
Intramolecular Carbene C-H Insertion Reactions of 2-Diazo-2-sulfamoylacetamides



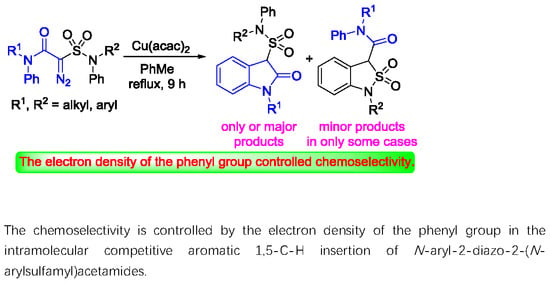
Abstract
:
1. Introduction
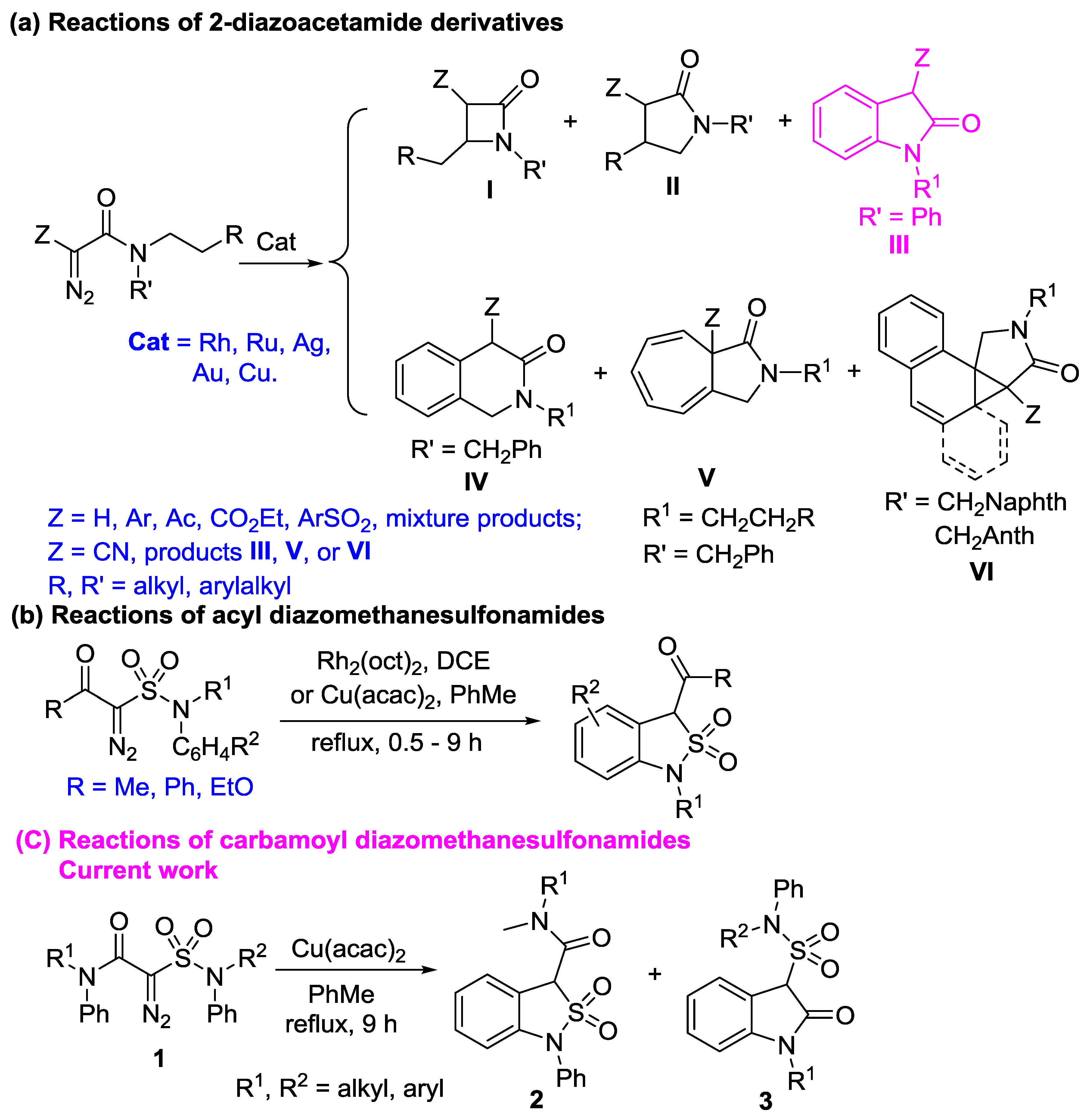
2. Results and Discussion
2.1. Synthesis of 2-Diazo-2-sulfamoylacetamides 1
2.2. Intramolecular Carbene C-H Insertion Reactions of 2-Diazo-2-sulfamoylacetamides 1
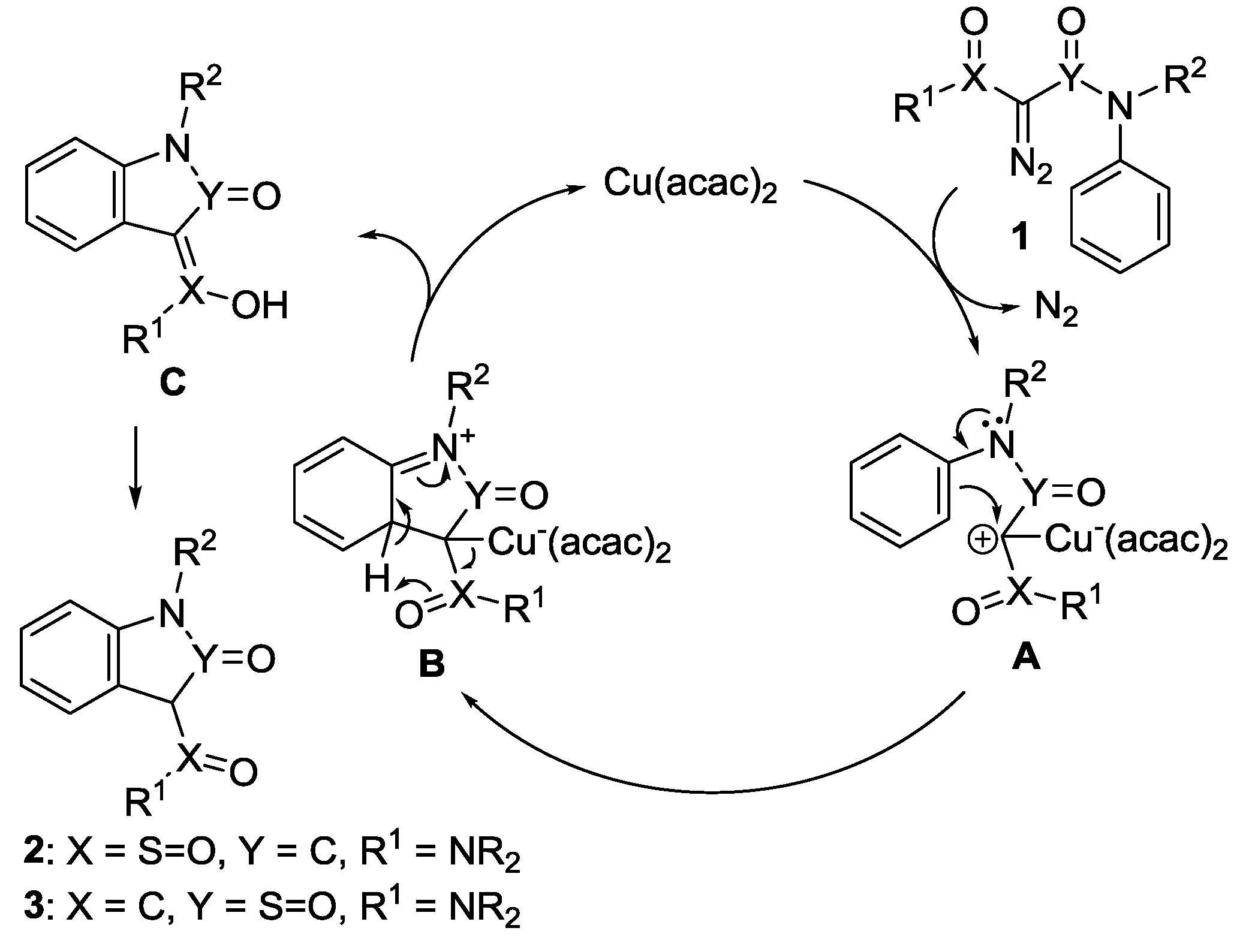
2.3. Mechanism on the Aromatic 1,5-C-H Insertion
2.4. Rationale on the Chemoselectivity in the Aromatic 1,5-C-H Insertions of N-aryl-2-diazo-2-(N-arylsulfamoyl)acetamides
3. Materials and Methods
3.1. Materials and Instruments
3.2. General Procedure for the Preparation of Chloroacetamides 5
3.2.1. 2-Chloro-N,N-dimethylacetamide (5a)
3.2.2. 2-Chloro-N-methyl-N-phenylacetamide (5c)
3.2.3. 2-Chloro-N,N-diphenylacetamide (5d)
3.3. General Procedure for the Synthesis of Sulfamoylacetamides 7
3.3.1. 2-(N-Benzyl-N-butylsulfamoyl)-N-methyl-N-phenylacetamide (7c)
3.3.2. 2-(N-Benzyl-N-butylsulfamoyl)-N,N-diphenylacetamide (7d)
3.3.3. N-Methyl-2-(N-methyl-N-phenylsulfamoyl)-N-phenylacetamide (7e)
3.3.4. N-Methyl-N-phenyl-2-(N,N-diphenylsulfamoyl)acetamide (7f)
3.3.5. 2-(N-Methyl-N-phenylsulfamoyl)-N,N-diphenylacetamide (7g)
3.3.6. N,N-Diphenyl-2-(N,N-diphenylsulfamoyl)acetamide (7h)
3.4. General Procedure for the Synthesis of 2-Diazo-2-sulfamoylacetamides 1
3.4.1. 2-Diazo-N,N-dimethyl-2-(N,N-diphenylsulfamoyl)acetamide (1b)
3.4.2. 2-(N-Benzyl-N-butylsulfamoyl)-2-diazo-N-methyl-N-phenyl acetamide (1c)
3.4.3. 2-(N-Benzyl-N-butylsulfamoyl)-2-diazo-N,N-diphenyl acetamide (1d)
3.4.4. 2-Diazo-N-methyl-2-(N-methyl-N-phenylsulfamoyl)-N-phenylacetamide (1e)
3.4.5. 2-Diazo-N-methyl-N-phenyl-2-(N,N-diphenylsulfamoyl)acetamide (1f)
3.4.6. 2-Diazo-2-(N-methyl-N-phenylsulfamoyl)-N,N-diphenylacetamide (1g)
3.4.7. 2-Diazo-N,N-diphenyl-2-(N,N-diphenylsulfamoyl)acetamide (1h)
3.5. General Procedure for the Reaction of 2-Diazo-2-sulfamoylacetamides 1
