, S.; Fiorillo, A.; Chemi, G.; Butini, S.; Lalle, M.; Ilari, A.; Gemma, S.; Campiani, G. Structural characterization of Giardia duodenalis thioredoxin reductase (g TrxR) and computational analysis of its interaction with NBDHEX. Eur. J. Med. Chem. 2017, 135, 479–490. [Google Scholar] [CrossRef]
- Muzzarelli, K.M.; Kuiper, B.; Spellmon, N.; Brunzelle, J.; Hackett, J.; Amblard, F.; Zhou, S.; Liu, P.; Kovari, I.A.; Yang, Z.; et al. Structural and Antiviral Studies of the Human Norovirus GII.4 Protease. Biochemistry 2019, 58, 900–907. [Google Scholar] [CrossRef]
- Chowdhury, S.R.; Kennedy, S.; Zhu, K.; Mishra, R.; Chuong, P.; Nguyen, A.; Kathman, S.G.; Statsyuk, A.V. Discovery of covalent enzyme inhibitors using virtual docking of covalent fragments. Bioorg. Med. Chem. Lett. 2019, 29, 36–39. [Google Scholar] [CrossRef]
- Vasaturo, M.; Fiengo, L.; De Tommasi, N.; Sabatino, L.; Ziccardi, P.; Colantuoni, V.; Bruno, M.; Cerchia, C.; Novellino, E.; Lupo, A.; et al. A compound-based proteomic approach discloses 15-ketoatractyligenin methyl ester as a new PPARγ partial agonist with anti-proliferative ability. Sci. Rep. 2017, 7, 41273. [Google Scholar] [CrossRef]
- Glide; Schrödinger Release 2018-4; Schrödinger, LLC: New York, NY, USA, 2018; Available online: https://www.schrodinger.com/.
- Prime; Schrödinger Release 2018-4; Schrödinger, LLC: New York, NY, USA, 2018; Available online: https://www.schrodinger.com/.
- Bull, S.D.; Davidson, M.G.; van den Elsen, J.M.H.; Fossey, J.S.; Jenkins, A.T.A.; Jiang, Y.-B.; Kubo, Y.; Marken, F.; Sakurai, K.; Zhao, J.; et al. Exploiting the Reversible Covalent Bonding of Boronic Acids: Recognition, Sensing, and Assembly. Acc. Chem. Res. 2013, 46, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Groll, M.; Berkers, C.R.; Ploegh, H.L.; Ovaa, H. Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure 2006, 14, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Marinaro, W.A.; Prankerd, R.; Kinnari, K.; Stella, V.J. Interaction of Model Aryl- and Alkyl-Boronic Acids and 1,2-Diols in Aqueous Solution. J. Pharm. Sci. 2015, 104, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.; Hawley, R.C.; Lynch, S.M.; Narayanan, A. Substituted Thiazole Compounds. WO 2014086701 A1, 12 June 2014. [Google Scholar]
- Huber, E.M.; Heinemeyer, W.; de Bruin, G.; Overkleeft, H.S.; Groll, M. A humanized yeast proteasome identifies unique binding modes of inhibitors for the immunosubunit β5i. EMBO J. 2016, 35, 2602–2613. [Google Scholar] [CrossRef] [PubMed]
- Huber, E.M.; Basler, M.; Schwab, R.; Heinemeyer, W.; Kirk, C.J.; Groettrup, M.; Groll, M. Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell 2012, 148, 727–738. [Google Scholar] [CrossRef]
- Muchamuel, T.; Basler, M.; Aujay, M.A.; Suzuki, E.; Kalim, K.W.; Lauer, C.; Sylvain, C.; Ring, E.R.; Shields, J.; Jiang, J.; et al. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med. 2009, 15, 781–787. [Google Scholar] [CrossRef]
- RDKit: Open-Source Cheminformatics. Release 2019.03.1. Available online: http://www.rdkit.org.
- Protein Preparation Wizard; Schrödinger Release 2018-4; Schrödinger, LLC: New York, NY, USA, 2018; Available online: https://www.schrodinger.com/.
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- JChem for Office 19.1.0.421, 2019; ChemAxon: Budapest, Hungary, 2019; Available online: http://www.chemaxon.com.
- Maestro; Schrödinger Release 2018-4; Schrödinger, LLC: New York, NY, USA, 2018; Available online: https://www.schrodinger.com/.
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | Residual Activity (%) at 100 μM Compound |
|---|---|---|
| 1 |  | 2 ± 4 |
| 2 |  | 1 ± 4 |
| 3 |  | 55 ± 9 |
| 4 |  | 62 ± 2 |
| 5 |  | 77 ± 15 |
| Compound | Kd (μM) | IC50 (μM) iCP | IC50 (μM) cCP | |
|---|---|---|---|---|
| Pre-Incubation Time: 0 min | Pre-Incubation Time: 30 min | Pre-Incubation Time: 30 min | ||
| 1 | 22.4 ± 5.1 | 60 ± 7 | 34 ± 2 | 102 ± 1 |
| 2 | 41.1 ± 0.6 | 59 ± 6 | 45 ± 1 | 105 ± 5 |
| Property Name | Property Value |
|---|---|
| Heavy atoms count | 10–30 |
| Rotatable bonds | ≤ 10 |
| Stereocenters | ≤ 3 |
| Rings count | 1–5 |
| Atoms allowed | C, H, O, N, S, B, P, F, Cl, Br, I |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scarpino, A.; Bajusz, D.; Proj, M.; Gobec, M.; Sosič, I.; Gobec, S.; Ferenczy, G.G.; Keserű, G.M. Discovery of Immunoproteasome Inhibitors Using Large-Scale Covalent Virtual Screening. Molecules 2019, 24, 2590. https://doi.org/10.3390/molecules24142590
Scarpino A, Bajusz D, Proj M, Gobec M, Sosič I, Gobec S, Ferenczy GG, Keserű GM. Discovery of Immunoproteasome Inhibitors Using Large-Scale Covalent Virtual Screening. Molecules. 2019; 24(14):2590. https://doi.org/10.3390/molecules24142590
Chicago/Turabian StyleScarpino, Andrea, Dávid Bajusz, Matic Proj, Martina Gobec, Izidor Sosič, Stanislav Gobec, György G. Ferenczy, and György M. Keserű. 2019. "Discovery of Immunoproteasome Inhibitors Using Large-Scale Covalent Virtual Screening" Molecules 24, no. 14: 2590. https://doi.org/10.3390/molecules24142590
APA StyleScarpino, A., Bajusz, D., Proj, M., Gobec, M., Sosič, I., Gobec, S., Ferenczy, G. G., & Keserű, G. M. (2019). Discovery of Immunoproteasome Inhibitors Using Large-Scale Covalent Virtual Screening. Molecules, 24(14), 2590. https://doi.org/10.3390/molecules24142590