2.1. Esterification
Initially, benzoic acid (
1) was chosen as a model substrate to optimize the esterification reaction protocol. In the typical experimental procedure, the mixture of
1 and methanol (MeOH) was heated at 70 °C in the presence of different catalysts (
Figure 2).
The results of this optimization are presented in
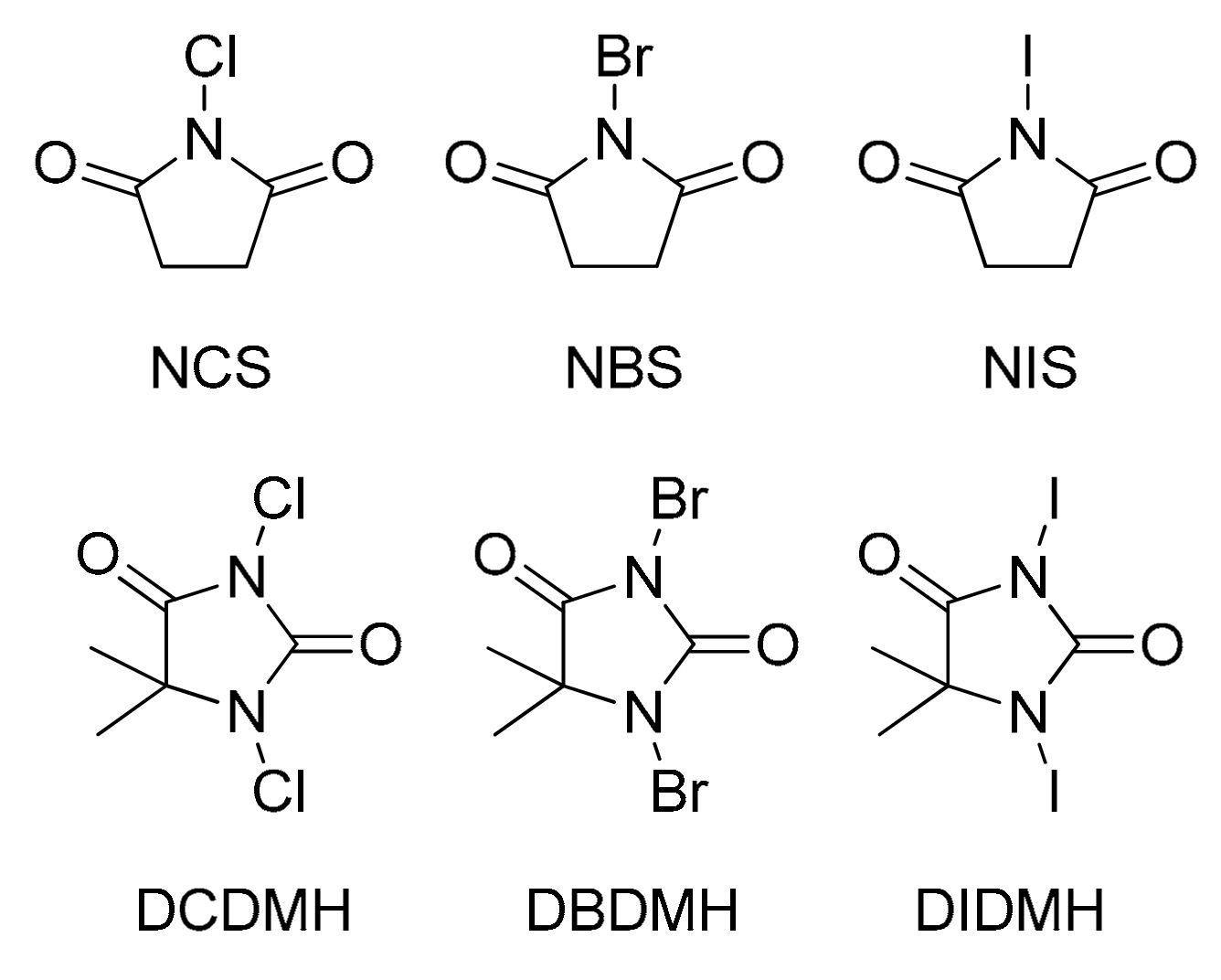
Table 1. Among the studied catalysts (
N-halosuccinimides, 1,3-dihalo-5,5-dimethylhydantoins, halogen mineral acids, molecular bromine and iodine), DBDMH exhibited the highest efficiency (Entry 3). In contrast, no conversion was noticed in the absence of catalyst (Entry 1). Furthermore, the effect of DBDMH loading was examined by changing its amount from a 3.5 molar percentage (mol%) to 7 mol% (
Table 1, Entries 3, 7, and 8). No competing reaction was noticed, and although an excellent conversion was obtained even at 3.5 mol% (92%, Entry 8), a quantitative transformation to methyl benzoate was observed in the presence of 7 mol% of DBDMH (Entry 3). Therefore, further optimization was performed with 7 mol% of DBDMH. Furthermore, varying the temperature from 30 °C to 70 °C (Entries 3, 10, and 11) showed a considerable impact on esterification efficiency, with the optimal temperature being 70 °C (Entry 3). Under dry reaction conditions, the efficiency of esterification dropped for a substantial 20% (Entry 9), which suggests a water-assisted mechanism, as discussed below (
Scheme 1).