3.5.1. N,N-Dimethyl-2,2-dioxido-1-phenyl-1,3-dihydrobenzo[c]isothiazole-3-carboxamide (2b)
3.5.2. N-Benzyl-N-butyl-2-oxo-1-methylindoline-3-sulfonamide (3c)
3.5.3. N-Benzyl-N-butyl-2-oxo-1-phenylindoline-3-sulfonamide (3d)
3.5.4. N,1-Dimethyl-2-oxo-N-phenylindoline-3-sulfonamide (3e)
3.5.5. N-Methyl-2,2-dioxido-N,1-diphenyl-1,3-dihydrobenzo[c]isothiazole-3-carboxamide (2f)
3.5.6. 1-Methyl-2-oxo-N,N-diphenylindoline-3-sulfonamide (3f)
3.5.7. N-Methyl-2-oxo-N,1-diphenylindoline-3-sulfonamide (3g)
3.5.8. 2-Oxo-N,N,1-triphenylindoline-3-sulfonamide (3h)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Padwa, A.; Krunpe, K.E. Application of Intramolecular Carbenoid Reactions in Organic Synthesis. Tetrahedron 1992, 48, 5385–5453. [Google Scholar] [CrossRef]
- Doyle, M.P.; McKervey, M.A.; Ye, T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds; John Willey and Sons: New York, NY, USA, 2008. [Google Scholar]
- Doyle, M.P.; Duffy, R.; Ratnikov, M.; Zhou, L. Catalytic Carbene Insertion into C−H Bonds. Chem. Rev. 2010, 110, 704–724. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.E.; Walvoord, R.R.; Padilla-Salinas, R.; Kozlowski, M.C. Aerobic Copper-Catalyzed Organic Reactions. Chem. Rev. 2013, 113, 6234–6258. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.P.; Hu, W.; Wee, A.G.H.; Wang, Z.; Duncan, S.C. Influences of Catalyst Configuration and Catalyst Loading on Selectivities in Reactions of Diazoacetamides. Barrier to Equilibrium between Diastereomeric Conformations. Org. Lett. 2003, 5, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.H.; Niagle, A.; Chen, C.; Gandhi, D.; Jung, K.W. γ-Lactam Synthesis via C−H Insertion: Elaboration of N-Benzyl Protecting Groups for High Regioselectivity toward the Total Synthesis of Rolipram. Org. Lett. 2003, 5, 2259–2262. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.K.W.; Yu, W.Y.; Che, C.M. Ruthenium-Catalyzed Stereoselective Intramolecular Carbenoid C−H Insertion for β- and γ-Lactam Formations by Decomposition of α-Diazoacetamides. Org. Lett. 2005, 7, 1081–1084. [Google Scholar] [CrossRef] [PubMed]
