Alkene Difunctionalization Using Hypervalent Iodine Reagents: Progress and Developments in the Past Ten Years


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
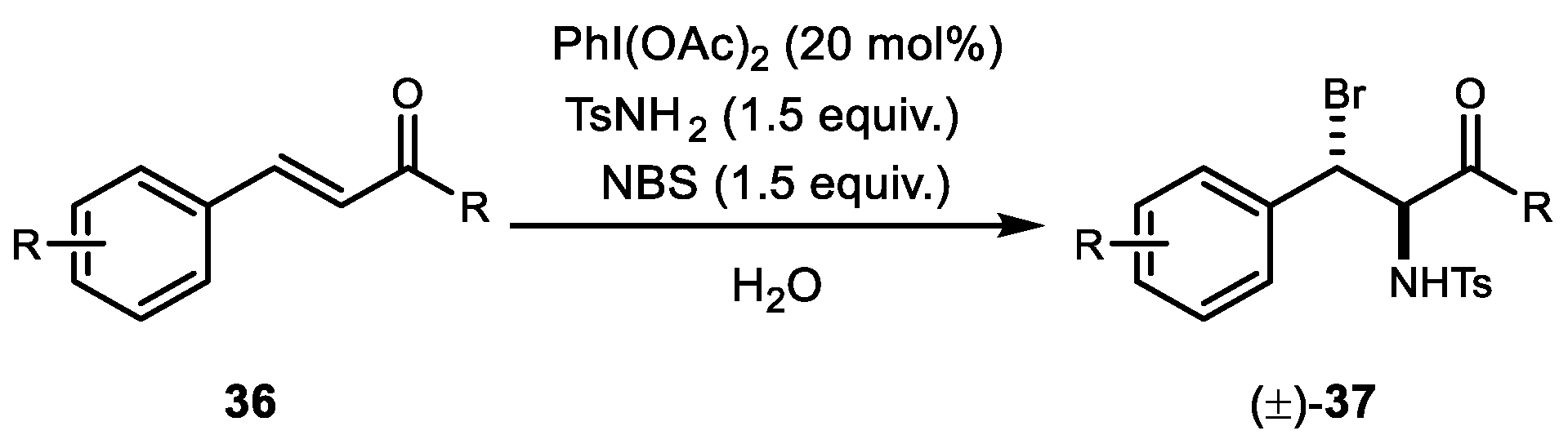{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
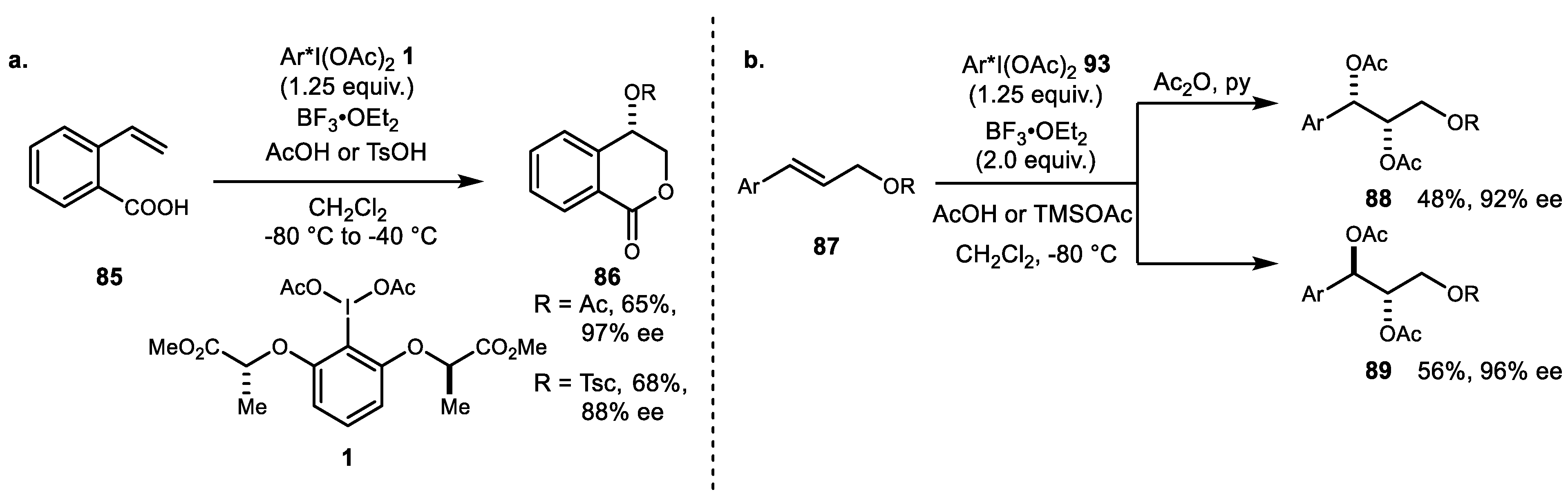{kind=link}
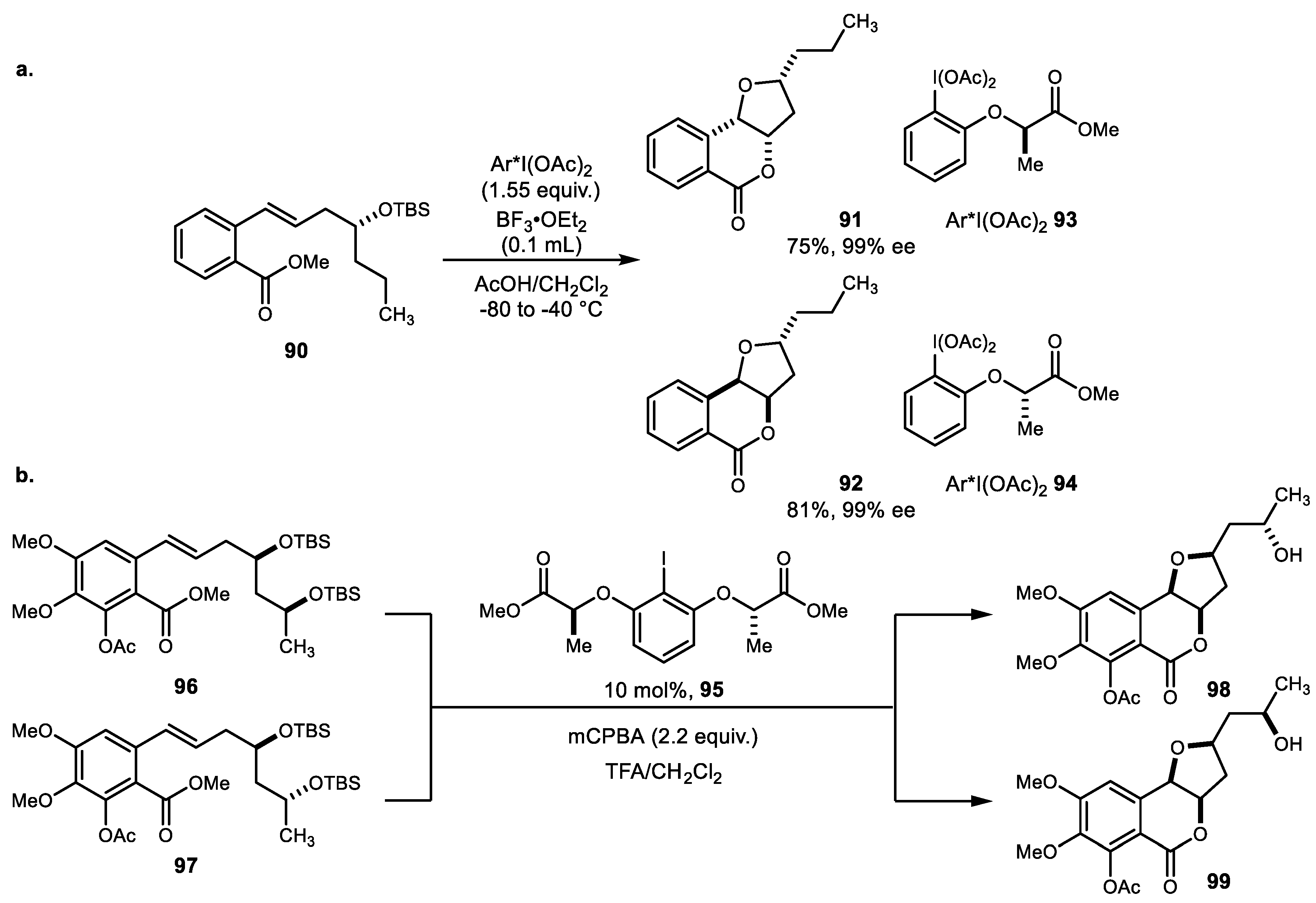{kind=link}
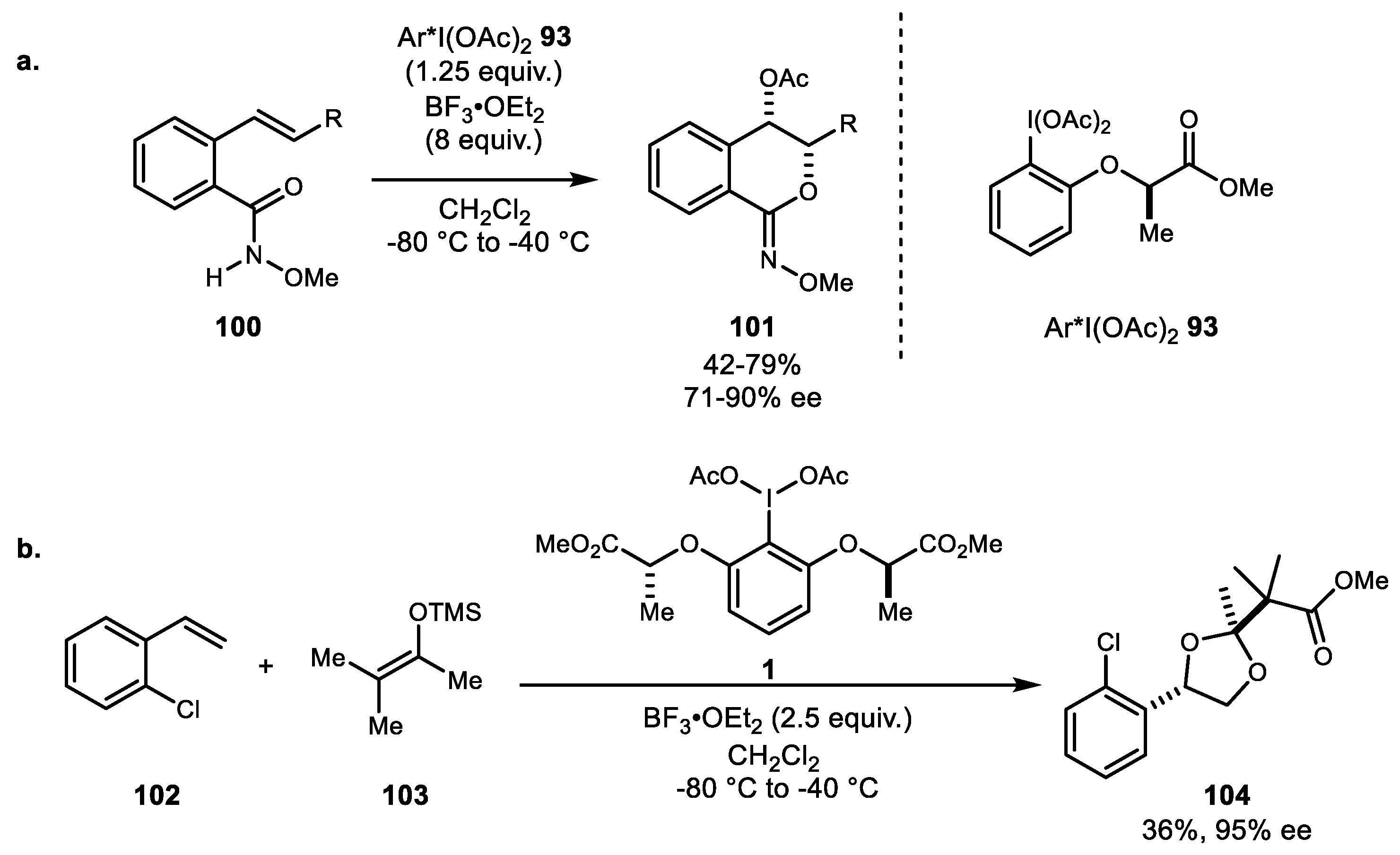{kind=link}
{kind=link}
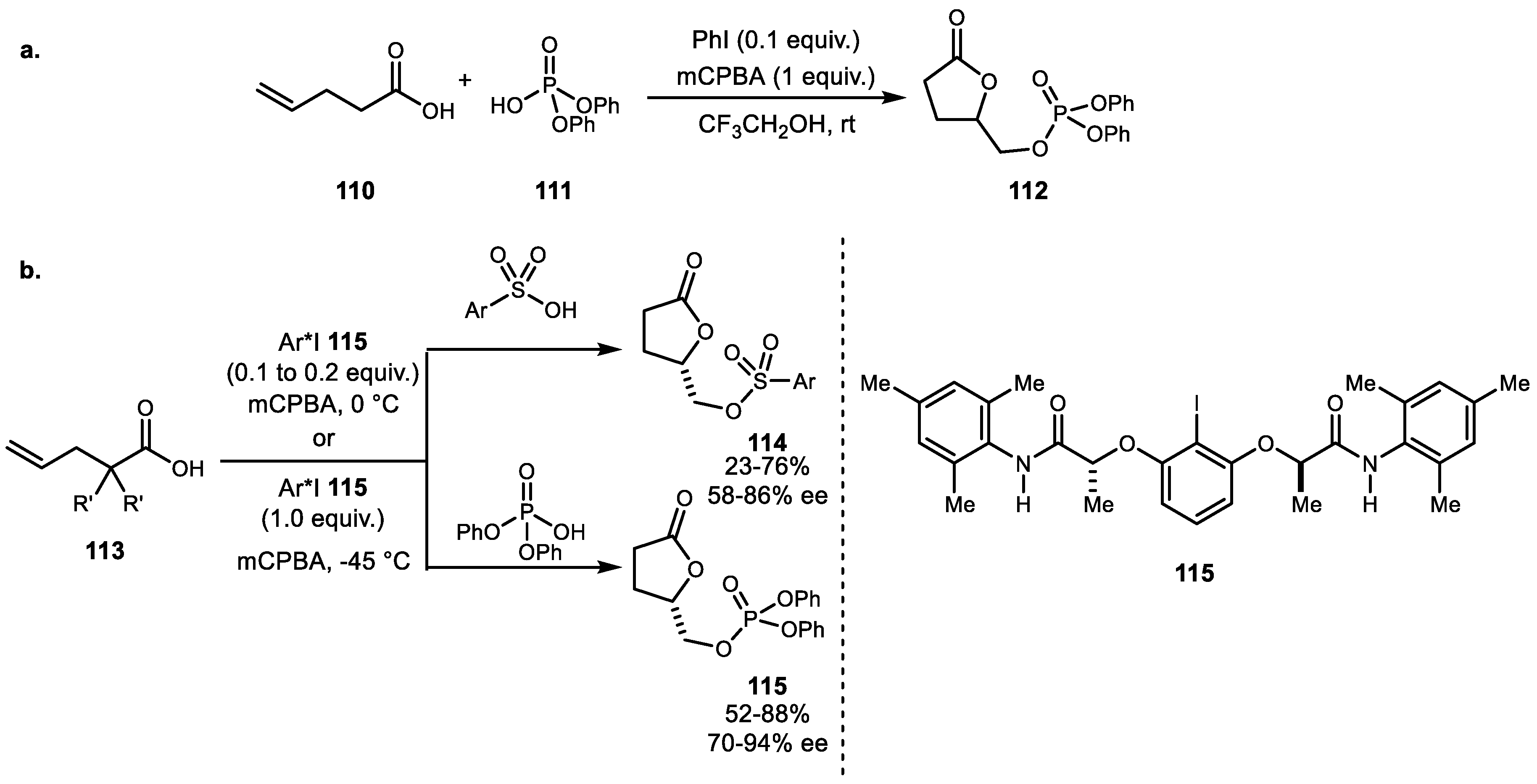{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
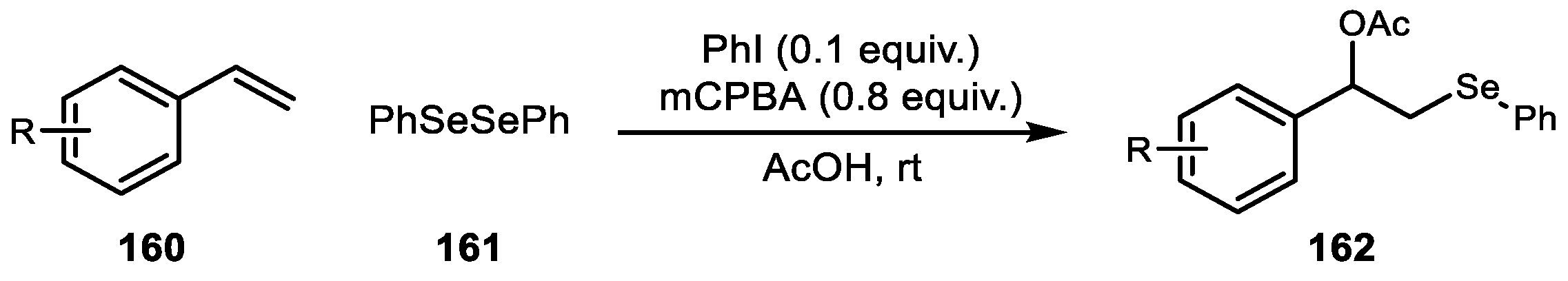{kind=link}
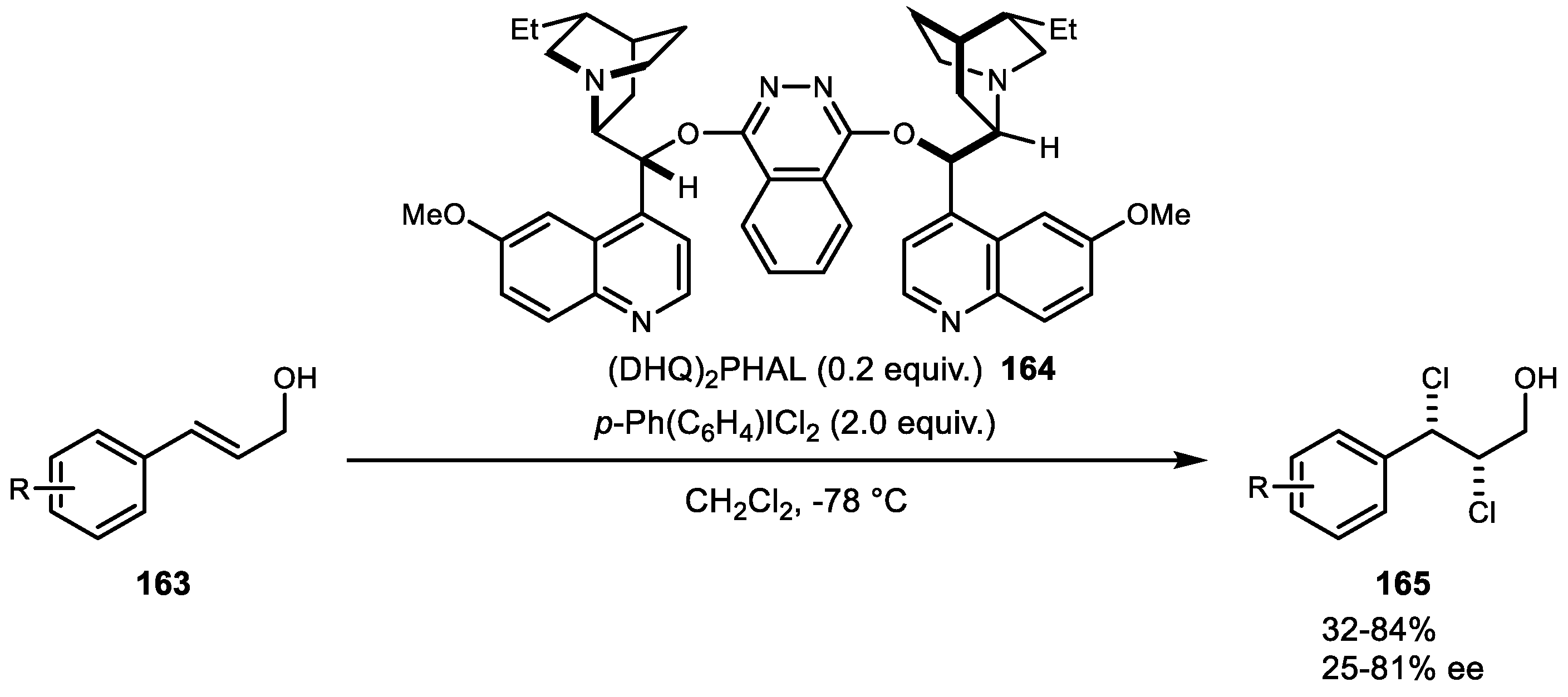{kind=link}
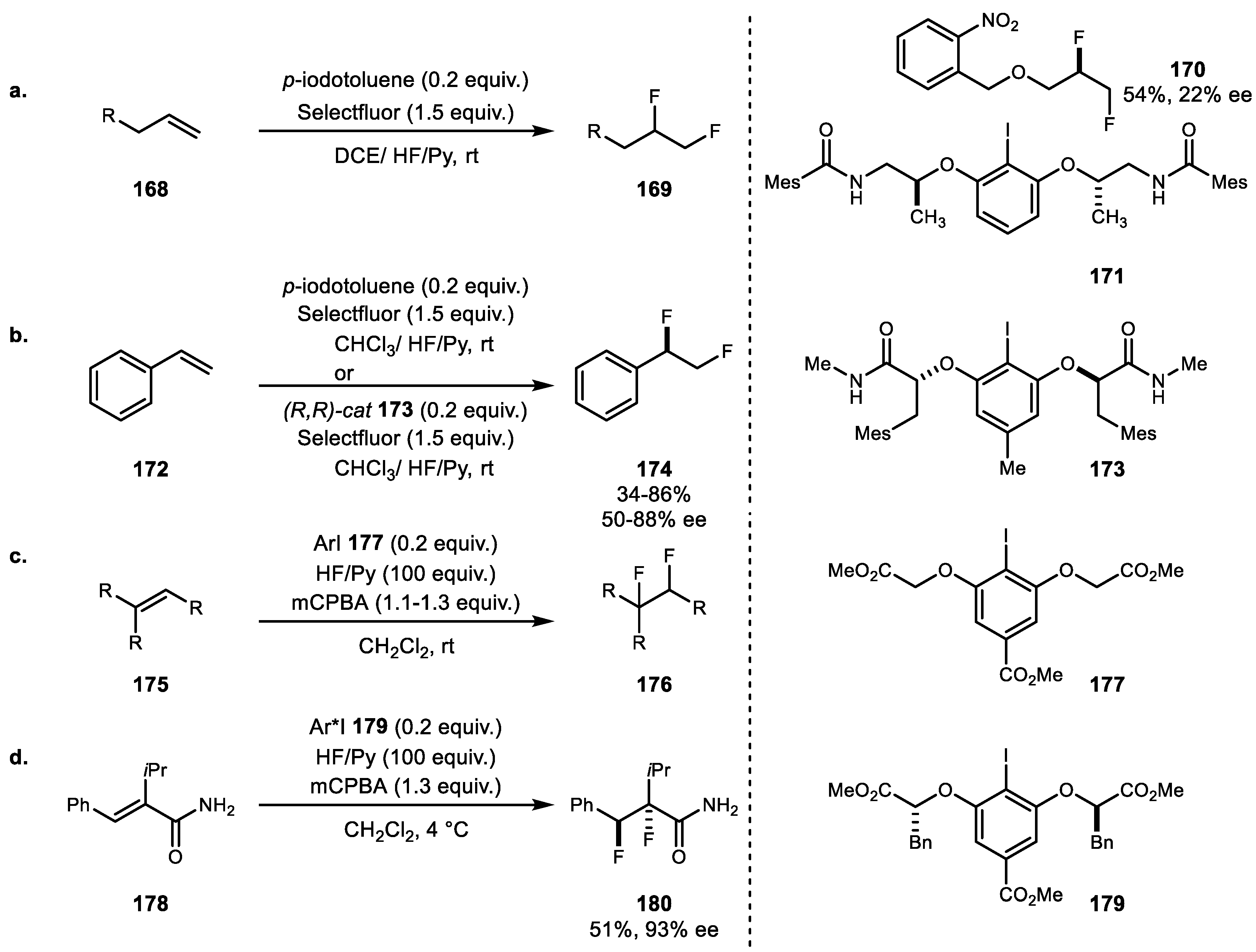{kind=link}
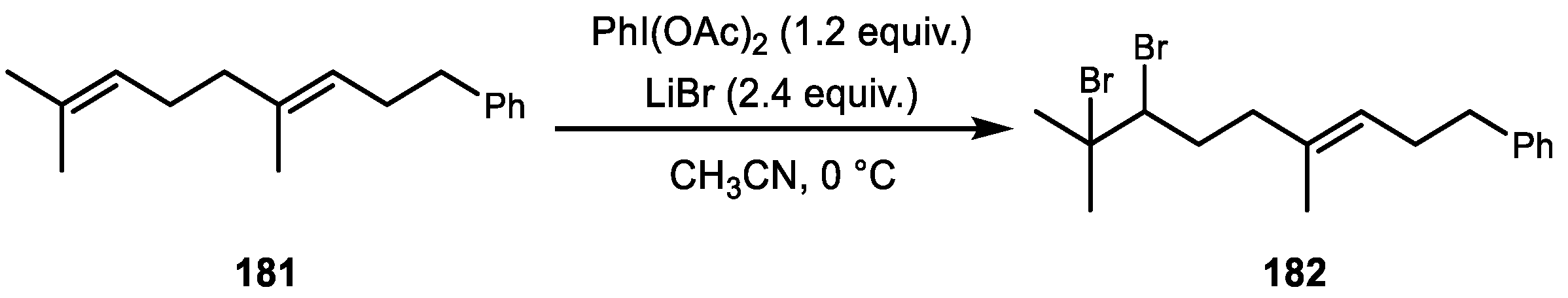{kind=link}
Abstract
1. Introduction
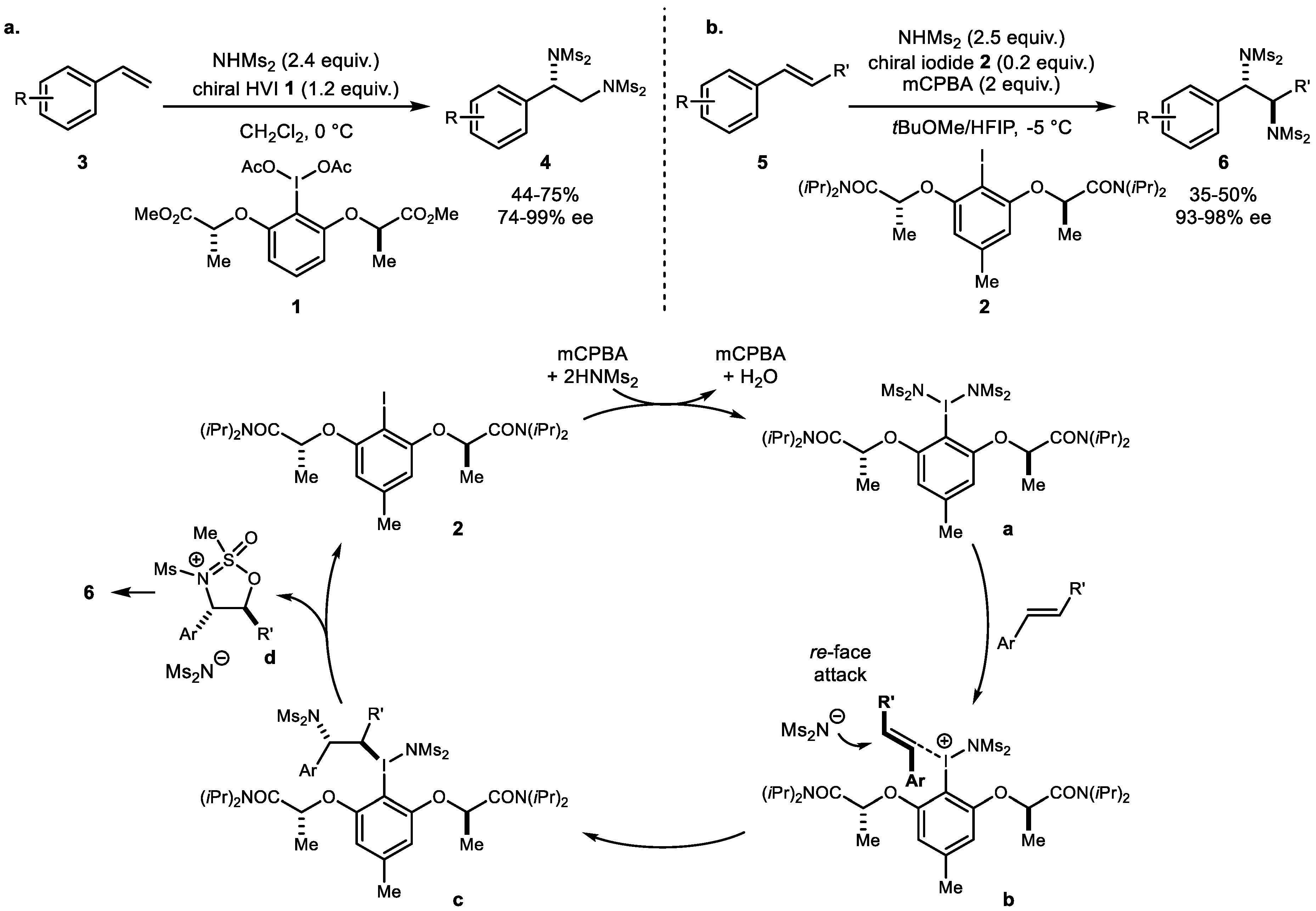
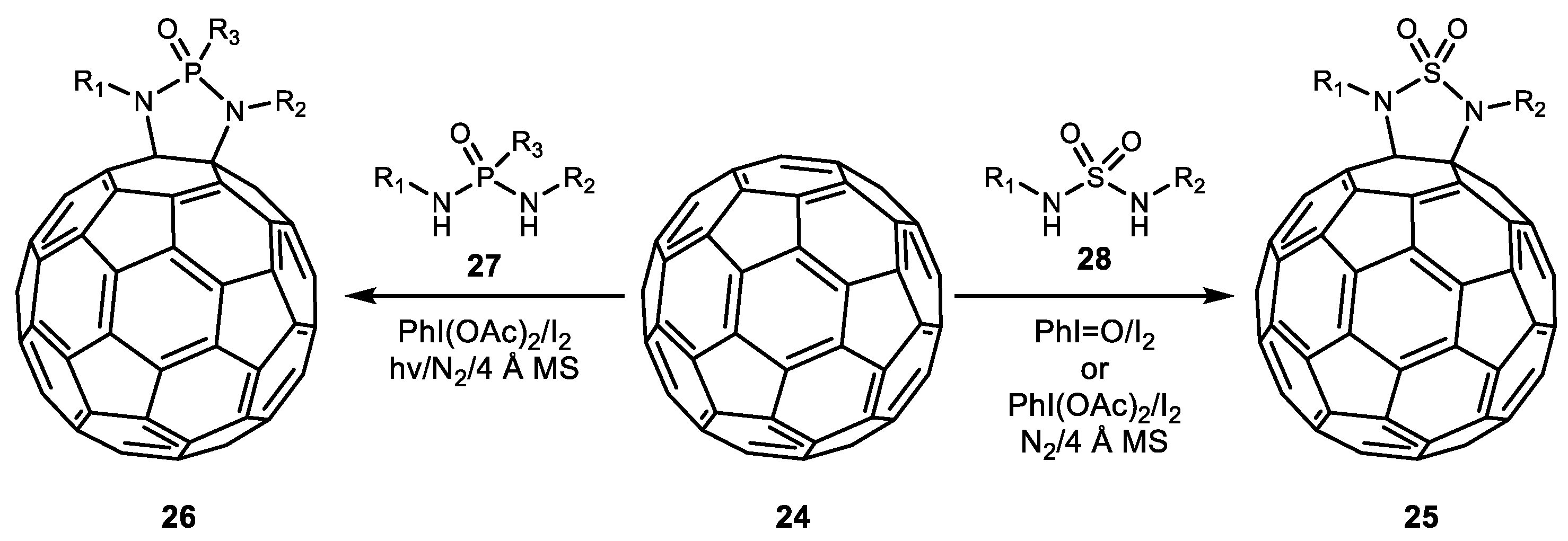
2. Alkene Diamination
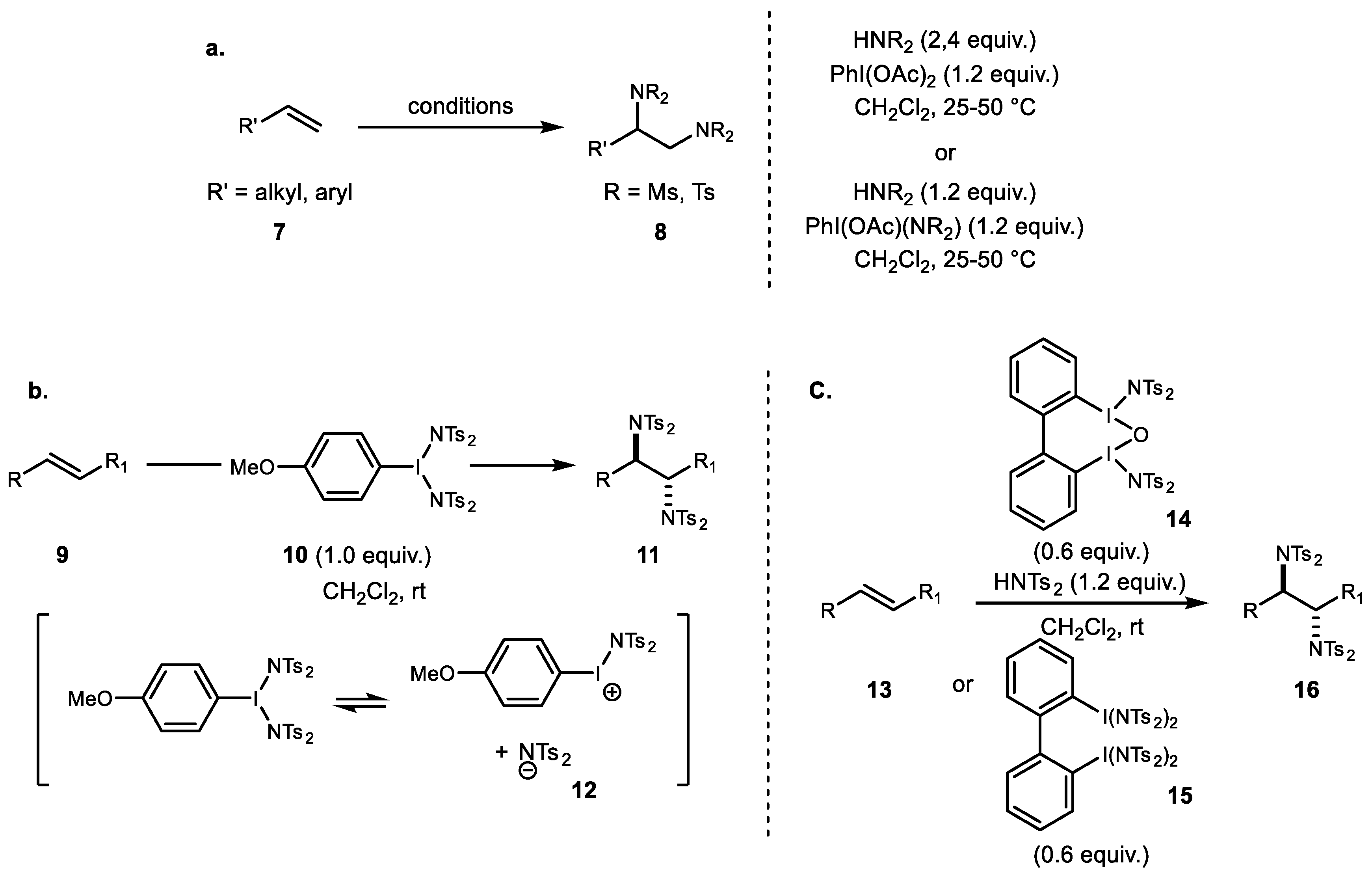
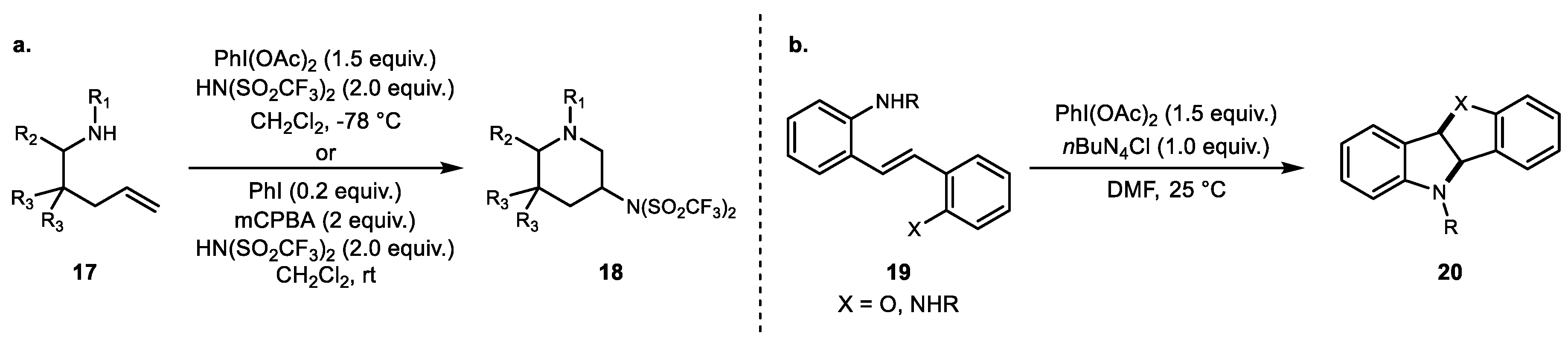
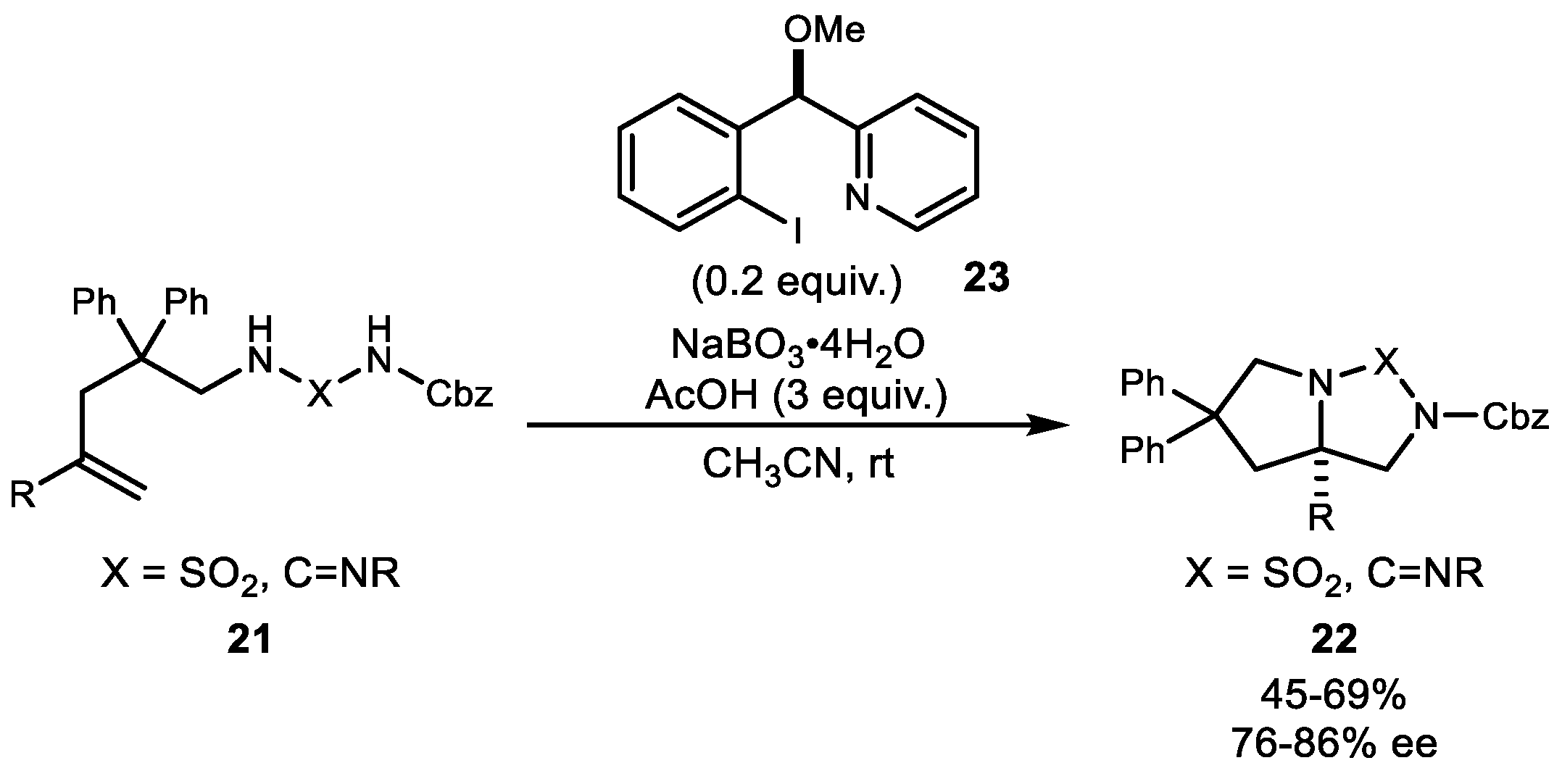
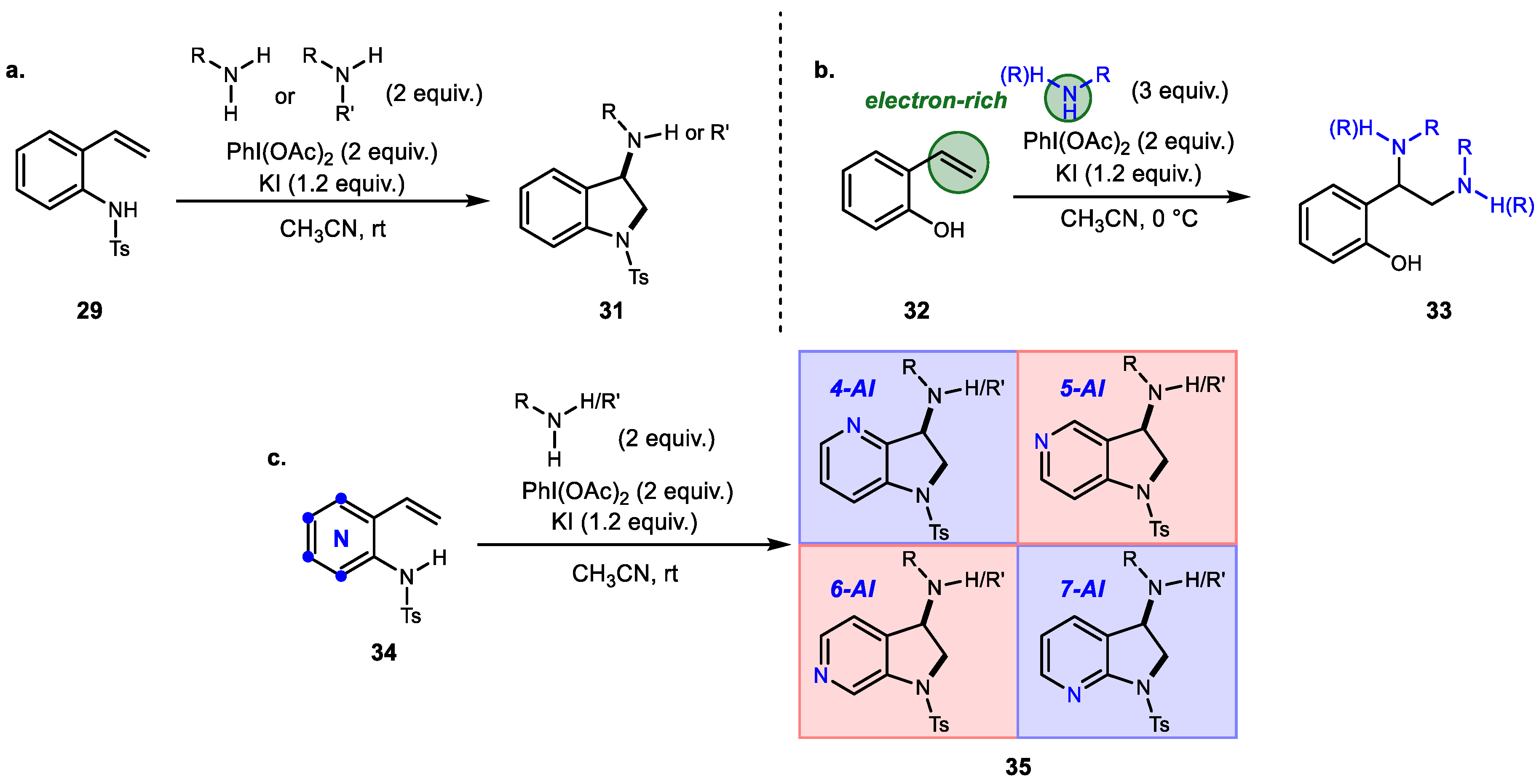
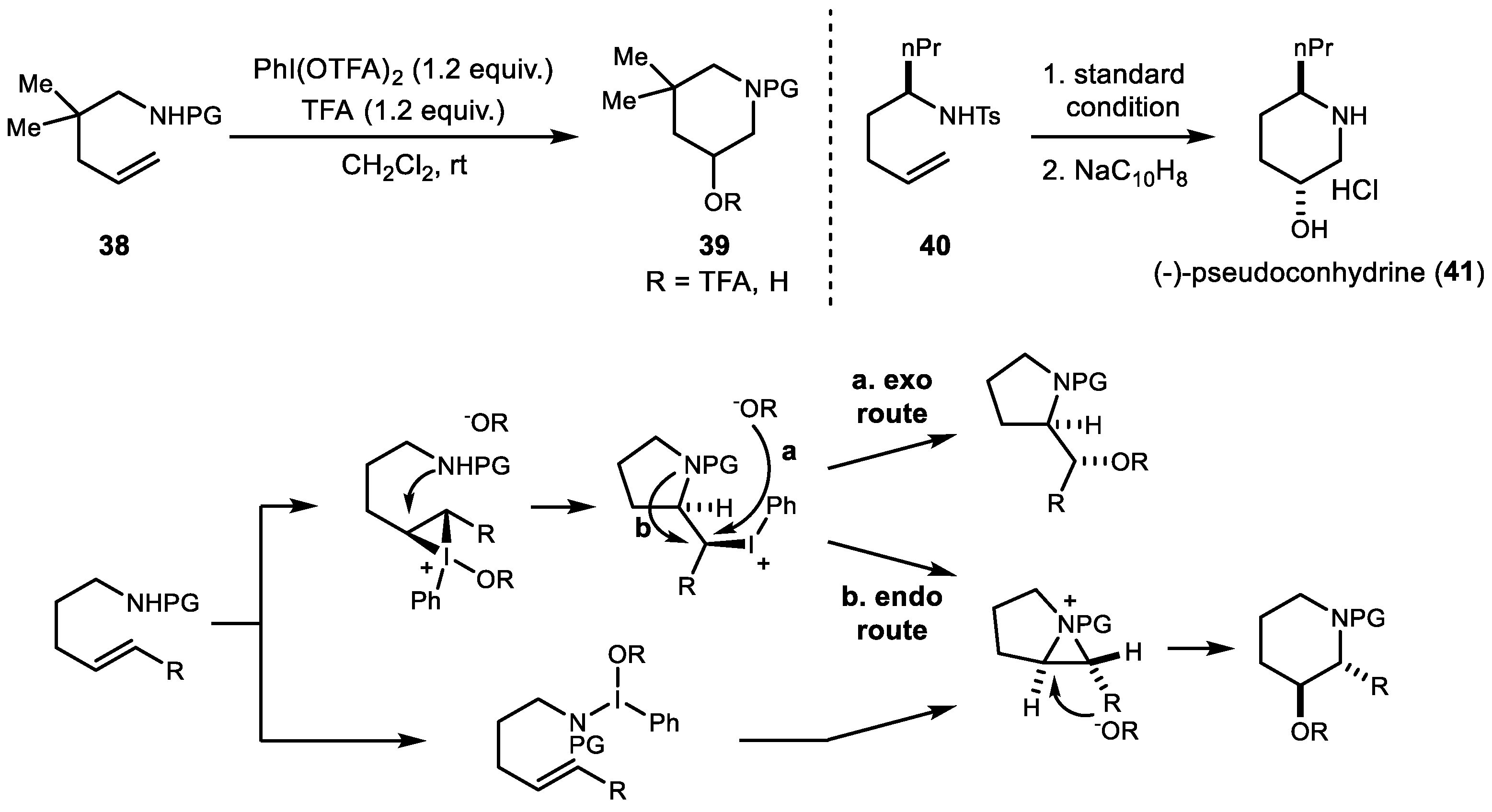
3. Alkene Aminofunctionalization
4. Alkene Diacetoxylation
5. Alkene Oxyfunctionalization
6. Alkene Dihalogenation
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wirth, T.; Hirt, U.H. Hypervalent iodine compounds. Recent advances in synthetic applications. Synthesis 1999, 1999, 1271–1287. [Google Scholar] [CrossRef]
- Wirth, T. Hypervalent iodine chemistry in synthesis: Scope and new directions. Angew. Chem. Int. Ed. 2005, 44, 3656–3665. [Google Scholar] [CrossRef] [PubMed]
- Ladziata, U.; Zhdankin, V.V. Hypervalent iodine(V) reagents in organic synthesis. Arkivoc 2006, 9, 26–58. [Google Scholar] [CrossRef]
- Zhdankin, V.V.; Stang, P.J. Chemistry of Polyvalent Iodine. Chem. Rev. 2008, 108, 5299–5358. [Google Scholar] [CrossRef] [PubMed]
- Dohi, T.; Kita, Y. Hypervalent iodine reagents as a new entrance to organocatalysts. Chem. Commun. 2009, 2073–2085. [Google Scholar] [CrossRef]
- Zhdankin, V.V. Hypervalent iodine(III) reagents in organic synthesis. Arkivoc 2009, 1–62. [Google Scholar]
