Annular Tautomerism of 3(5)-Disubstituted-1H-pyrazoles with Ester and Amide Groups


Abstract
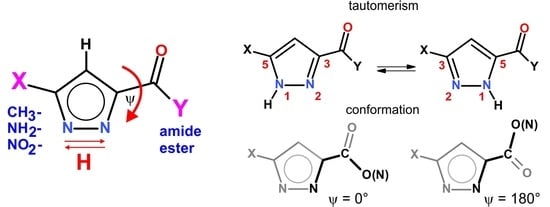
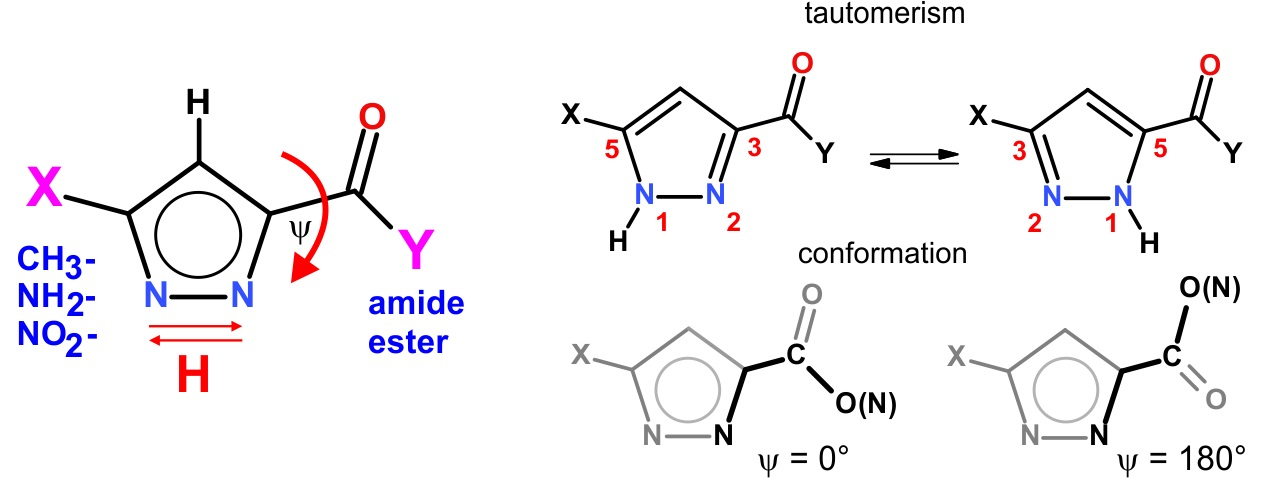
1. Introduction
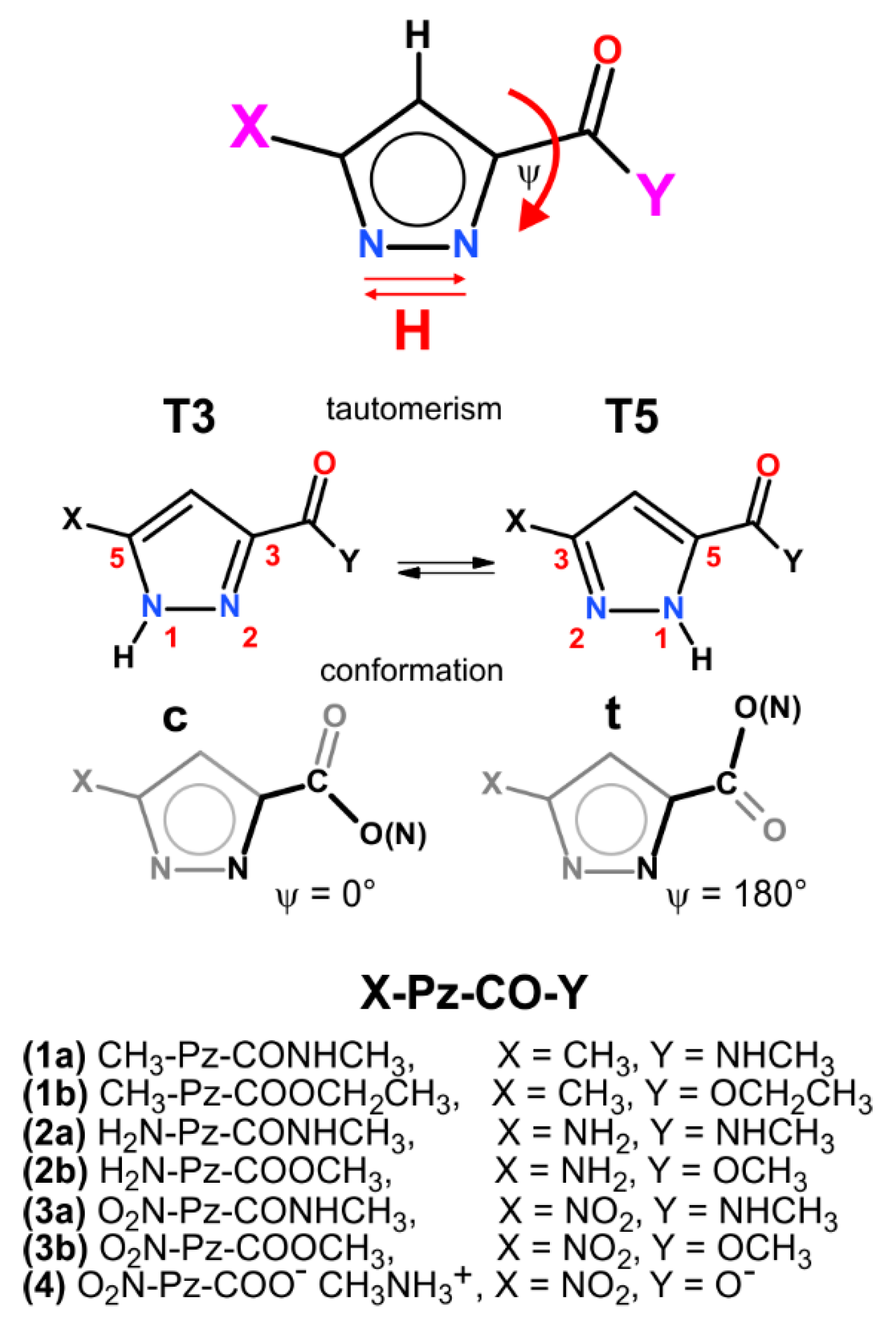
2. Results and Discussion
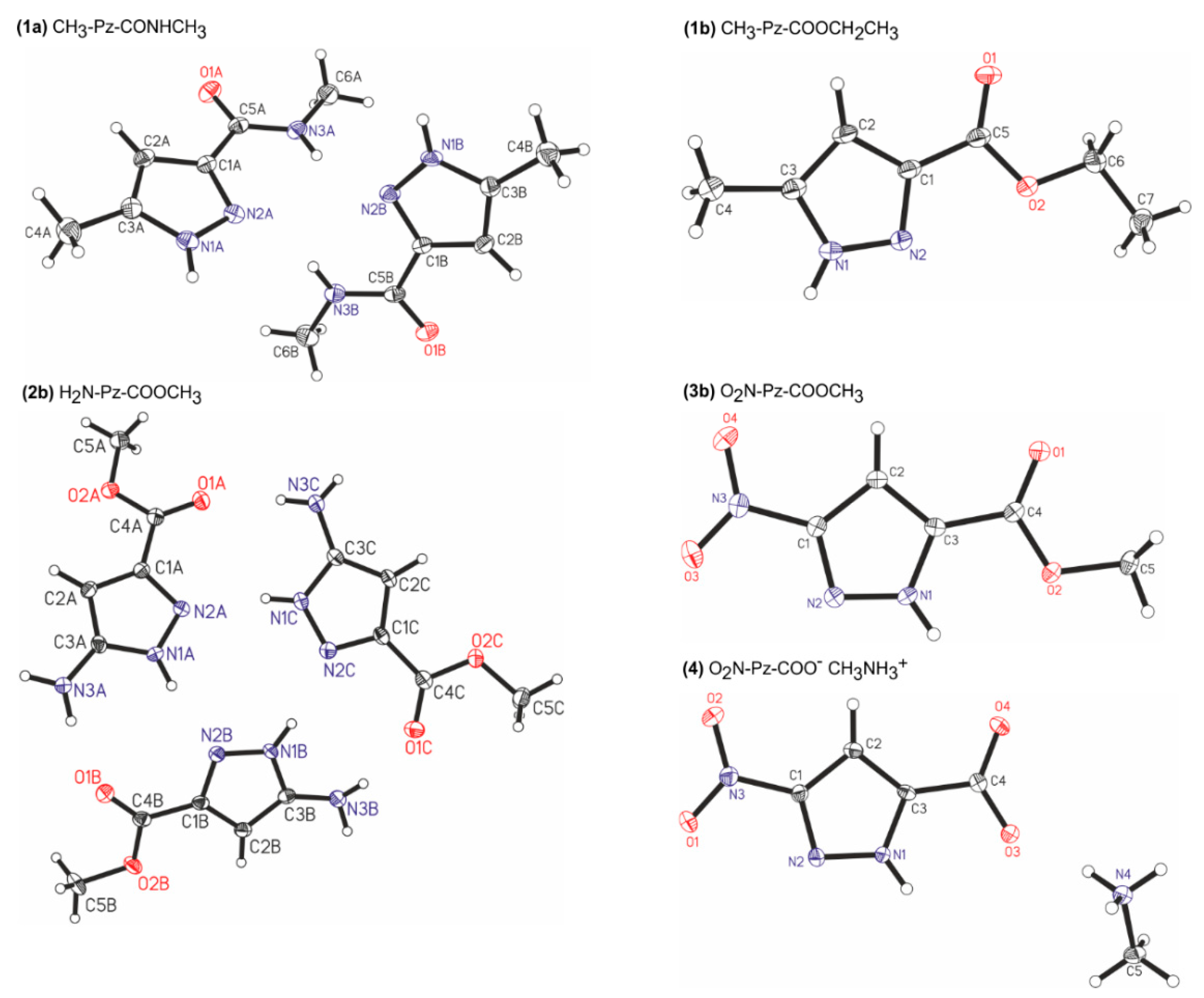
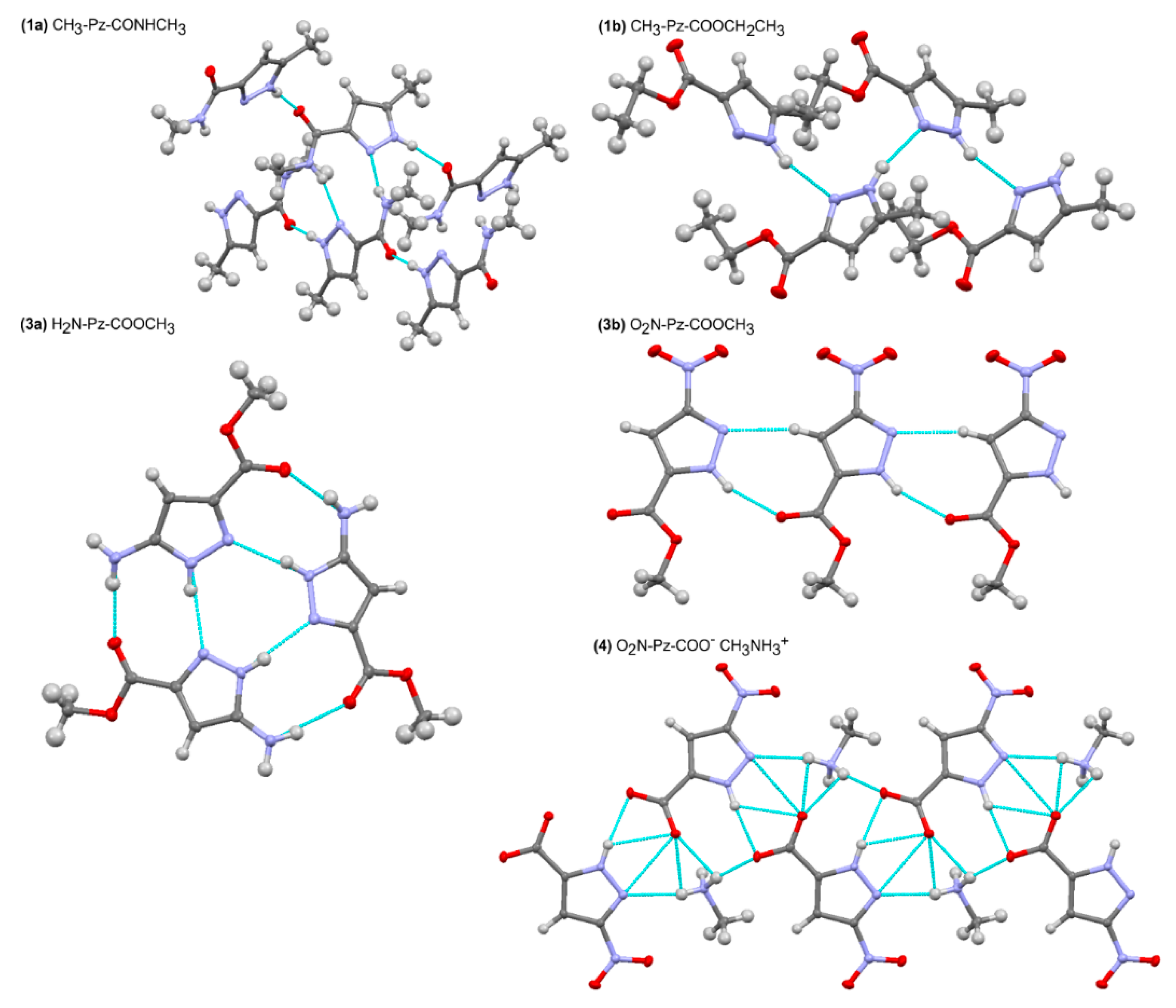
2.1. X-Ray Results
2.2. Theoretical Calculations
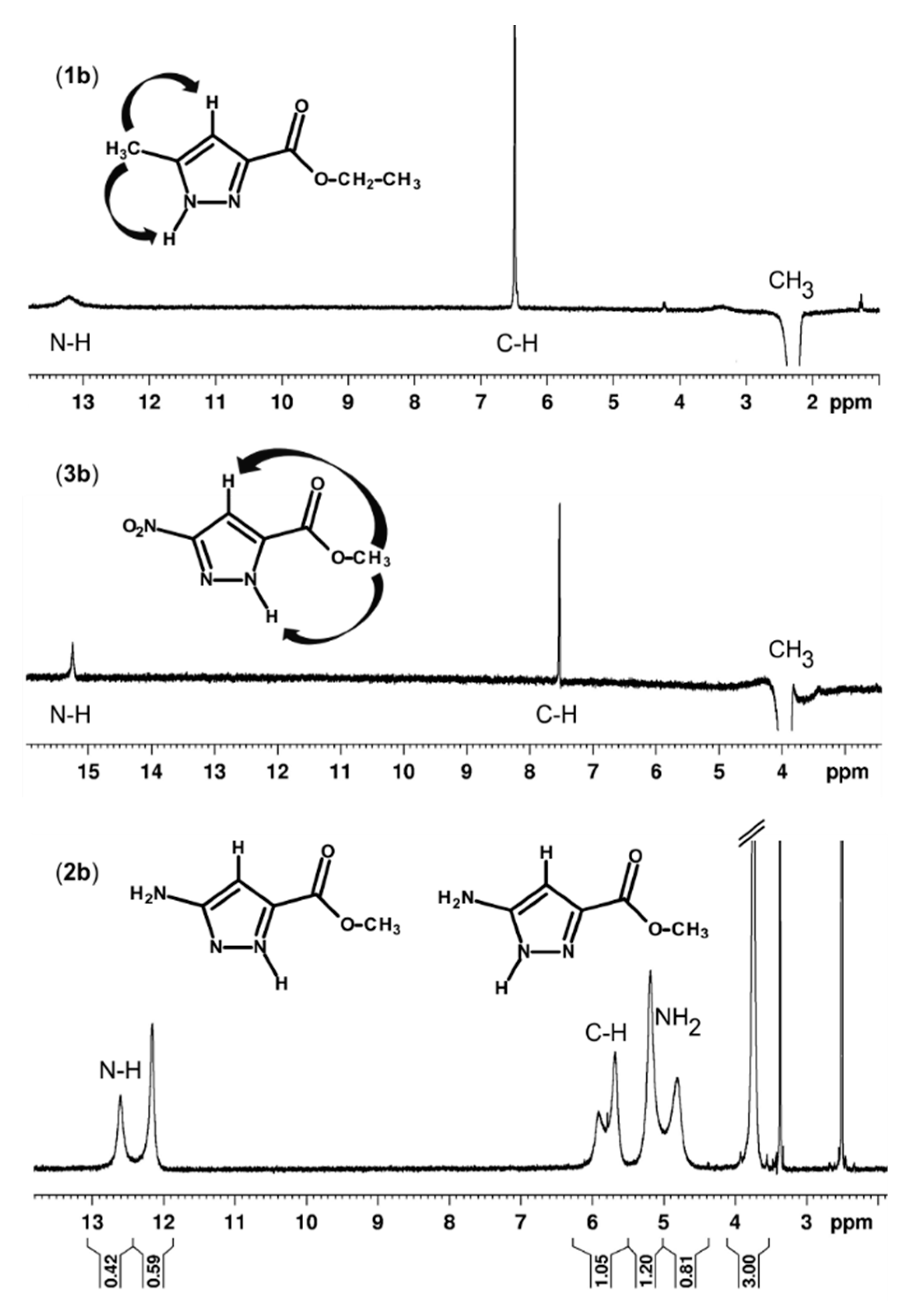
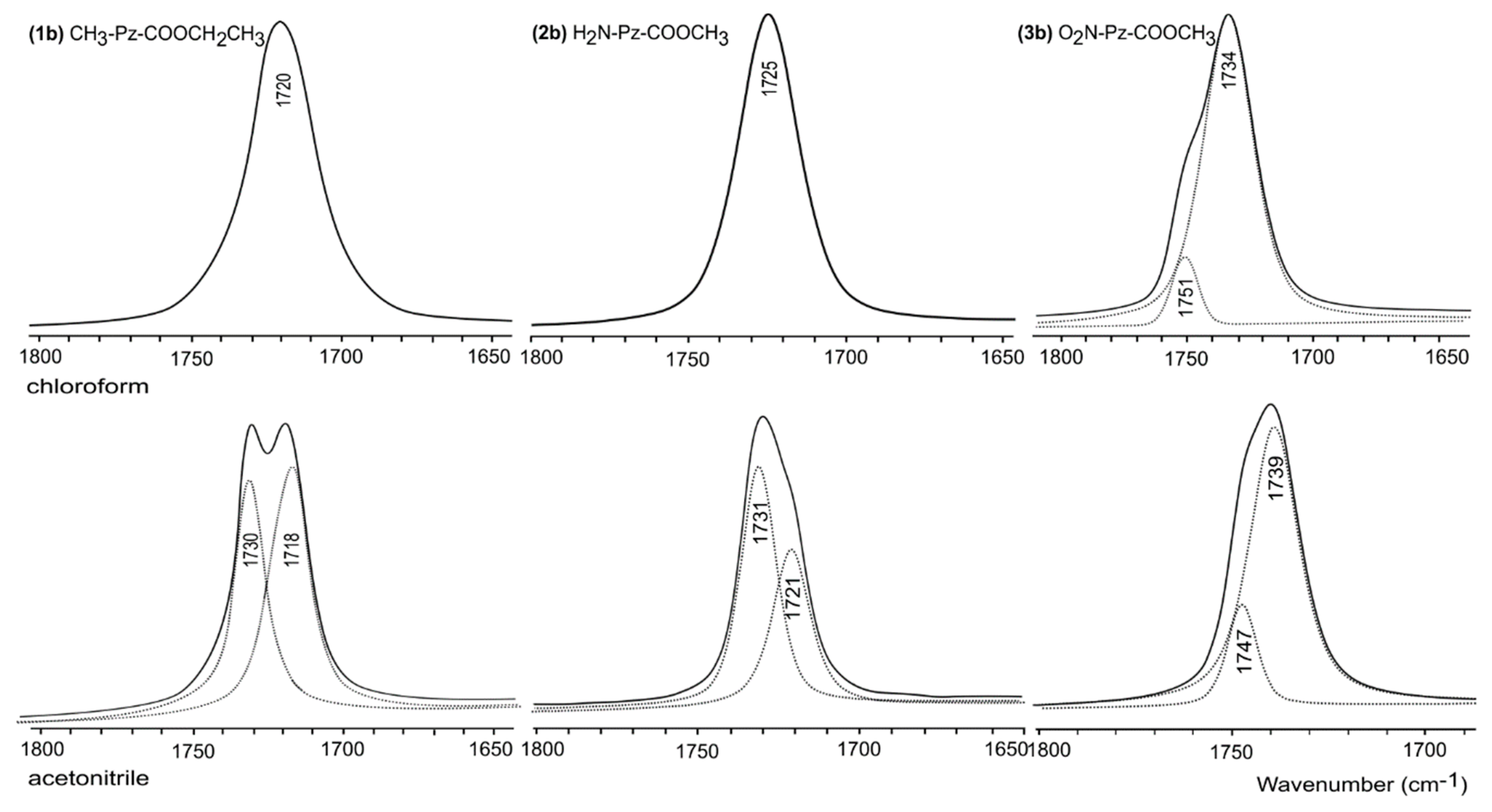
2.3. Study in Solution
3. Experimental Section
3.1. Synthesis
3.2. X-Ray
3.3. Computational Procedures
3.4. NMR Spectroscopy
3.5. FT-IR Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ugono, O.; Ratha, N.P.; Beatty, A.M. Exceptions to the rule: New hydrogen-bonded networks from an old reliable. CrystEngComm 2011, 13, 753–758. [Google Scholar] [CrossRef]
- Li, J.; Li, B.; Pan, M.; Liu, B.; Cheng, J.; Li, R.; Gao, X.; Wang, S.; Hou, H.; Liu, Z. Five Multidimensional Co(II)-Complexes (Zero-Dimensional to Three-Dimensional) Derived from an Asymmetric 5-(Pyridin-3-yl)-1H-pyrazole-3-carboxylic Acid: Syntheses, Structures, and Magnetic Properties. Cryst. Growth Des. 2017, 17, 2975–2986. [Google Scholar] [CrossRef]
- Cheng, M.-L.; Han, W.; Liu, Q.; Bao, J.-T.; Li, Z.-F.; Chen, L.-T.; Sun, X.-Q.; Xi, H.-T. Synthesis, crystal structures, and luminescent properties of Pb(II) and Sr(II) coordination polymers constructed by 5-methyl-1H-pyrazole-3-carboxylic acid. J. Coord. Chem. 2014, 67, 215–226. [Google Scholar] [CrossRef]
- Camurlu, P.; Gültekin, C.; Gürbulak, V. Optoelectronic Properties and Electrochromic Device Application of Novel Pyrazole Based Conducting Polymers. J. Macromol. Sci. A 2013, 50, 588–595. [Google Scholar] [CrossRef]
- Cegłowski, M.; Schroeder, G. Removal of heavy metal ions with the use of chelating polymers obtained by grafting pyridine–pyrazole ligands onto polymethylhydrosiloxane. Chem. Eng. J. 2015, 259, 885–893. [Google Scholar] [CrossRef]
- Yu, L.; Wang, X.; Cheng, M.; Rong, H.; Song, Y.; Liu, Q. A Three-Dimensional Copper Coordination Polymer Constructed by 3-Methyl-1H-pyrazole-4-carboxylic Acid with Higher Capacitance for Supercapacitors. Cryst. Growth Des. 2018, 18, 280–285. [Google Scholar] [CrossRef]
- Harit, T.; Malek, F.; Ameduri, B. Fluorinated polymers based on pyrazole groups for fuel cell. Membranes. Eur. Polym. J. 2016, 79, 72–81. [Google Scholar] [CrossRef]
- Cheng, M.; Wang, Q.; Bao, J.; Wu, Y.; Sun, L.; Yang, B.; Liu, Q. Synthesis and structural diversity of d10 metal coordination polymers constructed from new semi-rigid bis(3-methyl-1H-pyrazole-4-carboxylic acid)alkane ligands. New J. Chem. 2017, 41, 5151–5160. [Google Scholar] [CrossRef]