To gain a clearer insight into the course of the reaction pathway, esterification of benzoic acid with methanol in the presence of 7 mol% of DBDMH under optimal reaction conditions was performed, with the reaction mixture being sampled every 10–15 min for the first 75 min of the reaction time and the spectra of each sample subsequently measured by
1H-NMR (
Figure 3). Moreover, the pH of the reaction mixture was also measured after 10, 30, and 60 min. The results presented in
Figure 3 show the decomposition of DBDMH to 5,5-dimethylhydantoin (DMH) over the first 60 min. Interestingly, esterification started after the complete conversion of DBDMH to DMH, after 75 min. Furthermore, no conversion to the ester product was observed when a control experiment was performed by heating a mixture of benzoic acid and methanol under optimal reaction conditions at 70 °C in the presence of 7 mol% DMH, suggesting that the formation of bromine specie(s) during DBDMH decomposition is crucial for the esterification reaction and that the role of DBDMH is precatalytic rather than catalytic (
Table 2, Entry 3).
Figure 3 shows a considerable drop in pH value from 4.0–4.5 to 0–0.5 over 60 min, which stayed unchanged throughout the remainder of the reaction (24 h).
These promising results encouraged us to investigate the efficiency of DBDMH in mediating the direct esterification of carboxylic acids with alcohols (
Table 3,
Table 4,
Table 5,
Table 6 and
Table 7). In all cases, octanoic acid (
2) showed significantly higher activity towards esterification than benzoic acid (
1), yielding higher conversions (
Table 3). Since, unlike in the case of octanoic acid, the carbonyl C-atom in the electron accepting carboxyl group of benzoic acid can receive electron density from resonance stabilization of the phenyl ring, benzoic acid is less reactive toward the nucleophile attack than octanoic acid. As reaction-limiting factors were found to include the nucleophilic character and steric properties of the substrate, different alcohols were tested (
a–
j,
Table 3). The elongation of the alcohol alkyl chain (
a,
d, and
f) had a stronger impact on the esterification efficiency of benzoic acid than on that of octanoic acid. In general, DBDMH mediation exhibited significantly higher conversions relative to
N-bromosuccinimide (NBS), especially in the case of the secondary alcohols isopropanol (
c) and cyclopentanol (
g), as NBS mediation yielded only trace products or no conversion at all [
42]. The reaction limitation was observed for the bulky tertiary alcohols
t-BuOH (
e) and adamantanol (
i), as well as for phenol (
h). However, if the sterically-demanding adamantyl group was separated from the alcohol group with a CH
2 unit, the conversion increased to 79% for benzoic acid and 99% for alkyl acid (
Table 3,
1j,
2j). The unreactivity in the case of the phenol molecule can be explained by the aromatic phenol structure, which possesses lower nucleophilicity due to the resonance delocalization of the oxygen electron pair in the OH group.