- Candeias, N.R.; Gois, P.M.P.; Afonso, C.A.M. Rh(II)-Catalyzed Intramolecular C−H Insertion of Diazo Substrates in Water: Scope and Limitations. J. Org. Chem. 2006, 71, 5489–5497. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, D.G.; Schmidt, P.J.; Geigle, S.N.; Chougnet, A.; Woggon, W.-D.; Gillingham, D.G. Modular Ligands for Dirhodium Complexes Facilitate Catalyst Customization. Adv. Synth. Catal. 2015, 357, 2033–2038. [Google Scholar] [CrossRef] [Green Version]
- Etkin, N.; Babu, S.D.; Fooks, C.J.; Durst, T. Preparation of 3-acetyl-2-hydroxyindoles via rhodium carbenoid aromatic carbon-hydrogen insertion. J. Org. Chem. 1990, 55, 1093–1096. [Google Scholar] [CrossRef]
- Wee, A.G.H.; Liu, B.; Zhang, L. Dirhodium tetraacetate catalyzed carbon-hydrogen insertion reaction in N-substituted .alpha.-carbomethoxy-.alpha.-diazoacetanilides and structural analogs. Substituent and conformational effects. J. Org. Chem. 1992, 57, 4404–4414. [Google Scholar] [CrossRef]
- Wang, H.L.; Li, Z.; Wang, G.W.; Yang, S.D. Silver catalyzed intramolecular cyclization for synthesis of 3-alkylideneoxindoles via C–H functionalization. Chem. Commun. 2011, 47, 11336–11338. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.S.; Elliott, M.C.; Moody, C.J.; Mowlem, T.J.; Marino, J.P., Jr.; Padwa, A. Ligand Effects in the Rhodium(II)-Catalyzed Reactions of .alpha.-Diazoamides. Oxindole Formation is Promoted by the Use of Rhodium(II) Perfluorocarboxamide Catalysts. J. Org. Chem. 1994, 59, 2447–2455. [Google Scholar] [CrossRef]
- Mo, S.Y.; Yang, Z.H.; Xu, J.X. Aqueous copper nitrate catalyzed synthesis of 3-alkylideneoxindoles from α-diazo-β-ketoanilides. Eur. J. Org. Chem. 2014, 18, 3923–3929. [Google Scholar] [CrossRef]