- Farid, U.; Wirth, T. Stereoselective Synthesis with Hypervalent Iodine Reagents; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; pp. 197–203. [Google Scholar]
- Brown, M.; Farid, U.; Wirth, T. Hypervalent iodine reagents as powerful electrophiles. Synlett 2013, 24, 424–431. [Google Scholar]
- Singh, F.V.; Wirth, T. Oxidative Functionalization with Hypervalent Halides; Elsevier B.V.: Amsterdam, The Netherlands, 2014; pp. 880–933. [Google Scholar]
- Yoshimura, A.; Zhdankin, V.V. Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev. 2016, 116, 3328–3435. [Google Scholar] [CrossRef]
- Quideau, S. Hypervalent iodine chemistry: Recent advances and applications; Elsevier: Amsterdam, The Netherlands, 2010. [Google Scholar]
- Zhdankin, V.V. Hypervalent Iodine Chemistry Preparation, Structure and Synthetic Applications of Polyvalent Iodine Compounds; John Wiley & Sons: Chichester/West Sussex, UK, 2014. [Google Scholar]
- Wirth, T. Hypervalent Iodine Chemistry; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Moriarty, R.M.; Prakash, O. Oxidation of phenolic compounds with organohypervalent iodine reagents. Org. React. 2001, 57, 327–415. [Google Scholar]
- Rodriguez, S.; Wipf, P. Oxidative spiroacetalizations and spirolactonizations of arenes. Synthesis 2004, 2767–2783. [Google Scholar] [CrossRef]
- Zhdankin, V.V. Benziodoxole-based hypervalent iodine reagents in organic synthesis. Curr. Org. Synth. 2005, 2, 121–145. [Google Scholar] [CrossRef]
- Ciufolini, M.A.; Braun, N.A.; Canesi, S.; Ousmer, M.; Chang, J.; Chai, D. Oxidative amidation of phenols through the use of hypervalent iodine reagents. Development and applications. Synthesis 2007, 3759–3772. [Google Scholar] [CrossRef]
- Pouysegu, L.; Deffieux, D.; Quideau, S. Hypervalent iodine-mediated phenol dearomatization in natural product synthesis. Tetrahedron 2010, 66, 2235–2261. [Google Scholar] [CrossRef]
- Fernandez Gonzalez, D.; Benfatti, F.; Waser, J. Asymmetric Organocatalysis Meets Hypervalent Iodine Chemistry for the α-Functionalization of Carbonyl Compounds. ChemCatChem 2012, 4, 955–958. [Google Scholar] [CrossRef]
- Parra, A.; Reboredo, S. Chiral Hypervalent Iodine Reagents: Synthesis and Reactivity. Chem. Eur. J. 2013, 19, 17244–17260. [Google Scholar] [CrossRef] [PubMed]
