Genome-Wide and Functional View of Proteolytic and Lipolytic Bacteria for Efficient Biogas Production through Enhanced Sewage Sludge Hydrolysis

 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Physiological Characterization of Rummeliibacillus sp. POC4, Brevundimonas sp. LPMIX5 and Ochrobactrum sp. POC 9
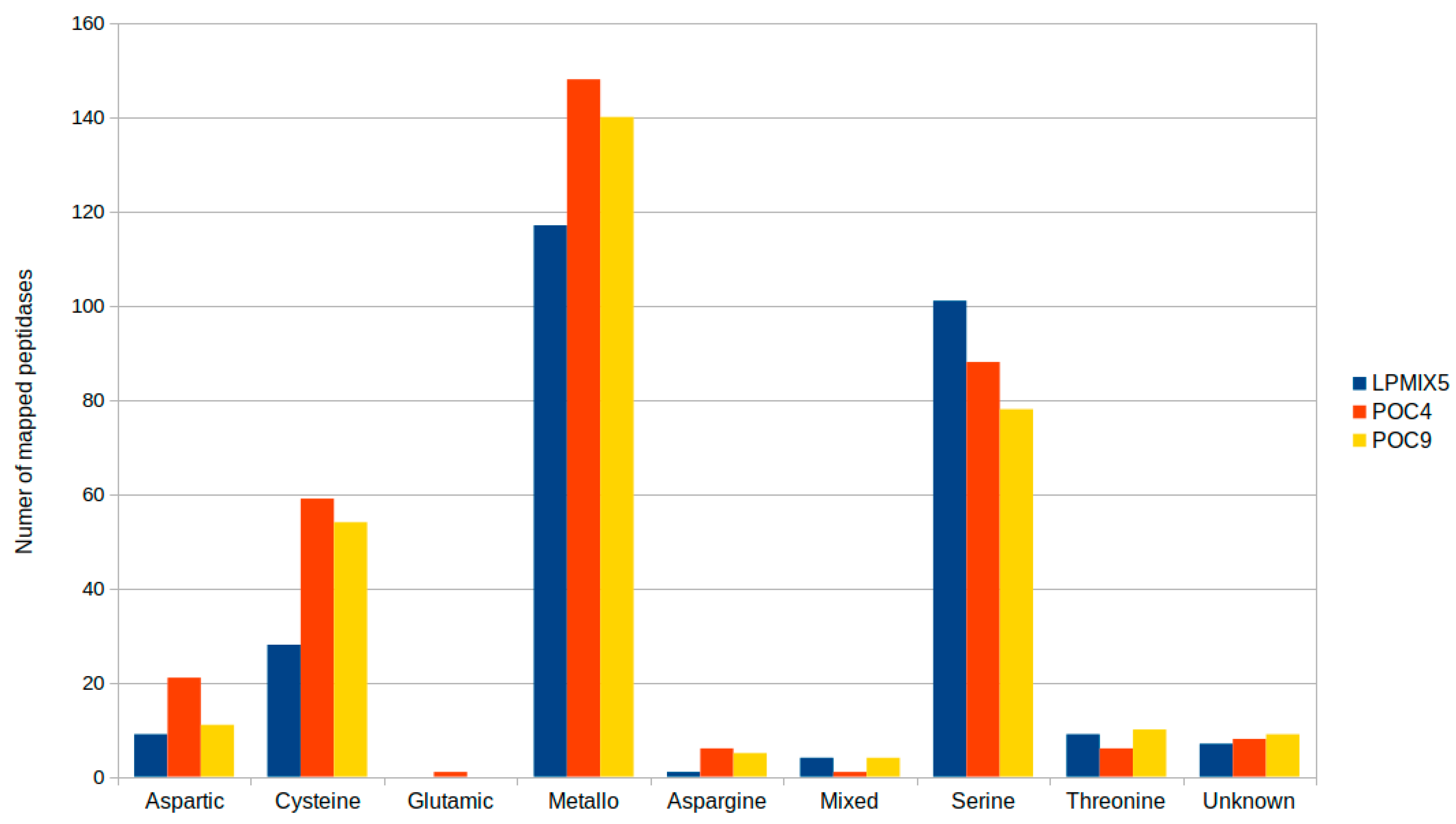
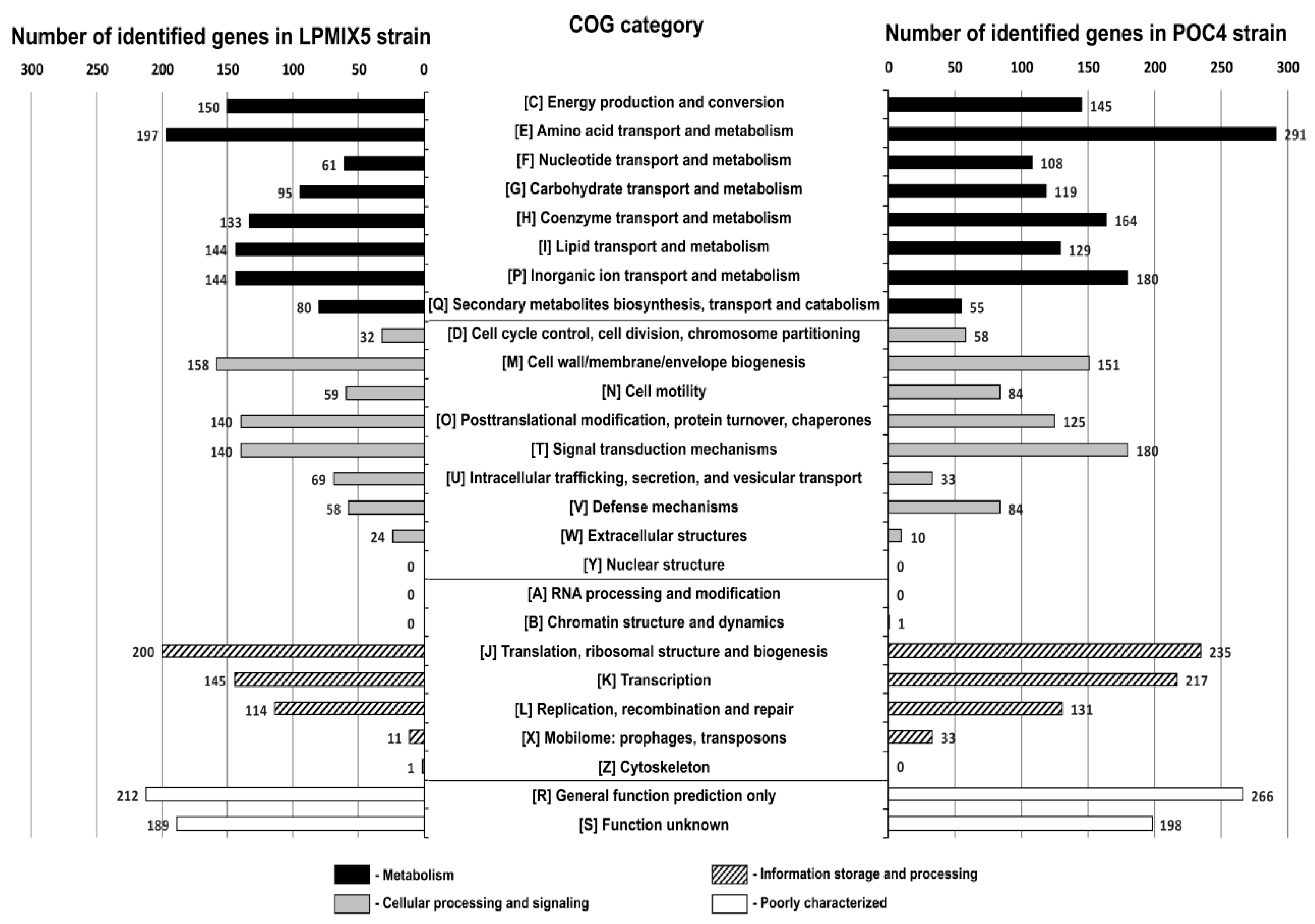
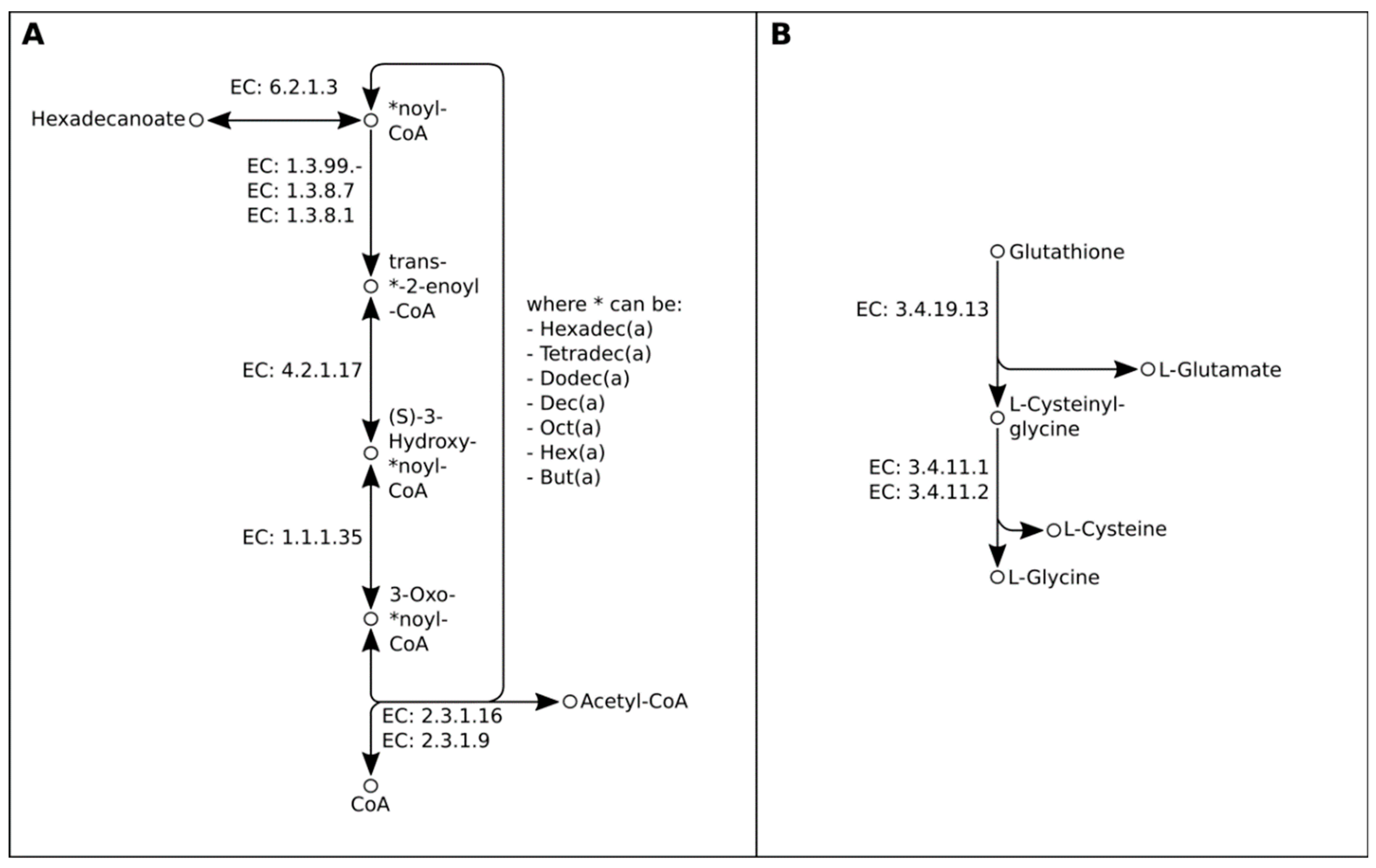
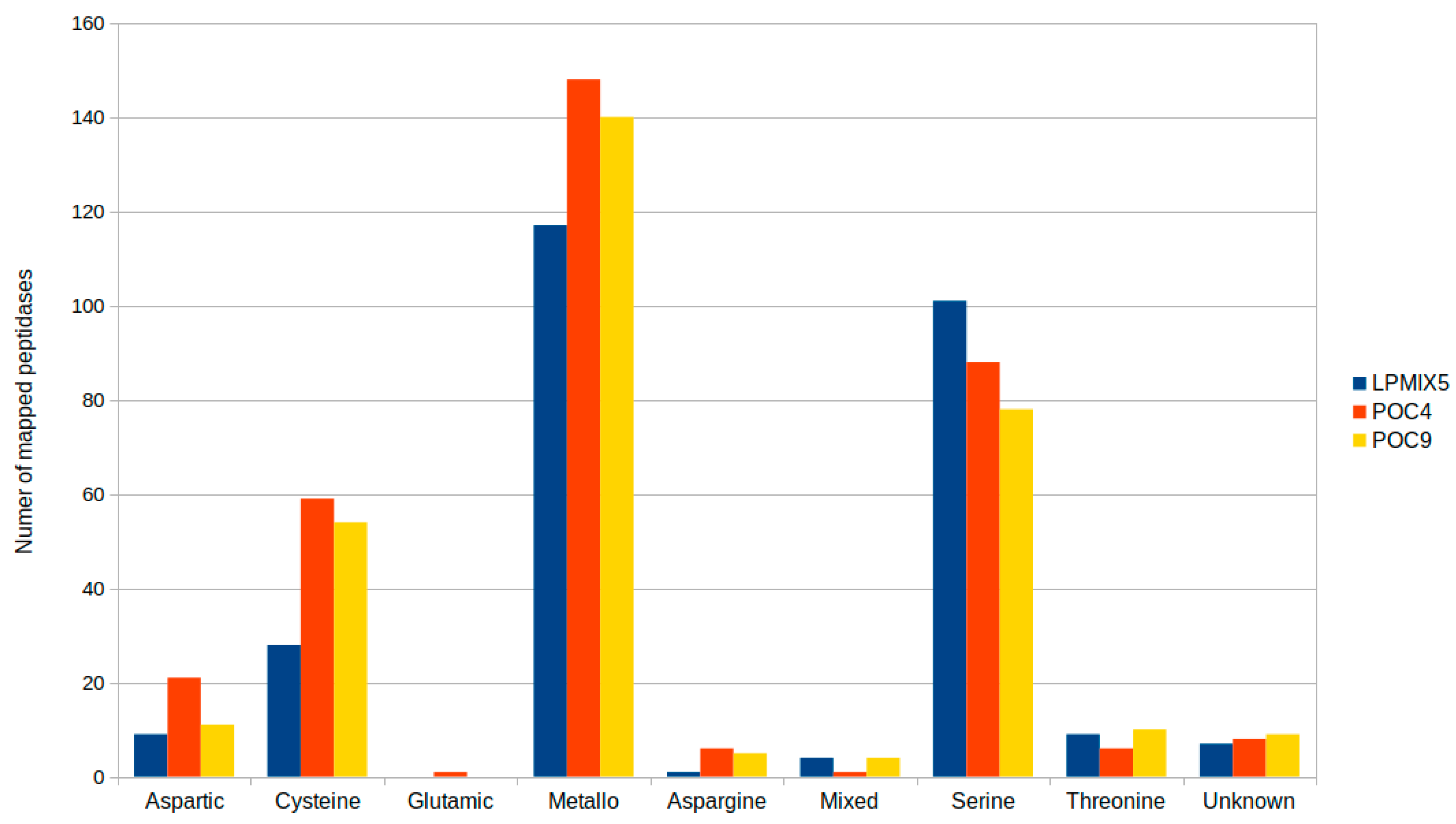
2.2. Genome-Guided Exploration of Rummeliibacillus sp. POC4, Brevundimonas sp. LPMIX5 and Ochrobactrum sp. POC 9 as Bioaugmentation Candidates
2.3. Bioaugmentation of Anaerobic Digestion with Rummeliibacillus sp. POC4, Brevundimonas sp. LPMIX5 and Ochrobactrum sp. POC 9 for Enhanced Biogas Production
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains
4.2. Culture Conditions and Enzyme Activity Assays
4.3. Determination of the Optimal Culture Conditions and the Minimal Inhibitory Concentrations of Metals
4.4. BIOLOG™ Test and Bacterial Growth on Waste Substrates
4.5. Batch Assay of the Anaerobic Digestion of Sewage Sludge
4.6. Analytical Methods
4.7. DNA Manipulations and PCR Conditions
4.8. Draft Genome Sequencing and Assembly
4.9. Bioinformatics
4.10. Nucleotide Sequence Accession Numbers
4.11. Statistical Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Parawira, W. Enzyme research and applications in biotechnological intensification of biogas production. Crit. Rev. Biotechnol. 2012, 32, 172–186. [Google Scholar] [CrossRef]
- Ariunbaatar, J.; Panico, A.; Esposito, G.; Pirozzi, F.; Lens, P.N. Pretreatment methods to enhance anaerobic digestion of organic solid waste. Appl. Energy 2014, 123, 143–156. [Google Scholar] [CrossRef]
- Wagner, A.O.; Lackner, N.; Mutschlechner, M.; Prem, E.M.; Markt, R.; Illmer, P. Biological Pretreatment Strategies for Second-Generation Lignocellulosic Resources to Enhance Biogas Production. Energies 2018, 11, 1797. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Singh, P.K.; Dash, S.; Pattnaik, R. Microbial pretreatment of lignocellulosic biomass for enhanced biomethanation and waste management. 3 Biotech 2018, 8, 458. [Google Scholar] [CrossRef]