- Faria, J.V.; Vegi, P.F.; Miguita, A.G.C.; dos Santos, M.S.; Boechat, N.; Bernardino, A.M.R. Recently reported biological activities of pyrazole compounds. Bioorg. Med. Chem. 2017, 25, 5891–5903. [Google Scholar] [CrossRef]
- Wang, Y.; Zhi, Y.; Jin, Q.; Lu, S.; Lin, G.; Yuan, H.; Yang, T.; Wang, Z.; Yao, C.; Ling, J.; et al. Discovery of 4-((7H-Pyrrolo [2,3-d]pyrimidin-4-yl)amino)-N-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrazole-3-carboxamide (FN-1501), an FLT3- and CDK-Kinase Inhibitor with Potentially High Efficiency against Acute Myelocytic Leukemia. J. Med. Chem. 2018, 61, 1499–1518. [Google Scholar] [CrossRef]
- Wright, S.W.; Arnold, E.P.; Yang, X. Steric redirection of alkylation in 1H-pyrazole-3-carboxylate esters. Tetrahedron Lett. 2018, 59, 402–405. [Google Scholar] [CrossRef]
- Markovic, V.; Joksovic, M.D. “On water” synthesis of N-unsubstituted pyrazoles: Semicarbazide hydrochloride as an alternative to hydrazine for preparation of pyrazole-3-carboxylate derivatives and 3,5-disubstituted pyrazoles. Green Chem. 2015, 17, 842–847. [Google Scholar] [CrossRef]
- Mykhailiuk, P.K. New Life for Diazoacetonitrile (N2CHCN): In situ Generation and Practical Synthesis of CN-Pyrazoles. Eur. J. Org. Chem. 2015, 33, 7235–7239. [Google Scholar] [CrossRef]
- Dishington, A.; Feron, J.L.; Gill, K.; Graham, M.A.; Hollingsworth, I.; Pink, J.H.; Roberts, A.; Simpson, I.; Tatton, M. Synthesis of Functionalized Cyanopyrazoles via Magnesium Bases. Org. Lett. 2014, 16, 6120–6123. [Google Scholar] [CrossRef] [PubMed]
- Lim, F.P.L.; Tan, L.Y.; Tiekink, E.R.T.; Dolzhenko, A.V. Synthesis of 3-(5-amino-1H-1,2,4-triazol-3-yl) propanamides and their tautomerism. RSC Adv. 2018, 8, 22351–22360. [Google Scholar] [CrossRef]
- Lim, F.P.L.; Hu, L.M.; Tiekink, E.R.T.; Dolzhenko, A.V. One-pot, microwave-assisted synthesis of polymethylene-bridged bis (1H-1,2,4-triazol-5(3)-amines) and their tautomerism. Tetrahedron Lett. 2018, 59, 3792–3796. [Google Scholar] [CrossRef]
- Lim, F.P.L.; Tan, K.C.; Luna, G.; Tiekink, E.R.T.; Dolzhenko, A.V. A new practical synthesis of 3-amino-substituted 5-aminopyrazoles and their tautomerism. Tetrahedron 2019, 75, 2314–2321. [Google Scholar] [CrossRef]
- Nagarapu, L.; Mateti, J.; Gaikwad, H.K.; Bantu, R.; Rani, M.S.; Subhashini, N.J.P. Synthesis and anti-inflammatory activity of some novel 3-phenyl-N-[3-(4-phenylpiperazin-1yl)propyl]-1H-pyrazole-5-carboxamide derivatives. Bioorg. Med. Chem. Lett. 2011, 21, 4138–4140. [Google Scholar] [CrossRef]