- Singh, F.V.; Wirth, T. Oxidative rearrangements with hypervalent iodine reagents. Synthesis 2013, 45, 2499–2511. [Google Scholar]
- Romero, R.M.; Woeste, T.H.; Muniz, K. Vicinal Difunctionalization of Alkenes with Iodine (III) Reagents and Catalysts. Chem. Asian J. 2014, 9, 972–983. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, J.; Fruh, N.; Togni, A. Electrophilic Trifluoromethylation by Use of Hypervalent Iodine Reagents. Chem. Rev. 2015, 115, 650–682. [Google Scholar] [CrossRef]
- Kumar, R.; Wirth, T. Asymmetric synthesis with hypervalent iodine reagents. Top. Curr. Chem. 2016, 373, 243–262. [Google Scholar]
- Muniz, K. Aminations with hypervalent iodine. Top. Curr. Chem. 2016, 373, 105–134. [Google Scholar]
- Waser, J. Alkynylation with hypervalent iodine reagents. Top. Curr. Chem. 2016, 373, 187–222. [Google Scholar] [PubMed]
- Boelke, A.; Finkbeiner, P.; Nachtsheim, B.J. Atom-economical group-transfer reactions with hypervalent iodine compounds. Beilstein J. Org. Chem. 2018, 14, 1263–1280. [Google Scholar] [CrossRef] [PubMed]
- Reddy Kandimalla, S.; Prathima Parvathaneni, S.; Sabitha, G.; Subba Reddy, B.V. Recent Advances in Intramolecular Metal-Free Oxidative C-H Bond Aminations Using Hypervalent Iodine (III) Reagents. Eur. J. Org. Chem. 2019, 2019, 1687–1714. [Google Scholar] [CrossRef]
- Xing, L.; Zhang, Y.; Du, Y. Hypervalent Iodine-Mediated Synthesis of Spiroheterocycles via Oxidative Cyclization. Curr. Org. Chem. 2019, 23, 14–37. [Google Scholar] [CrossRef]
- Röben, C.; Souto, J.A.; González, Y.; Lishchynskyi, A.; Muñiz, K. Enantioselective Metal-Free Diamination of Styrenes. Angew. Chem. Int. Ed. 2011, 50, 9478–9482. [Google Scholar] [CrossRef] [PubMed]
- Muñiz, K.; Barreiro, L.; Romero, R.M.; Martínez, C. Catalytic Asymmetric Diamination of Styrenes. J. Am. Chem. Soc. 2017, 139, 4354–4357. [Google Scholar] [CrossRef] [PubMed]