- Romano, R.T.; Zhang, R. Co-digestion of onion juice and wastewater sludge using an anaerobic mixed biofilm reactor. Bioresour. Technol. 2008, 99, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Deflaun, M.F.; Steffan, R.J. Bioaugmentation. In Encyclopedia of Environmental Microbiology; Bitton, G., Ed.; Wiley-Interscience: New York, NY, USA, 2002; Volume 1, pp. 434–442. [Google Scholar]
- Herrero, M.; Stuckey, D.; Stuckey, D. Bioaugmentation and its application in wastewater treatment: A review. Chemosphere 2015, 140, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Poszytek, K.; Ciężkowska, M.; Skłodowska, A.; Drewniak, Ł. Microbial Consortium with High Cellulolytic Activity (MCHCA) for Enhanced Biogas Production. Front. Microbiol. 2016, 7, 548. [Google Scholar] [CrossRef] [PubMed]
- Zuroff, T.R.; Xiques, S.B.; Curtis, W.R. Consortia-mediated bioprocessing of cellulose to ethanol with a symbiotic Clostridium phytofermentans/yeast co-culture. Biotechnol. Biofuels 2013, 6, 59. [Google Scholar] [CrossRef] [PubMed]
- Lebiocka, M.; Montusiewicz, A.; Cydzik-Kwiatkowska, A. Effect of Bioaugmentation on Biogas Yields and Kinetics in Anaerobic Digestion of Sewage Sludge. Int. J. Environ. Res. Public Heal. 2018, 15, 1717. [Google Scholar] [CrossRef]
- Angelidaki, I.; Ahring, B. Methods for increasing the biogas potential from the recalcitrant organic matter contained in manure. Water Sci. Technol. 2000, 41, 189–194. [Google Scholar] [CrossRef]
- Nielsen, H.B.; Mladenovska, Z.; Ahring, B.K. Bioaugmentation of a two-stage thermophilic (68C/55C) anaerobic digestion concept for improvement of the methane yield from cattle manure. Biotechnol. Bioeng. 2000, 97, 1638–1643. [Google Scholar] [CrossRef] [PubMed]
- Savant, D.V.; Ranade, D.R. Application of Methanobrevibacter acididurans in anaerobic digestion. Water Sci. Technol. 2004, 50, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Cirne, D.G.; Björnsson, L.; Alves, M.; Mattiasson, B. Effects of bioaugmentation by an anaerobic lipolytic bacterium on anaerobic digestion of lipid-rich waste. J. Chem. Technol. Biotechnol. 2006, 81, 1745–1752. [Google Scholar] [CrossRef] [Green Version]