- Wen, J.; Niu, Q.; Liu, J.; Bao, Y.; Yang, J.; Luan, S.; Fan, Y.; Liu, D.; Zhao, L. Novel thiol-based histone deacetylase inhibitors bearing 3-phenyl-1H-pyrazole-5-carboxamide scaffold as surface recognition motif: Design, synthesis and SAR study. Bioorg. Med. Chem. Lett. 2016, 26, 375–379. [Google Scholar] [CrossRef]
- Sribalan, R.; Banuppriya, G.; Kirubavathi, M.; Jayachitra, A.; Vediappen, P. Multiple biological activities and molecular docking studies of newly synthesized 3-(pyridin-4-yl)-1H-pyrazole-5-carboxamide chalcone hybrids. Bioorg. Med. Chem. Lett. 2016, 26, 5624–5630. [Google Scholar] [CrossRef]
- Ribeiro, N.; Di Paolo, R.E.; Galvão, A.M.; Marques, F.; Pessoa, J.C.; Correia, I. Photophysical properties and biological evaluation of a Zinc (II)-5-methyl-1I-pyrazole Schiff base complex. Spectrochim. Acta A 2018, 204, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Dales, N.A.; Winther, M.D. Opportunities and Challenges in Developing Stearoyl-Coenzyme A Desaturase-1 Inhibitors as Novel Therapeutics for Human Disease. J. Med. Chem. 2014, 57, 5039–5056. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.-H.; Kim, B.M. Discovery of Novel Transcription Factor Inhibitors Using a Pyrazole-based Small Molecule Library. Bull. Korean Chem. Soc. 2008, 29, 323–327. [Google Scholar] [CrossRef]
- Tamura, S.Y.; Richman, J.G.; Connolly, D.T.; Semple, G. 5-N,N-Disubstituted 5-aminopyrazole-3-carboxylic acids are highly potent agonists of GPR109b. Bioorg. Med. Chem. Lett. 2009, 19, 4207–4209. [Google Scholar] [CrossRef]
- Rzepecki, P.; Geib, N.; Peifer, M.; Biesemeier, F.; Schrader, T. Synthesis and Binding Studies of Alzheimer Ligands on Solid Suport. J. Org. Chem. 2007, 72, 3614–3624. [Google Scholar] [CrossRef] [PubMed]
- Rzepecki, P.; Gallmeier, H.; Geib, N.; Černovská, K.; König, B.; Schrader, T. New heterocyclic β-sheet ligands with peptidic recognition elements. J. Org. Chem. 2004, 69, 5168–5178. [Google Scholar] [CrossRef] [PubMed]
- Černovská, K.; Kemter, M.; Gallmeier, H.; Rzepecki, P.; Schrader, T.; König, B. PEG-supported synthesis of pyrazole oligoamides with peptide β-sheet affinity. Org. Biomol. Chem. 2004, 2, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- Rzepecki, P.; Schrader, T. β-Sheet Ligands in Action: KLVFF Recognition by Aminopyrazole Hybrid Receptors in Water. J. Am. Chem. Soc. 2005, 127, 3016–3025. [Google Scholar] [CrossRef]
- Kusakiewicz-Dawid, A.; Porada, M.; Ochędzan-Siodłak, W.; Broda, M.A.; Bujak, M.; Siodłak, D. Pyrazole amino acids: Hydrogen bonding directed conformations of 3-amino-1H-pyrazole-5-carboxylic acid residue. J. Pept. Sci. 2017, 23, 716–726. [Google Scholar] [CrossRef]