Moreover, a modest limitation of the substrate scope was observed for monosubstituted benzoic acids, as the reaction only failed in the case of 2-methoxybenzoic acid (
Table 4) and hydroxy derivatives. The lower reactivity in case of ortho-substituted benzoic acids may be attributed to the steric hindrance of the ortho-positioned groups in the phenyl ring. The more the ortho-positioned group is sterically abundant, lower is the conversion. On the other hand, unreactivity of hydroxy-substituted derivatives is caused by the electron-donating properties of the OH group, which gives rise to low electron deficiency on the carbonyl C-atom, and consequently, lower susceptibility of carbonyl group to nucleophile attack. Bader charges for the carbon and oxygen atoms of the carbonyl group were calculated and the analysis confirmed that the carbonyl C-atom in the case of
p-OH-benzoic acid was not as positively charged, as in the case of benzoic acid (
Figure S1 and Table S1). When benzoic acid derivatives bore multiple electron-rich moieties on the aromatic ring, esterification was more limited, as only 3,4-dimethoxybenzoic acid produced an acceptable yield (
Table 4, Compound
18a). In the case of isonicotinic acid (Product
19a), the unreactivity probably originated from the higher basicity and higher nucleophilicity of the nitrogen atom in the pyridine ring relative to the basicity (nucleophilicity) of the carbonyl oxygen atom in the carboxyl group. The nitrogen atom in the pyridine ring could therefore take a role of a Lewis base in a strong halogen bond interaction. [
49] In net effect, it could therefore act as a Br
+ scavenger, preventing the activation of carboxylic acid toward the nucleophile (alcohol) attack in the esterification reaction.
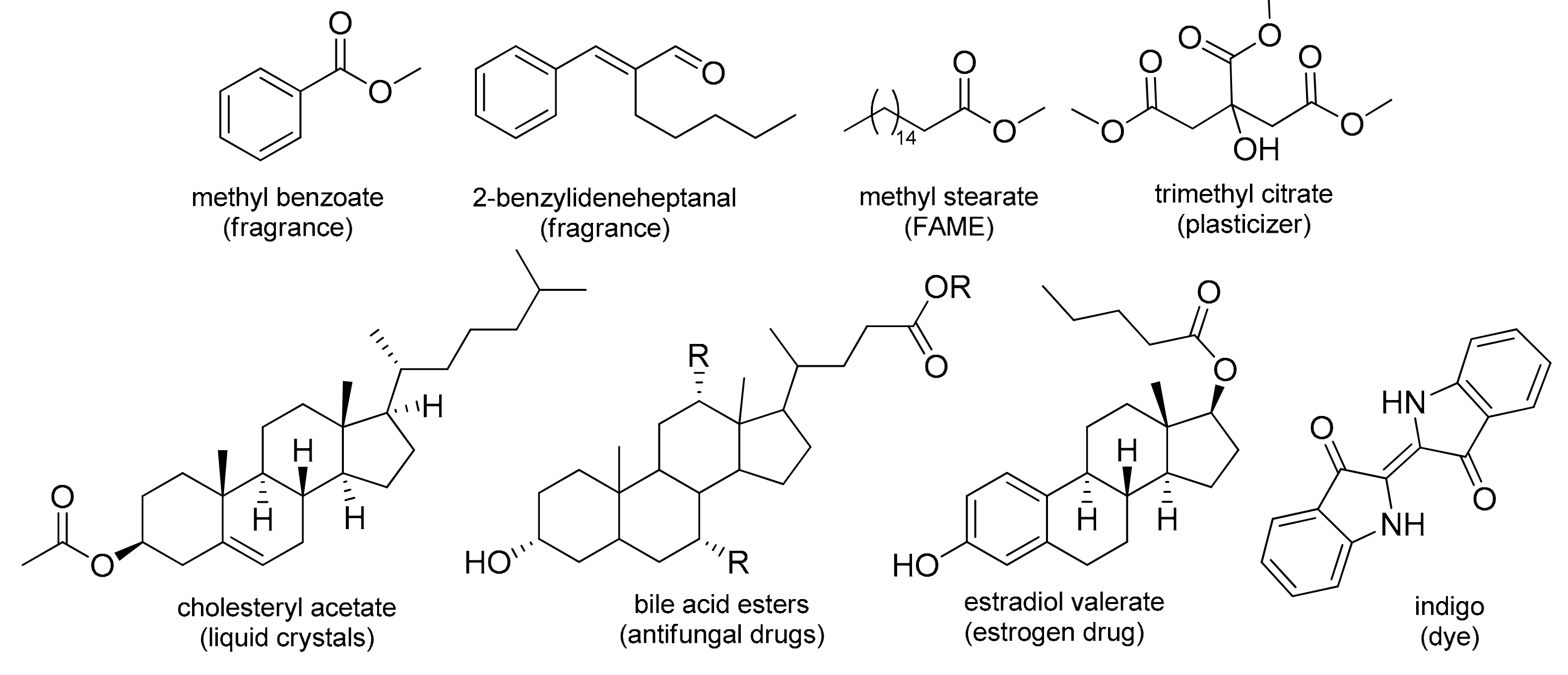
The commercial importance of certain methyl esters in biodiesel production (FAME) and perfumery (methyl benzoate) encouraged us to test the esterification of different types of alkyl carboxylic acids with MeOH under optimal reaction conditions in the presence of DBDMH (
Table 5). No limitations were observed, and excellent yields were achieved, even in the case of polycarboxy derivatives.