- Weis, S.; Tauber, M.; Somitsch, W.; Meincke, R.; Müller, H.; Berg, G.; Guebitz, G. Enhancement of biogas production by addition of hemicellulolytic bacteria immobilised on activated zeolite. Water Res. 2010, 44, 1970–1980. [Google Scholar] [CrossRef] [PubMed]
- Tsapekos, P.; Kougias, P.; Vasileiou, S.; Treu, L.; Campanaro, S.; Lyberatos, G.; Angelidaki, I. Bioaugmentation with hydrolytic microbes to improve the anaerobic biodegradability of lignocellulosic agricultural residues. Bioresour. Technol. 2017, 234, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Poszytek, K.; Pyzik, A.; Sobczak, A.; Lipinski, L.; Sklodowska, A.; Drewniak, L. The effect of the source of microorganisms on adaptation of hydrolytic consortia dedicated to anaerobic digestion of maize silage. Anaerobe 2017, 46, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, M.; da Fonseca, M.M.; de Carvalho, C.C. Bioaugmentation and biostimulation strategies to improve the effectiveness of bioremediation processes. Biodegradation 2011, 22, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Boon, N.; Goris, J.; De Vos, P.; Verstraete, W.; Top, E.M. Bioaugmentation of Activated Sludge by an Indigenous 3-Chloroaniline-Degrading Comamonas testosteroni Strain, I2gfp. Appl. Environ. Microbiol. 2000, 66, 2906–2913. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, S.; Shiota, N.; Katsura, K.; Akashi, A. Solubilization of organic sludge by thermophilic aerobic bacteria as a pretreatment for anaerobic digestion. Water Sci. Technol. 2011, 41, 163–169. [Google Scholar] [CrossRef]
- Singer, A.C.; Van Der Gast, C.J.; Thompson, I.P. Perspectives and vision for strain selection in bioaugmentation. Trends Biotechnol. 2005, 23, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Poszytek, K.; Karczewska-Golec, J.; Ciok, A.; Decewicz, P.; Dziurzynski, M.; Gorecki, A.; Jakusz, G.; Krucon, T.; Lomza, P.; Romaniuk, K.; et al. Genome-Guided Characterization of Ochrobactrum sp. POC9 Enhancing Sewage Sludge Utilization—Biotechnological Potential and Biosafety Considerations. Int. J. Environ. Res. Public Health 2018, 15, 1501. [Google Scholar] [CrossRef] [PubMed]