- Souto, J.A.; González, Y.; Iglesias, A.; Zian, D.; Lishchynskyi, A.; Muñiz, K. Iodine (III)-Promoted Intermolecular Diamination of Alkenes. Chem. Asian J. 2012, 7, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Souto, J.A.; Martínez, C.; Velilla, I.; Muñiz, K. Defined Hypervalent Iodine (III) Reagents Incorporating Transferable Nitrogen Groups: Nucleophilic Amination through Electrophilic Activation. Angew. Chem. Int. Ed. 2013, 52, 1324–1328. [Google Scholar] [CrossRef]
- Romero, R.M.; Souto, J.A.; Muñiz, K. Substitution Effects of Hypervalent Iodine (III) Reagents in the Diamination of Styrene. J. Org. Chem. 2016, 81, 6118–6122. [Google Scholar] [CrossRef]
- Röben, C.; Souto, J.A.; Escudero-Adán, E.C.; Muñiz, K. Oxidative Diamination Promoted by Dinuclear Iodine (III) Reagents. Org. Lett. 2013, 15, 1008–1011. [Google Scholar] [CrossRef]
- Kong, A.; Blakey, S.B. Intramolecular Olefin Diamination for the Stereoselective Synthesis of 3-Aminopiperidines. Synthesis 2012, 44, 1190–1198. [Google Scholar]
- Kim, H.J.; Cho, S.H.; Chang, S. Intramolecular Oxidative Diamination and Aminohydroxylation of Olefins under Metal-Free Conditions. Org. Lett. 2012, 14, 1424–1427. [Google Scholar] [CrossRef] [PubMed]
- Muñiz, K. Advancing Palladium-Catalyzed C–N Bond Formation: Bisindoline Construction from Successive Amide Transfer to Internal Alkenes. J. Am. Chem. Soc. 2007, 129, 14542–14543. [Google Scholar] [CrossRef] [PubMed]
- Mizar, P.; Laverny, A.; El-Sherbini, M.; Farid, U.; Brown, M.; Malmedy, F.; Wirth, T. Enantioselective Diamination with Novel Chiral Hypervalent Iodine Catalysts. Chem. Eur. J. 2014, 20, 9910–9913. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-T.; Lu, X.-W.; Xing, M.-L.; Sun, X.-Q.; Miao, C.-B. Hypervalent Iodine Reagent Mediated Diamination of [60] Fullerene with Sulfamides or Phosphoryl Diamides. Org. Lett. 2014, 16, 5882–5885. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.B.; Johnston, J.N. Alkene Diamination Using Electron-Rich Amines: Hypervalent Iodine-Promoted Inter-/Intramolecular C–N Bond Formation. Org. Lett. 2014, 16, 3804–3807. [Google Scholar] [CrossRef]
- Danneman, M.W.; Hong, K.B.; Johnston, J.N. Oxidative Inter-/Intermolecular Alkene Diamination of Hydroxy Styrenes with Electron-Rich Amines. Org. Lett. 2015, 17, 2558–2561. [Google Scholar] [CrossRef]