- Mo, S.Y.; Tu, J.Z.; Xu, J.X. Chemospecific synthesis of 2-acetoxyindole-3-carbonitriles catalyzed by Cu(acac)2. Russ. Chem. Bull. 2016, 65, 1773–1778. [Google Scholar] [CrossRef]
- Merlic, C.A.; Zechman, A.L.; Miller, M.M. Reactivity of (η6-Arene)tricarbonylchromium Complexes with Carbenoids: Arene Activation or Protection? J. Am. Chem. Soc. 2001, 123, 11101–11102. [Google Scholar] [CrossRef] [PubMed]
- Park, C.P.; Nagle, A.; Yoon, C.H.; Chen, C.L.; Jung, K.W. Formal Aromatic C−H Insertion for Stereoselective Isoquinolinone Synthesis and Studies on Mechanistic Insights into the C−C Bond Formation. J. Org. Chem. 2009, 74, 6231–6236. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Stefane, B.; Jaber, D.; Smith, J.A.I.; Vickery, C.; Diop, M.; Sintim, H.O. Remote C-H Functionalization: Using the N-O Moiety as an Atom-Economical Tether to Obtain 1,5-and the Rare 1,7-C-H Insertions. Angew. Chem. Int. Ed. 2010, 49, 3964–3968. [Google Scholar] [CrossRef] [PubMed]
- Lo, V.K.Y.; Guo, Z.; Choi, M.K.W.; Yu, W.Y.; Huang, J.S.; Che, C.-M. Highly Selective Intramolecular Carbene Insertion into Primary C–H Bond of α-Diazoacetamides Mediated by a (p-Cymene) ruthenium(II) Carboxylate Complex. J. Am. Chem. Soc. 2012, 134, 7588–7591. [Google Scholar] [CrossRef]
- Mo, S.Y.; Xu, J.X. Chemospecific intramolecular Büchner reaction catalyzed by copper(II) acetylacetonate. ChemCatChem 2014, 6, 1679–1683. [Google Scholar] [CrossRef]
- Mo, S.Y.; Li, X.H.; Xu, J.X. In situ generated iodonium ylides as safe carbene precursors for the chemoselective intramolecular Buchner reaction. J. Org. Chem. 2014, 79, 9186–9195. [Google Scholar] [CrossRef]
- Dai, M.; Danishefsky, S.J. The Total Synthesis of Spirotenuipesines A and B. J. Am. Chem. Soc. 2007, 129, 3498–3499. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Danishefsky, S.J. Total Synthesis of (±)-Aplykurodinone-1: Traceless Stereochemical Guidance. J. Am. Chem. Soc. 2010, 132, 9567–9569. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.H.; Xu, J.X. Synthesis of benzo-γ-sultams via the Rh-catalyzed aromatic C-H functionalization of diazosulfonamides. Chem. Commun. 2014, 50, 3616–3618. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.P.; Yang, Z.H.; Xu, J.X. Specific intramolecular aromatic C−H insertion of diazosulfonamides. Tetrahedron 2017, 73, 3255–3265. [Google Scholar] [CrossRef]
- Hrytsak, M.; Etkin, N.; Durst, T. Intramolecular rhodium carbenoid insertions into aromatic C-H bonds. Preparation of 1-carboalkoxy-1,3-dihydrobenzo[c]thiophene 2,2-dioxides. Tetrahedron Lett. 1986, 27, 5679–5682. [Google Scholar] [CrossRef]
- John, J.P.; Novikov, A.V. Selective Formation of Six-Membered Cyclic Sulfones and Sulfonates by C−H Insertion. Org. Lett. 2007, 9, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Wolckenhauer, S.A.; Devlin, A.S.; Du Bois, J. δ-Sultone Formation Through Rh-Catalyzed C−H Insertion. Org. Lett. 2007, 9, 4363–4366. [Google Scholar] [CrossRef] [PubMed]
- Flynn, C.J.; Elcoate, C.J.; Lawrence, S.E.; Maguire, A.R. Highly Enantioselective Intramolecular Copper Catalyzed C−H Insertion Reactions of α-Diazosulfones. J. Am. Chem. Soc. 2010, 132, 1184–1185. [Google Scholar] [CrossRef] [PubMed]
- Jungon, C.S.; Novikov, A.V. Asymmetric intramolecular C–H insertion of sulfonyldiazoacetates catalyzed by Rh(II). Tetrahedron Asymmetry 2013, 24, 151–155. [Google Scholar] [CrossRef]
- Clarke, L.A.; Ring, A.; Ford, A.; Sinha, A.S.; Lawrence, S.E.; Maguire, A.R. Enantioselective copper catalysed C–H insertion reaction of 2-sulfonyl-2-diazoacetamides to form γ-lactams. Org. Biomol. Chem. 2014, 12, 7612–7628. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Xu, X.; Jin, L.-M.; Wojtas, L.; Zhang, X.P. Stereoselective radical C–H alkylation with acceptor/acceptor-substituted diazo reagents via Co(II)-based metalloradical catalysis. Chem. Sci. 2015, 6, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.B.; March, J. Advanced Organic Chemistry, 5th ed.; John Wiley: New York, NY, USA, 2004; p. 370. [Google Scholar]