- Sadhu, S.; Maiti, T.K. Cellulase Production by Bacteria: A Review. Br. Microbiol. Res. J. 2013, 3, 235–258. [Google Scholar] [CrossRef] [Green Version]
- Anson, M.L. The estimation of pepsin, trypsin, papain, and cathepsin with hemoglobin. J. Gen. Physiol. 1938, 22, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Cupp-Enyard, C. Sigma’s Non-specific Protease Activity Assay—Casein as a Substrate. J. Vis. Exp. 2008, 19, 899. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Samant, K.; Sahu, A. Isolation of Cellulose-Degrading Bacteria and Determination of Their Cellulolytic Potential. Int. J. Microbiol. 2012, 2012, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mongkolthanaruk, W.; Dharmsthiti, S. Biodegradation of lipid-rich wastewater by a mixed bacterial consortium. Int. Biodeterior. Biodegradation 2002, 50, 101–105. [Google Scholar] [CrossRef]
- Jadhav, V.V.; Pote, S.S.; Yadav, A.; Shouche, Y.S.; Bhadekar, R.K. Extracellular cold active lipase from the psychrotrophic Halomonas sp. BRI 8 isolated from the Antarctic sea water. Songklanakarin J. Sci. Technol. 2013, 5, 623–630. [Google Scholar]
- Gryta, A.; Frąc, M.; Oszust, K. The Application of the Biolog EcoPlate Approach in Ecotoxicological Evaluation of Dairy Sewage Sludge. Appl. Biochem. Biotechnol. 2014, 174, 1434–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krawczyk, P.S.; Lipinski, L.; Dziembowski, A. PlasFlow: predicting plasmid sequences in metagenomic data using genome signatures. Nucleic Acids Res. 2018, 46, e35. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: an updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Côté, N.; Fleury, A.; Dumont-Blanchette, E.; Fukamizo, T.; Mitsutomi, M.; Brzezinski, R. Two exo-beta-d-glucosaminidases/exochitosanases from actinomycetes define a new subfamily within family 2 of glycoside hydrolases. Biochem. J. 2006, 394 Pt 3, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2017, 46, D624–D632. [Google Scholar] [CrossRef] [PubMed]