- Danneman, M.W.; Hong, K.B.; Johnston, J.N. A Unified Approach to the Four Azaindoline Families by Inter-/Intramolecular Annulative Diamination of Vinylpyridines. Org. Lett. 2015, 17, 3806–3809. [Google Scholar] [CrossRef]
- Wu, X.-L.; Wang, G.-W. Hypervalent iodine-mediated aminobromination of olefins in water. Tetrahedron 2009, 65, 8802–8807. [Google Scholar] [CrossRef]
- Lovick, H.M.; Michael, F.E. Metal-Free Highly Regioselective Aminotrifluoroacetoxylation of Alkenes. J. Am. Chem. Soc. 2010, 132, 1249–1251. [Google Scholar] [CrossRef]
- Farid, U.; Wirth, T. Highly Stereoselective Metal-Free Oxyaminations Using Chiral Hypervalent Iodine Reagents. Angew. Chem. Int. Ed. 2012, 51, 3462–3465. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Kaga, A.; Chiba, S. Diastereoselective Aminooxygenation and Diamination of Alkenes with Amidines by Hypervalent Iodine (III) Reagents. Org. Lett. 2014, 16, 6136–6139. [Google Scholar] [CrossRef] [PubMed]
- Sanjaya, S.; Chiba, S. Copper-Catalyzed Aminooxygenation of N-Allylamidines with PhI(OAc)2. Org. Lett. 2012, 14, 5342–5345. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-F.; Zhu, X.; Chiba, S. Copper-Catalyzed Aerobic [3 + 2]-Annulation of N-Alkenyl Amidines. J. Am. Chem. Soc. 2012, 134, 3679–3682. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Kaga, A.; Chiba, S. Anti-Selective aminofluorination of alkenes with amidines mediated by hypervalent iodine (iii) reagents. Org. Biomol. Chem. 2016, 14, 5481–5485. [Google Scholar] [CrossRef] [PubMed]
- Wardrop, D.J.; Bowen, E.G.; Forslund, R.E.; Sussman, A.D.; Weerasekera, S.L. Intramolecular Oxamidation of Unsaturated O-Alkyl Hydroxamates: A Remarkably Versatile Entry to Hydroxy Lactams. J. Am. Chem. Soc. 2010, 132, 1188–1189. [Google Scholar] [CrossRef] [PubMed]
- Bowen, E.G.; Wardrop, D.J. Diastereoselective Nitrenium Ion-Mediated Cyclofunctionalization: Total Synthesis of (+)-Castanospermine. Org. Lett. 2010, 12, 5330–5333. [Google Scholar] [CrossRef] [PubMed]