- Aguilar-Parrilla, F.; Cativiela, C.; de Villegas, M.D.D.; Elguero, J.; Foces-Foces, C.; Laureiro, J.G.; Cano, F.H.; Limbach, H.-H.; Smithe, J.A.S.; Toiron, C. The Tautomerism of 3(5)-Phenylpyrazoles: An Experimental (1H, 13C, 15N NMR and X-Ray Crystallography) Study. J. Chem. Soc. Perkin Trans. 1992, 2, 1737–1742. [Google Scholar] [CrossRef]
- Lopez, C.; Claramunt, R.M.; Trofimenko, S.; Elguero, J. A 13C NMR spectroscopy study of the structure of N-H pyrazoles and indazoles. Can. J. Chem. 1993, 71, 678–684. [Google Scholar] [CrossRef]
- Puello, J.Q.; Obando, B.I.; Foces-Foces, C.; Infantes, L.; Claramunt, R.M.; Cabildo, P.; Jiménez, J.A. Structure and tautomerism of 3(5)-amino-5(3)-arylpyrazoles in the solid state and in solution: An X-ray and NMR study. Tetrahedron 1997, 53, 10783–10802. [Google Scholar] [CrossRef]
- Foces-Foces, C.; Llamas-Saiz, A.L.; Menéndez, M.; Jagerovic, N.; Elguero, J. Structure of 3-nitropyrazole in solution and in the solid state. J. Phys. Org. Chem. 1997, 10, 637–645. [Google Scholar] [CrossRef]
- Emelina, E.E.; Petrov, A.A.; Filyukov, D.V. Structure and Tautomerism of 4-Substituted 3(5)-Aminopyrazoles in Solution and in the Solid State: NMR Study and Ab Initio Calculations. Russ. J. Org. Chem. 2014, 50, 412–421. [Google Scholar] [CrossRef]
- Claramunt, R.M.; Garcia, M.A.; Concepcion, L.; Trofimenko, S.; Yap, G.P.A.; Alkorta, I.; Elguero, J. The tautomerism of 1H-pyrazole-3(5)-(N-tert-butyl)carboxamide in the solid state and in solution. Magn. Reson. Chem. 2005, 43, 89–91. [Google Scholar] [CrossRef]
- Nieto, C.I.; Cabildo, M.P.; Cornago, M.P.; Sanz, D.; Claramunt, R.M.; Alkorta, I.; Elguero, J.; García, J.A.; Lopez, A.; Acuna-Castroviejo, D. Synthesis, structure and biological activity of 3(5)-trifluoromethyl-1Hpyrazoles derived from hemicurcuminoids. J. Mol. Sruct. 2015, 1100, 518–529. [Google Scholar] [CrossRef]
- Jarończyk, M.; Dobrowolski, J.C.; Mazurek, A.P. Theoretical studies on tautomerism and IR spectra of pyrazole derivatives. J. Mol. Struc. Theochem 2004, 673, 17–28. [Google Scholar] [CrossRef]
- Marín-Luna, M.; Alkorta, I.; Elguero, J. A theoretical study of the gas phase (proton affinity) and aqueous (pKa) basicity of a series of 150 pyrazoles. New J. Chem. 2015, 39, 2861–2871. [Google Scholar] [CrossRef]
- Radula-Janik, K.; Kupka, T.; Ejsmont, K.; Daszkiewicz, Z.; Sauer, S.P.A. DFT and experimental studies on structure and spectroscopic parameters of 3,6-diiodo-9-ethyl-9H-carbazole. Struct. Chem. 2016, 27, 199–207. [Google Scholar] [CrossRef]