- Grass, G.; Otto, M.; Fricke, B.; Haney, C.J.; Rensing, C.; Nies, D.H.; Munkelt, D. FieF (YiiP) from Escherichia coli mediates decreased cellular accumulation of iron and relieves iron stress. Arch. Microbiol. 2005, 183, 9–18. [Google Scholar] [CrossRef]
- Ackerley, D.F.; Gonzalez, C.F.; Park, C.H.; Blake, R.; Keyhan, M.; Matin, A.C. Chromate-Reducing Properties of Soluble Flavoproteins from Pseudomonas putida and Escherichia coli. Appl. Environ. Microbiol. 2004, 70, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, A.H.; Moreno-Sánchez, R.; Cervantes, C. Chromate Efflux by Means of the ChrA Chromate Resistance Protein from Pseudomonas aeruginosa. J. Bacteriol. 1999, 181, 7398–7400. [Google Scholar] [PubMed]
- Cha, J.S.; Cooksey, D.A. Copper resistance in Pseudomonas syringae mediated by periplasmic and outer membrane proteins. Proc. Natl. Acad. Sci. USA 1991, 88, 8915–8919. [Google Scholar] [CrossRef] [PubMed]
- Grass, G.; Große, C.; Nies, D.H. Regulation of the cnr Cobalt and Nickel Resistance Determinant from Ralstonia sp. Strain CH34. J. Bacteriol. 2000, 182, 1390–1398. [Google Scholar] [CrossRef]
- Schmidt, T.; Schlegel, H.G. Combined nickel-cobalt-cadmium resistance encoded by the ncc locus of Alcaligenes xylosoxidans 31A. J. Bacteriol. 1994, 176, 7045–7054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borremans, B.; Provoost, A.; Van Der Lelie, D.; Hobman, J.L.; Brown, N.L. Cloning and functional analysis of the pbr lead resistance determinant of Ralstonia metallidurans CH34. J. Bacteriol. 2001, 183, 5651–5658. [Google Scholar] [CrossRef]
- Brémond, U.; De Buyer, R.; Steyer, J.-P.; Bernet, N.; Carrère, H. Biological pretreatments of biomass for improving biogas production: an overview from lab scale to full-scale. Renew. Sustain. Energy Rev. 2018, 90, 583–604. [Google Scholar] [CrossRef]
- Nzila, A. Mini review: Update on bioaugmentation in anaerobic processes for biogas production. Anaerobe 2017, 46, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Nigam, P.S. Microbial Enzymes with Special Characteristics for Biotechnological Applications. Biomolecules 2013, 3, 597–611. [Google Scholar] [CrossRef] [PubMed]