- Wardrop, D.J.; Bowen, E.G. Nitrenium Ion-Mediated Alkene Bis-Cyclofunctionalization: Total Synthesis of (−)-Swainsonine. Org. Lett. 2011, 13, 2376–2379. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Gerasimov, M.V.; DeJong, S.; Wardrop, D.J. Oxamidation of Unsaturated O-Alkyl Hydroxamates: Synthesis of the Madangamine Diazatricylic (ABC Rings) Skeleton. Org. Lett. 2017, 19, 6570–6573. [Google Scholar] [CrossRef]
- Wang, Q.; Zhong, W.; Wei, X.; Ning, M.; Meng, X.; Li, Z. Metal-free intramolecular aminofluorination of alkenes mediated by PhI(OPiv)2/hydrogen fluoride–pyridine system. Org. Biomol. Chem. 2012, 10, 8566–8569. [Google Scholar] [CrossRef]
- Kong, W.; Feige, P.; de Haro, T.; Nevado, C. Regio- and Enantioselective Aminofluorination of Alkenes. Angew. Chem. Int. Ed. 2013, 52, 2469–2473. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Kamo, T.; Fukushi, K.; Hiramatsu, T.; Tokunaga, E.; Dohi, T.; Kita, Y.; Shibata, N. Iodoarene-catalyzed fluorination and aminofluorination by an Ar-I/HF·pyridine/mCPBA system. Chem. Sci. 2014, 5, 2754–2760. [Google Scholar] [CrossRef]
- Cui, J.; Jia, Q.; Feng, R.-Z.; Liu, S.-S.; He, T.; Zhang, C. Boron Trifluoride Etherate Functioning as a Fluorine Source in an Iodosobenzene-Mediated Intramolecular Aminofluorination of Homoallylic Amines. Org. Lett. 2014, 16, 1442–1445. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Miyake, A.; Muta, K.; Oyamada, A.J. Hypervalent Iodine/HF Reagents for the Synthesis of 3-Fluoropyrrolidines. J. Org. Chem. 2017, 82, 11721–11726. [Google Scholar] [CrossRef] [PubMed]
- Mennie, K.M.; Banik, S.M.; Reichert, E.C.; Jacobsen, E.N. Catalytic Diastereo- and Enantioselective Fluoroamination of Alkenes. J. Am. Chem. Soc. 2018, 140, 4797–4802. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.-Q.; Li, Y.-M. Regioselective (Diacetoxyiodo)benzene-Promoted Halocyclization of Unfunctionalized Olefins. J. Org. Chem. 2014, 79, 10094–10109. [Google Scholar] [CrossRef]
- Wang, H.; Frings, M.; Bolm, C. Halocyclizations of Unsaturated Sulfoximines. Org. Lett. 2016, 18, 2431–2434. [Google Scholar] [CrossRef]
- Daniel, M.; Blanchard, F.; Nocquet-Thibault, S.; Cariou, K.; Dodd, R.H. Halocyclization of Unsaturated Guanidines Mediated by Koser’s Reagent and Lithium Halides. J. Org. Chem. 2015, 80, 10624–10633. [Google Scholar] [CrossRef]