- CrysAlis CCD; Oxford Diffraction Ltd.: Abingdon, UK, 2002.
- CrysAlis RED; Oxford Diffraction Ltd.: Abingdon, UK, 2002.
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. New features added to the refinement program SHELXL since 2008 are described and explained. Acta. Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0–new features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Accounts 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Schwabe, T.; Grimme, S. Double-hybrid density functionals with longrange dispersion corrections: Higher accuracy and extended applicability. Phys. Chem. Chem. Phys. 2007, 9, 3397–3406. [Google Scholar] [CrossRef]
- Schleyer, P.; Maerker, C.; Dransfeld, A.; Jiao, H.; Hommes, N.J.R. Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef]
- Stanger, A. Nucleus-Independent Chemical Shifts (NICS): Distance Dependence and Revised Criteria for Aromaticity and Antiaromaticity. J. Org. Chem. 2006, 71, 883–893. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1a, 1-3b, and 4 are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1a | 1b | 2b | 3b | 4 | |
|---|---|---|---|---|---|
| Chemical formula | C6H9N3O | C7H10N2O2 | C5H7N3O2 | C5H5N3O4 | C5H8N4O4 |
| Mr | 139.16 | 154.17 | 141.14 | 171.12 | 188.15 |
| Crystal system, space group | Monoclinic, P21/c | Monoclinic, P21/n | Monoclinic, P21/n | Monoclinic, P21/m | Monoclinic, P21/n |
| a, b, c (Å) | 14.7736 (4), 7.2104 (2), 13.6295 (4) | 9.7186 (7), 6.0828 (4), 13.4976 (11) | 10.8040 (5), 13.6444 (5), 13.8883 (6) | 5.5232 (3), 6.1484 (3), 10.8318 (5) | 3.7803 (1), 22.9836 (7), 9.0425 (3) |
| β (°) | 90.800 (3) | 102.810 (7) | 110.173 (5) | 102.679 (5) | 98.082 (3) |
| V (Å3) | 1451.72 (7) | 778.07 (10) | 1921.74 (15) | 358.87 (3) | 777.85 (4) |
| Z | 8 | 4 | 12 | 2 | 4 |
| µ (mm−1) | 0.09 | 0.1 | 0.12 | 0.14 | 0.14 |
| Crystal size (mm) | 0.5 × 0.4 × 0.2 | 0.5 × 0.3 × 0.1 | 0.4 × 0.3 × 0.2 | 0.5 × 0.4 × 0.2 | 0.4 × 0.3 × 0.2 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 9524, 2826, 2196 | 5041, 1523, 1130 | 9264, 3742, 2660 | 2439, 776, 685 | 5234, 1530, 1186 |
| Rint | 0.024 | 0.037 | 0.027 | 0.013 | 0.027 |
| (sin θ/λ)max (Å−1) | 0.617 | 0.616 | 0.617 | 0.617 | 0.617 |
| Refinement | |||||
| R[F2 > 2σ(F2)], wR(F2), S | 0.037, 0.103, 1.08 | 0.038, 0.103, 0.88 | 0.034, 0.085, 0.77 | 0.027, 0.076, 1.11 | 0.029, 0.070, 0.95 |
| No. of reflections | 2826 | 1523 | 3742 | 776 | 1530 |
| No. of parameters | 185 | 102 | 292 | 79 | 120 |