- Sagar, K.; Bashir, Y.; Phukan, M.M.; Kamwar, B.K. Isolation of lipolytic bacteria from waste contaminated soil: A study with regard to process optimization for lipase. Int. J. Sci. Technol. Res. 2013, 2, 214–218. [Google Scholar]
- Park, I.J.; Yoon, J.C.; Park, S.J.; Kim, E.H.; Cho, Y.J.; Shin, K.S. Characterization of the proteolytic bacteria isolated from a rotating biological contactor. J. Microbiol. 2003, 41, 73–77. [Google Scholar]
- Bramucci, M.G.; Nagarajan, V. Industrial wastewater bioreactors: sources of novel microorganisms for biotechnology. Trends Biotechnol. 2000, 18, 501–505. [Google Scholar] [CrossRef]
- Campanaro, S.; Treu, L.; Kougias, P.G.; De Francisci, D.; Valle, G.; Angelidaki, I.; Francisci, D. Metagenomic analysis and functional characterization of the biogas microbiome using high throughput shotgun sequencing and a novel binning strategy. Biotechnol. Biofuels 2016, 9, 5478. [Google Scholar] [CrossRef]
- Parawira, W.; Murto, M.; Read, J.; Mattiasson, B. Profile of hydrolases and biogas production during two-stage mesophilic anaerobic digestion of solid potato waste. Process. Biochem. 2005, 40, 2945–2952. [Google Scholar] [CrossRef]
- Guedon, E.; Desvaux, M.; Petitdemange, H. Improvement of Cellulolytic Properties of Clostridium cellulolyticum by Metabolic Engineering. Appl. Environ. Microbiol. 2002, 68, 53–58. [Google Scholar] [CrossRef]
- Lü, F.; Li, T.; Wang, T.; Shao, L.; He, P. Improvement of sludge digestate biodegradability by thermophilic bioaugmentation. Appl. Microbiol. Biotechnol. 2014, 98, 969–977. [Google Scholar] [CrossRef]
- Miah, M.S.; Tada, C.; Sawayama, S. Enhancement of Biogas Production from Sewage Sludge with the Addition of Geobacillus sp. Strain AT1 Culture. Jpn. J. Water Treat. Boil. 2004, 40, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.; Fan, H.; Liu, S.; Liu, Y.; Liu, Z. A bioaugmentation failure caused by phage infection and weak biofilm formation ability. J. Environ. Sci. 2009, 21, 1153–1161. [Google Scholar] [CrossRef]