- Mizar, P.; Niebuhr, R.; Hutchings, M.; Farooq, U.; Wirth, T. Thioamination of Alkenes with Hypervalent Iodine Reagents. Chem. Eur. J. 2016, 22, 1614–1617. [Google Scholar] [CrossRef]
- Jeon, H.; Kim, D.; Lee, J.H.; Song, J.; Lee, W.S.; Kang, D.W.; Kang, S.; Lee, S.B.; Choi, S.; Hong, K.B. Hypervalent Iodine-Mediated Alkene Functionalization: Oxazoline and Thiazoline Synthesis via Inter-/Intramolecular Aminohydroxylation and Thioamination. Adv. Synth. Catal. 2018, 360, 779–783. [Google Scholar] [CrossRef]
- Park, S.W.; Kim, S.-H.; Song, J.; Park, G.Y.; Kim, D.; Nam, T.-G.; Hong, K.B. Hypervalent iodine-mediated Ritter-type amidation of terminal alkenes: The synthesis of isoxazoline and pyrazoline cores. Beilstein J. Org. Chem. 2018, 14, 1028–1033. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.-M.; Cai, C. Iodine(iii)-mediated intramolecular sulfeno- and selenofunctionalization of β,γ-unsaturated tosyl hydrazones and oximes. Org. Biomol. Chem. 2018, 16, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Yoshida, Y.; Miyata, K.; Wakisaka, A.; Sugimura, T. Enantiodifferentiating endo-Selective Oxylactonization of ortho-Alk-1-enylbenzoate with a Lactate-Derived Aryl-λ3-Iodane. Angew. Chem. Int. Ed. 2010, 49, 7068–7071. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Wakita, M.; Sugimura, T. Enantioselective Prévost and Woodward reactions using chiral hypervalent iodine(iii): Switchover of stereochemical course of an optically active 1,3-dioxolan-2-yl cation. Chem. Commun. 2011, 47, 3983–3985. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Mori, K.; Shimogaki, M.; Sugimura, T. Asymmetric Synthesis of 4,8-Dihydroxyisochroman-1-one Polyketide Metabolites Using Chiral Hypervalent Iodine(III). Org. Lett. 2012, 14, 1294–1297. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Mori, K.; Shimogaki, M.; Sugimura, T. Total synthesis of (12R)- and (12S)-12-hydroxymonocerins: Stereoselective oxylactonization using a chiral hypervalent iodine(iii) species. RSC Adv. 2013, 3, 17717–17725. [Google Scholar] [CrossRef]