In addition, the esterification of a selection of relevant bile acids was conducted to verify the synthetic applicability of this method on larger and more complex structures (
Table 6). Esterification with methanol (
a) and
n-butanol (
d) predominantly resulted in excellent yields, while the use of the secondary alcohol isopropanol (
c) resulted in fair conversion.
Moreover, the results regarding the acetylation of cholesterol and epiandrosterone are presented in
Table 7. Due to the high lipophilicity, and consequently, low solubility of these steroid alcohols in acetic acid, no esterification took place, and ethyl acetate was used as a reaction medium to obtain ester derivatives
34k and
34l in high yields. Using this approach, we also managed to demonstrate the potential of DBDMH as mediator in transesterification reactions. Additionally, to confirm the synthetic value of the presented methodology, scaled-up syntheses of methyl benzoate (
1a), methyl stearate (
21a), methyl citrate (
29a), and cholic acid methyl ester (
30a) up to 30 mmol were performed with excellent yields (93–100%).
The disinfection agency of DBDMH is known to originate from its ability to act as a source of Br
+ in the presence of water through the release of hypobromous acid (HOBr), which further reacts as an oxidant in the process of disinfection [
34]. However, as the substoichiometric amounts of DBDMH (7 mol%) used in the esterification method discussed herein were sufficient for high-yield, direct esterification between carboxylic acid and alcohol, Br
+ clearly acted as a catalytic species rather than an oxidant.
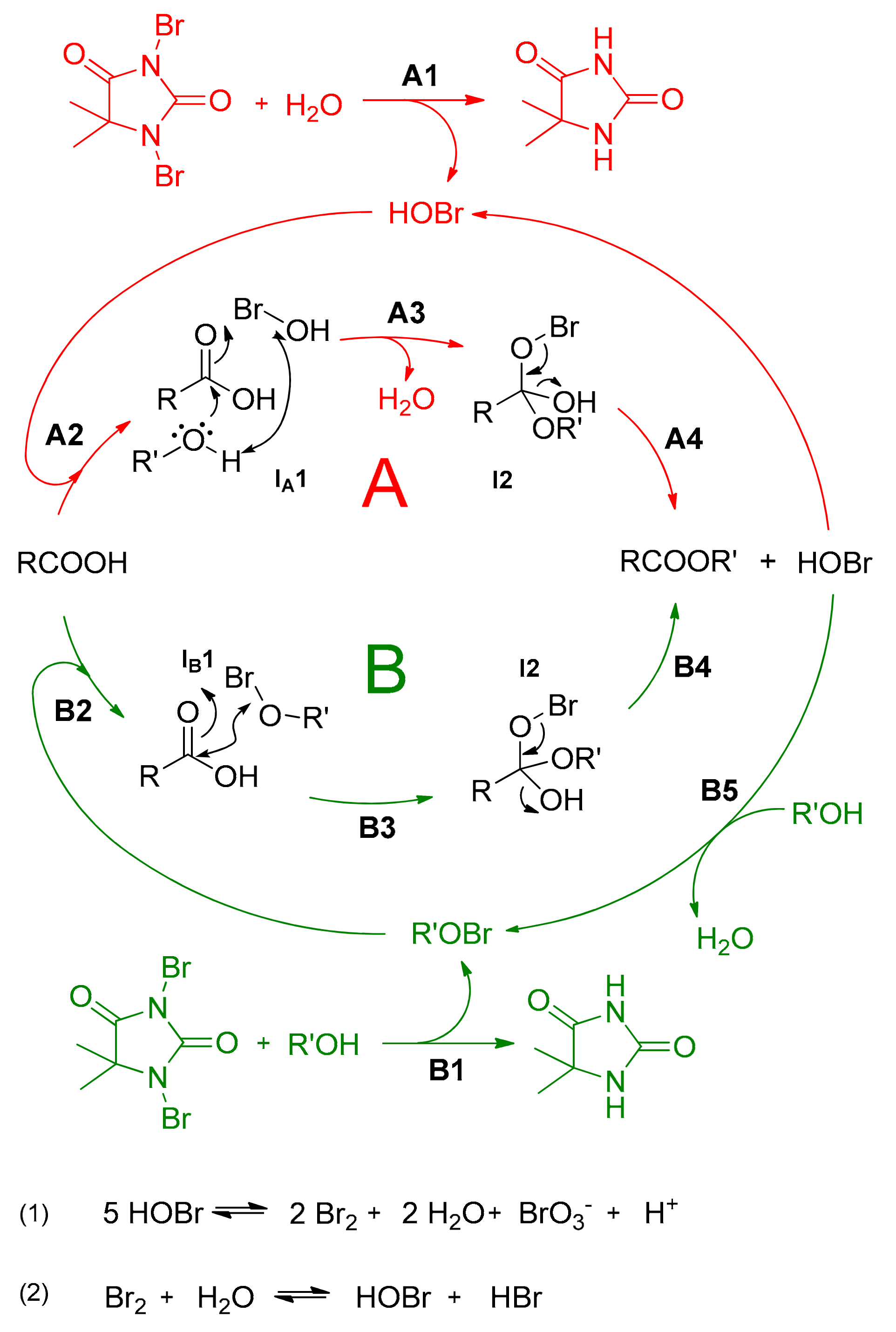
Based on the results of the control experiments presented in
Table 2 and
Figure 3, the proposed reaction pathway is presented in
Scheme 1. In general, DBDMH in the presence of traces of water decomposes to form HOBr (
A1), which catalyses the esterification reaction and regenerates
via path
A (steps
A2–
A4, depicted in red colour). However, as the reaction yields 80% conversion even under dry starting reaction conditions (
Table 2, Entry 7), the initial DBDMH decomposition most probably originates from its reaction with alcohol to form alkyl hypobromite [
50], which further promotes the esterification reaction via path
B (steps
B2–
B4, depicted in green colour). As a consequence of water formation during the esterification reaction (step