| H-atom treatment | H-atom parameters constrained | H-atom parameters constrained | H atoms treated by a mixture of independent and constrained refinement | H atoms treated by a mixture of independent and constrained refinement | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.26, −0.26 | 0.20, −0.27 | 0.17, −0.25 | 0.29, −0.19 | 0.24, −0.24 |
| X-Pz-CO- | Tautomer (T3/5)/Conformer (c/t) | |||
|---|---|---|---|---|
| X- | T3c | T3t | T5c | T5t |
| Me- | 7 | |||
| H- | 2 | 3 | ||
| Ph- | 12 | 1 | 3 | 2 |
| OOC-Pz-COOH | 11 | 5 | 1 | 2 |
| NC- | 1 | 3 | ||
| -CF- | 6 | 2 | ||
| HO- | 1 | |||
| In Vacuo | Chloroform | Acetonitrile | Water | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ψ | Energy | ΔE | ψ | Energy | ΔE | ψ | Energy | ΔE | ψ | Energy | ΔE | |
| Conformer Code↓ | (°) | (Ha) | (kcal/mol) | (°) | (Ha) | (kcal/mol) | (°) | (Ha) | (kcal/mol) | (°) | (Ha) | (kcal/mol) |
| (1a) CH3-Pz-CONHCH3 | ||||||||||||
| T3c | 0.0 | −473.322883 | 0.00 | 0.0 | −473.340504 | 0.00 | 0.0 | −473.344429 | 0.00 | 0.0 | −473.344742 | 0.00 |
| T3t | 180.0 | −473.312263 | 6.66 | 180.0 | −473.334178 | 3.97 | 180.0 | −473.340706 | 2.34 | 180.0 | −473.342131 | 1.64 |
| T5c | 24.4 | −473.314249 | 5.42 | 24.1 | −473.333208 | 4.58 | 17.6 | −473.338227 | 3.89 | 18.4 | −473.339003 | 3.60 |
| T5t | 180.0 | −473.321441 | 0.90 | 180.0 | −473.337598 | 1.82 | 180.0 | −473.341534 | 1.82 | 180.0 | −473.341632 | 1.95 |
| (1b) CH3-Pz-COOCH2CH3 | ||||||||||||
| T3c | 0.0 | −532.474789 | 1.83 | 0.0 | −532.492603 | 0.00 | 0.0 | −532.496648 | 0.00 | 0.0 | −532.494392 | 0.00 |
| T3t | 180.0 | −532.473347 | 2.73 | 180.0 | −532.491806 | 0.50 | 180.0 | −532.496280 | 0.23 | 180.0 | −532.494119 | 0.17 |
| T5c | 0.2 | −532.477188 | 0.32 | 0.4 | −532.491509 | 0.69 | 0.3 | −532.494031 | 1.64 | 0.0 | −532.490849 | 2.22 |
| T5t | 180.0 | −532.477703 | 0.00 | 180.0 | −532.491767 | 0.52 | 180.0 | −532.494172 | 1.55 | 180.0 | −532.490747 | 2.29 |
| (2a) H2N-Pz-CONHCH3 | ||||||||||||
| T3c | 0.0 | −489.374774 | 1.25 | 0.0 | −489.397950 | 0.00 | 0.0 | −489.404669 | 0.00 | 0.0 | −489.405337 | 0.00 |
| T3t | −179.9 | −489.365033 | 7.36 | −180.0 | −489.392782 | 3.24 | −180.0 | −489.402127 | 1.59 | −180.0 | −489.404202 | 0.71 |
| T5c | 24.2 | −489.371558 | 3.26 | 25.3 | −489.393897 | 2.54 | 18.1 | −489.400630 | 2.53 | 5.1 | −489.401874 | 2.17 |
| T5t | −180.0 | −489.376759 | 0.00 | 180.0 | −489.397510 | 0.28 | 180.0 | −489.403626 | 0.65 | −180.0 | −489.404197 | 0.72 |
| (2b) H2N-Pz-COOCH3 | ||||||||||||
| T3c | 0.0 | −509.246751 | 4.67 | 0.0 | −509.267576 | 1.80 | 0.0 | −509.274622 | 0.43 | 0.0 | −509.273901 | 0.27 |
| T3t | 180.0 | −509.245885 | 5.21 | 180.0 | −509.267586 | 1.80 | 180.0 | −509.275108 | 0.13 | 180.0 | −509.274337 | 0.00 |