- Pasquinucci, L.; Prezzavento, O.; Marrazzo, A.; Amata, E.; Ronsisvalle, S.; Georgoussi, Z.; Fourla, D.D.; Scoto, G.M.; Parenti, C.; Arico, G.; et al. Evaluation of N-substitution in 6,7-benzomorphan compounds. Bioor Med. Chem. 2010, 18, 4975–4982. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, E.J.; Buchwald, S.L. Synthesis of Substituted Oxindoles from α-Chloroacetanilides via Palladium-Catalyzed C−H Functionalization. J. Am. Chem. Soc. 2003, 125, 12084–12085. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 2 and 3 are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | R1 | R2 | R3 | R4 | Yield (%) | ||
|---|---|---|---|---|---|---|---|
| 5 | 7 | 1 | |||||
| a | Me | Me | Me | Ph | 90 | - | - |
| b | Me | Me | Ph | Ph | 25 | 16 | |
| c | Me | Ph | Bu | Bn | 90 | 59 | 45 |
| d | Ph | Ph | Bu | Bn | 96 | 28 | 50 |
| e | Me | Ph | Me | Ph | 90 | 74 | 88 |
| f | Me | Ph | Ph | Ph | 30 | 26 | |
| g | Ph | Ph | Me | Ph | 96 | 88 | 60 |
| h | Ph | Ph | Ph | Ph | 72 | 65 | |
 |
 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Que, C.; Huang, P.; Yang, Z.; Chen, N.; Xu, J. Intramolecular Carbene C-H Insertion Reactions of 2-Diazo-2-sulfamoylacetamides. Molecules 2019, 24, 2628. https://doi.org/10.3390/molecules24142628
Que C, Huang P, Yang Z, Chen N, Xu J. Intramolecular Carbene C-H Insertion Reactions of 2-Diazo-2-sulfamoylacetamides. Molecules. 2019; 24(14):2628. https://doi.org/10.3390/molecules24142628
Chicago/Turabian StyleQue, Chuqiang, Peipei Huang, Zhanhui Yang, Ning Chen, and Jiaxi Xu. 2019. "Intramolecular Carbene C-H Insertion Reactions of 2-Diazo-2-sulfamoylacetamides" Molecules 24, no. 14: 2628. https://doi.org/10.3390/molecules24142628
APA StyleQue, C., Huang, P., Yang, Z., Chen, N., & Xu, J. (2019). Intramolecular Carbene C-H Insertion Reactions of 2-Diazo-2-sulfamoylacetamides. Molecules, 24(14), 2628. https://doi.org/10.3390/molecules24142628