A3 or/and
B5), the reaction can further proceed both ways simultaneously, where the formation of HOBr acts as the ultimate driving force of the reaction.
As already mentioned, a considerable drop in pH from 4.0–4.5 to 0–0.5 was measured during the reaction, which could not be attributed to the acidity of HOBr because of its relatively high pK
a value (pK
a = 8.65). At the same time, the formation of a distinctive orange color and bromine odor was observed during the reaction, which readily disappeared upon introduction of a sodium thiosulphate solution, NaS
2O
3(aq), to the reaction mixture. All of this could be explained by the instability of HOBr and its decomposition at pH below 4 to form bromine (Br
2) and bromic acid (HOBr
3) with a considerably lower pK
a of −2 (
Scheme 1, Equation 1) [
51]. Bromine is well-known to form HOBr and HBr in reactions with water through an equilibrium reaction (
Scheme 1, Equation 2), thereby allowing the regeneration of HOBr for subsequent esterification catalysis. Therefore, the water formed through the esterification reaction is more than only an inevitable side product, it is an assisting component in catalysis, due to this equilibrium reaction according to Le Chatelier’s principle. The hypothesis of water-assistance in the proposed mechanism is supported by the fact that only an 80% conversion to the ester product was observed when the reaction was performed in the presence of a drying agent (Na
2SO
4), which is in contrast to the general requirement for water removal for efficient transformation in an acid-catalyzed Fischer esterification methodology (
Table 2, Entry 7). However, if the reaction was performed in water as a solvent, the hydrolysis of the ester product due to the carboxylic acid/ester equilibrium dominated the beneficial increased concentration of HOBr and the reaction resulted in no conversion. Furthermore, to eliminate the possibility of classic Brønsted acid catalysis acting as the major driving force of the quantitative esterification reaction (by the
in-
situ formed HBrO
3 and HBr), the reaction was conducted in the presence of 7 mol% of hydrobromic acid (pK
a = −9), and only 84 percent conversion to methyl benzoate was noticed (
Table 1, Entry 9).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}