| T5c | 0.0 | −509.253108 | 0.68 | 0.4 | −509.270059 | 0.25 | 0.4 | −509.275012 | 0.19 | 0.2 | −509.273864 | 0.30 |
| T5t | 180.0 | −509.254195 | 0.00 | 179.9 | −509.270450 | 0.00 | 179.8 | −509.275315 | 0.00 | −179.9 | −509.273994 | 0.22 |
| (3a) O2N-Pz-CONHCH3 | ||||||||||||
| T3c | 0.0 | −638.517709 | 0.63 | 0.0 | −638.535327 | 2.29 | 0.0 | −638.539811 | 3.59 | 0.0 | −638.538274 | 3.81 |
| T3t | −155.5 | −638.508452 | 6.43 | −174.4 | −638.529928 | 5.68 | −174.3 | −638.536869 | 5.44 | −170.8 | −638.536671 | 4.82 |
| T5c | 26.8 | −638.510067 | 5.42 | 24.8 | −638.533577 | 3.39 | 22.6 | −638.541327 | 2.64 | 20.4 | −638.540059 | 2.69 |
| T5t | 180.0 | −638.518707 | 0.00 | −177.3 | −638.538973 | 0.00 | 180.0 | −638.545531 | 0.00 | −179.9 | −638.544350 | 0.00 |
| (3b) O2N-Pz-COOCH3 | ||||||||||||
| T3c | 0.0 | −658.389922 | 2.95 | 0.0 | −658.404657 | 3.63 | 0.0 | −658.409316 | 3.72 | 0.0 | −658.405911 | 3.65 |
| T3t | 180.0 | −658.389303 | 3.34 | 180.0 | −658.404464 | 3.75 | 180.0 | −658.409578 | 3.56 | 180.0 | −658.406313 | 3.40 |
| T5c | 0.0 | −658.393160 | 0.92 | 0.0 | −658.409513 | 0.59 | 0.0 | −658.414409 | 0.53 | 0.0 | −658.411099 | 0.39 |
| T5t | 180.0 | −658.394621 | 0.00 | 180.0 | −658.410447 | 0.00 | 180.0 | −658.415251 | 0.00 | 180.0 | −658.411725 | 0.00 |
| Compound | NICS(0) | NICS(1) | NICS(1)zz |
|---|---|---|---|
| Pyrazole | −13.51 | −11.37 | −33.87 |
| 3-Methyl-1H-pyrazole | −12.57 | −13.22 | −31.38 |
| 5-Methyl-1H-pyrazole | −12.13 | −12.12 | −30.52 |
| 1H-pyrazol-3-amine (3-Aminopyrazole) | −11.97 | −8.93 | −26.42 |
| 1H-pyrazol-5-amine (5-Aminopyrazole) | −11.94 | −8.72 | −25.71 |
| 3-Nitro-1H-pyrazole | −13.50 | −10.63 | −29.74 |
| 5-Nitro-1H-pyrazole | −12.43 | −10.36 | −28.76 |
| N-Methyl-1H-pyrazole-3-carboxamide (ψ = 0°) | −12.81 | −10.89 | −30.98 |
| N-Methyl-1H-pyrazole-3-carboxamide (ψ = 180°) | −12.95 | −10.99 | −31.00 |
| N-Methyl-1H-pyrazole-5-carboxamide (ψ = 0°) | −12.61 | −10.90 | −32.07 |
| N-Methyl-1H-pyrazole-5-carboxamide (ψ = 180°) | −12.28 | −10.86 | −30.69 |
| Methyl 1H-pyrazole-3-carboxylate (ψ = 0°) | −12.88 | −10.98 | −31.14 |
| Methyl 1H-pyrazole-3-carboxylate (ψ = 180°) | −12.88 | −11.03 | −31.16 |
| Methyl 1H-pyrazole-5-carboxylate (ψ = 0°) | −12.29 | −10.89 | −30.85 |
| Methyl 1H-pyrazole-5-carboxylate (ψ = 180°) | −12.21 | −10.88 | −30.68 |
| Code | NICS(0) | NICS(1) | NICS(1)zz | ∆NICS(1)zz |
|---|---|---|---|---|
| (1a) CH3-Pz-CONHCH3 | ||||
| T3c | −11.57 | −10.08 | −27.86 | 0.00 |
| T3t | −11.68 | −10.12 | −27.88 | −0.02 |
| T5c | −11.57 | −10.05 | −27.94 | −0.08 |
| T5t | −11.35 | −10.23 | −28.09 | −0.23 |