- Stephenson, D.; Stephenson, T. Bioaugmentation for enhancing biological wastewater treatment. Biotechnol. Adv. 1992, 10, 549–559. [Google Scholar] [CrossRef]
- Qu, Y.-Y.; Zhou, J.-T.; Wang, J.; Xing, L.-L.; Jiang, N.; Gou, M.; Uddin, M.S. Population dynamics in bioaugmented membrane bioreactor for treatment of bromoamine acid wastewater. Bioresour. Technol. 2009, 100, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Bouchez, T.; Patureau, D.; Dabert, P.; Juretschko, S.; Doré, J.; Delgenès, P.; Moletta, R.; Wagner, M. Ecological study of a bioaugmentation failure. Environ. Microbiol. 2000, 2, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Lane, D.J. 16S/23S rRNA sequencing. In Nucleic Acid Techniques in Bacterial Systematic; Stackebrandt, E., Goodfellow, M., Eds.; John Wiley and Sons: Chichester, UK, 1991; pp. 115–175. [Google Scholar]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2001, 17, 10–12. [Google Scholar] [CrossRef]
- Rodriguez-R, L.M.; Gunturu, S.; Harvey, W.T.; Rosselló-Mora, R.; Tiedje, J.M.; Cole, J.R.; Konstantinidis, K.T. The Microbial Genomes Atlas (MiGA) webserver: Taxonomic and gene diversity analysis of Archaea and Bacteria at the whole genome level. Nucleic Acids Res. 2018, 46, W282–W288. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2016, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Chojnacki, S.; Cowley, A.; Lee, J.; Foix, A.; Lopez, R.; Lee, J. Programmatic access to bioinformatics tools from EMBL-EBI update: 2017. Nucleic Acids Res. 2017, 45, W550–W553. [Google Scholar] [CrossRef] [Green Version]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed]
- Pal, C.; Bengtsson-Palme, J.; Rensing, C.; Kristiansson, E.; Larsson, D.G.J. BacMet: Antibacterial biocide and metal resistance genes database. Nucleic Acids Res. 2013, 42, D737–D743. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using diamond. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: architecture and applications. BMC Bioinform. 2008, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org/ (accessed on 17 June 2019).