| (1b) CH3-Pz-COOCH2CH3 | ||||
| T3c | −11.61 | −10.06 | −27.93 | 0.00 |
| T3t | −11.65 | −10.19 | −28.02 | −0.08 |
| T5c | −11.37 | −10.76 | −28.3 | −0.36 |
| T5t | −11.33 | −10.29 | −28.12 | −0.18 |
| (2a) H2N-Pz-CONHCH3 | ||||
| T3c | −11.48 | -8.45 | -23.36 | 0.00 |
| T3t | −11.64 | -8.56 | -23.44 | -0.08 |
| T5c | −11.45 | -8.95 | -25.81 | -2.44 |
| T5t | −11.09 | -8.75 | -24.01 | -0.65 |
| (2b) H2N-Pz-COOCH3 | ||||
| T3c | −11.70 | −8.61 | −23.60 | 0.00 |
| T3t | −11.72 | −8.63 | −23.71 | −0.11 |
| T5c | −11.43 | −9.08 | −24.84 | −1.24 |
| T5t | −11.40 | −9.10 | −24.86 | −1.26 |
| (3a) O2N-Pz-CONHCH3 | ||||
| T3c | −12.12 | −10.00 | −26.33 | 0.00 |
| T3t | −12.50 | −10.28 | −27.40 | −1.07 |
| T5c | −13.06 | −10.29 | −27.99 | −1.66 |
| T5t | −12.72 | −10.33 | −27.16 | −0.82 |
| (3b) O2N-Pz-COOCH3 | ||||
| T3c | −12.30 | −10.17 | −26.60 | 0.00 |
| T3t | −12.36 | −10.22 | −26.72 | −0.12 |
| T5c | −12.77 | −10.33 | −27.28 | −0.68 |
| T5t | −12.76 | −10.35 | −27.22 | −0.62 |
| Code | Chloroform | Acetonitrile | ||||
|---|---|---|---|---|---|---|
| Calc. | Scaled a | Measured | Calc. | Scaled b | Measured | |
| (1b) CH3-Pz-COOCH2CH3 | ||||||
| T3c | 1803.76 | 1715 | 1720 | 1799.13 | 1716 | 1718 |
| T3t | 1824.47 | 1735 | 1814.27 | 1731 | 1730 | |
| (2b) H2N-Pz-COOCH3 | ||||||
| T3c | 1807.09 | 1718 | 1801.77 | 1719 | 1721 | |
| T3t | 1826.99 | 1737 | 1818.40 | 1735 | 1731 | |
| T5c | 1827.74 | 1737 | 1820.13 | 1736 | 1731 | |
| T5t | 1808.87 | 1720 | 1725 | 1805.58 | 1723 | 1721 |
| (3b) O2N-Pz-COOCH3 | ||||||
| T5c | 1840.61 | 1750 | 1751 | 1831.87 | 1748 | 1747 |
| T5t | 1824.01 | 1734 | 1734 | 1822.54 | 1739 | 1739 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kusakiewicz-Dawid, A.; Porada, M.; Dziuk, B.; Siodłak, D. Annular Tautomerism of 3(5)-Disubstituted-1H-pyrazoles with Ester and Amide Groups. Molecules 2019, 24, 2632. https://doi.org/10.3390/molecules24142632
Kusakiewicz-Dawid A, Porada M, Dziuk B, Siodłak D. Annular Tautomerism of 3(5)-Disubstituted-1H-pyrazoles with Ester and Amide Groups. Molecules. 2019; 24(14):2632. https://doi.org/10.3390/molecules24142632
Chicago/Turabian StyleKusakiewicz-Dawid, Anna, Monika Porada, Błażej Dziuk, and Dawid Siodłak. 2019. "Annular Tautomerism of 3(5)-Disubstituted-1H-pyrazoles with Ester and Amide Groups" Molecules 24, no. 14: 2632. https://doi.org/10.3390/molecules24142632
APA StyleKusakiewicz-Dawid, A., Porada, M., Dziuk, B., & Siodłak, D. (2019). Annular Tautomerism of 3(5)-Disubstituted-1H-pyrazoles with Ester and Amide Groups. Molecules, 24(14), 2632. https://doi.org/10.3390/molecules24142632