{kind=link}
{kind=link}
{kind=link}
| Strain | Temperature Range °C | The Optimal Temperature in °C | Growth pH Range | The Optimal pH | The minimal inhibitory Concentrations of Metals (MICs) [mM] | Enzymatic Activity | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cd | Cr | Cu | Zn | Ni | Pb | Protease | Lipase | Cellulase | Amylase | Xylanase | |||||
| Rummeliibacillus sp. POC4 | 7–8 | 1 | 5 | 7.5 | 5 | 5 | 10 | + | + | - | + | - | |||
| Ochrobactrum sp. POC9 | 5–7 | 2 | 10 | 10 | 5 | 7.5 | 10 | + | + | + | + | - | |||
| Brevundimonas sp. LPMIX5 | 6–10 | 2 | 5 | 7.5 | 2 | 5 | 10 | + | + | + | - | + | |||
| Carbon Source | Strain | ||
|---|---|---|---|
| POC4 | POC9 | LPMIX5 | |
| β-Methyl-d-glucoside | + | + | + |
| d-Galactonic acid-ɣ-lactone | + | + | - |
| l-Arginine | + | - | - |
| Pyruvic acid methyl ester | + | + | + |
| d-Xylose | + | + | + |
| d-Galacturonic acid | + | + | - |
| l-Asparagine | + | + | + |
| Tween 40 | + | + | + |
| i-Erythritol | + | + | - |
| 2-Hydroxybenzoic acid | - | - | - |
| l-Phenylalanine | - | - | - |
| Tween 80 | + | + | + |
| d-Mannitol | + | + | + |
| 4-Hydroxybenzoic acid | + | - | - |
| l-Serine | + | + | - |
| α-Cyclodextrin | - | - | - |
| N-acetyl-d-glucosamine | + | + | + |
| ɣ-Hydroxybutyric acid | - | + | - |
| l-Threonine | + | + | + |
| Glycogen | - | - | + |
| d-Glucosaminic acid | + | + | - |
| Itaconic acid | + | - | - |
| Glycyl-l-glutamic acid | + | + | + |
| d-Cellobiose | + | + | + |
| Glucose-1-phosphate | + | + | - |
| α-Ketobutyric acid | + | + | + |
| Phenylethylamine | + | - | - |
| α-d-Lactose | + | + | - |
| d,l-α-Glycerol phosphate | + | + | - |
| d-Malic acid | + | + | - |
| Putrescine | + | - | - |
| Parameters | Cumulative Biogas Production 1 | CH4 Content | VFAs 2 | sCOD 3 | |
|---|---|---|---|---|---|
| Units | L/kgvs | % | g/L | g/L | |
| 0 day | 1.97 ± 0.09 | 5.03 ± 0.06 | |||
| Control | 3 days | 229.58 ± 13.92 | 43.41 | 2.58 ± 0.07 | 6.53 ± 0.12 |
| 7 days | 61.34 | 2.15 ± 0.79 | 5.83 ± 0.95 | ||
| 30 days | 49.18 | 0.95 ± 0.09 | 3.67 ± 0.12 | ||
| POC4 | 3 days | 279.98 ± 13.58 | 47.45 | 2.98 ± 0.77 | 6.85 ± 1.04 |
| 7 days | 65.08 | 1.60 ± 1.43 | 5.30 ± 1.89 | ||
| 30 days | 66.88 | 0.89 ± 0.50 | 3.40 ± 0.59 | ||
| POC9 | 3 days | 294.58 ± 44.98 | 46.00 | 3.28 ± 0.45 | 6.97 ± 0.55 |
| 7 days | 66.48 | 1.78 ± 1.38 | 5.00 ± 2.19 | ||
| 30 days | 58.87 | 0.82 ± 0.61 | 3.30 ± 1.62 | ||
| LPMIX5 | 3 days | 245.87 ± 36.36 | 46.13 | 2.87 ± 0.32 | 6.70 ± 0.53 |
| 7 days | 65.91 | 1.67 ± 0.40 | 4.93 ± 0.59 | ||
| 30 days | 55.69 | 0.77 ± 0.25 | 2.70 ± 0.87 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poszytek, K.; Karczewska-Golec, J.; Dziurzynski, M.; Stepkowska-Kowalska, O.; Gorecki, A.; Decewicz, P.; Dziewit, L.; Drewniak, L. Genome-Wide and Functional View of Proteolytic and Lipolytic Bacteria for Efficient Biogas Production through Enhanced Sewage Sludge Hydrolysis. Molecules 2019, 24, 2624. https://doi.org/10.3390/molecules24142624
Poszytek K, Karczewska-Golec J, Dziurzynski M, Stepkowska-Kowalska O, Gorecki A, Decewicz P, Dziewit L, Drewniak L. Genome-Wide and Functional View of Proteolytic and Lipolytic Bacteria for Efficient Biogas Production through Enhanced Sewage Sludge Hydrolysis. Molecules. 2019; 24(14):2624. https://doi.org/10.3390/molecules24142624
Chicago/Turabian StylePoszytek, Krzysztof, Joanna Karczewska-Golec, Mikolaj Dziurzynski, Olga Stepkowska-Kowalska, Adrian Gorecki, Przemyslaw Decewicz, Lukasz Dziewit, and Lukasz Drewniak. 2019. "Genome-Wide and Functional View of Proteolytic and Lipolytic Bacteria for Efficient Biogas Production through Enhanced Sewage Sludge Hydrolysis" Molecules 24, no. 14: 2624. https://doi.org/10.3390/molecules24142624
APA StylePoszytek, K., Karczewska-Golec, J., Dziurzynski, M., Stepkowska-Kowalska, O., Gorecki, A., Decewicz, P., Dziewit, L., & Drewniak, L. (2019). Genome-Wide and Functional View of Proteolytic and Lipolytic Bacteria for Efficient Biogas Production through Enhanced Sewage Sludge Hydrolysis. Molecules, 24(14), 2624. https://doi.org/10.3390/molecules24142624