- Shimogaki, M.; Fujita, M.; Sugimura, T. Enantioselective Oxidation of Alkenylbenzoates Catalyzed by Chiral Hypervalent Iodine(III) To Yield 4-Hydroxyisochroman-1-ones. Eur. J. Org. Chem. 2013, 2013, 7128–7138. [Google Scholar] [CrossRef]
- Takesue, T.; Fujita, M.; Sugimura, T.; Akutsu, H. A Series of Two Oxidation Reactions of ortho-Alkenylbenzamide with Hypervalent Iodine(III): A Concise Entry into (3R,4R)-4-Hydroxymellein and (3R,4R)-4-Hydroxy-6-methoxymellein. Org. Lett. 2014, 16, 4634–4637. [Google Scholar] [CrossRef]
- Shimogaki, M.; Fujita, M.; Sugimura, T. Stereoselective Formation of Substituted 1,3-Dioxolanes through a Three-Component Assembly during the Oxidation of Alkenes with Hypervalent Iodine(III). Molecules 2015, 20, 17041. [Google Scholar] [CrossRef]
- Alhalib, A.; Kamouka, S.; Moran, W.J. Iodoarene-Catalyzed Cyclizations of Unsaturated Amides. Org. Lett. 2015, 17, 1453–1456. [Google Scholar] [CrossRef]
- Moon, N.G.; Harned, A.M. Iodine(III)-promoted synthesis of oxazolines from N-allylamides. Tetrahedron Lett. 2013, 54, 2960–2963. [Google Scholar] [CrossRef]
- Koser, G.F.; Lodaya, J.S.; Ray, D.G.; Kokil, P.B. Direct. alpha.-phosphoryloxylation of ketones and (phosphoryloxy) lactonization of pentenoic acids with [hydroxy[(bis(phenyloxy)phosphoryl)oxy]iodo]benzene. J. Am. Chem. Soc. 1988, 110, 2987–2988. [Google Scholar] [CrossRef]
- Zhou, Z.-S.; He, X.-H. A convenient phosphoryloxylactonization of pentenoic acids with catalytic hypervalent iodine(III) reagent. Tetrahedron Lett. 2010, 51, 2480–2482. [Google Scholar] [CrossRef]
- Gelis, C.; Dumoulin, A.; Bekkaye, M.; Neuville, L.; Masson, G. Chiral Hypervalent Iodine(III) Catalyst Promotes Highly Enantioselective Sulfonyl- and Phosphoryl-oxylactonizations. Org. Lett. 2017, 19, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Yang, J.; Meng, X.; Li, Z. BF3·OEt2-Promoted Diastereoselective Diacetoxylation of Alkenes by PhI(OAc)2. J. Org. Chem. 2011, 76, 9997–10004. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Liu, S.; Yang, J.; Meng, X.; Li, Z. Metal-Free, Organocatalytic Syn Diacetoxylation of Alkenes. Org. Lett. 2012, 14, 3336–3339. [Google Scholar] [CrossRef]
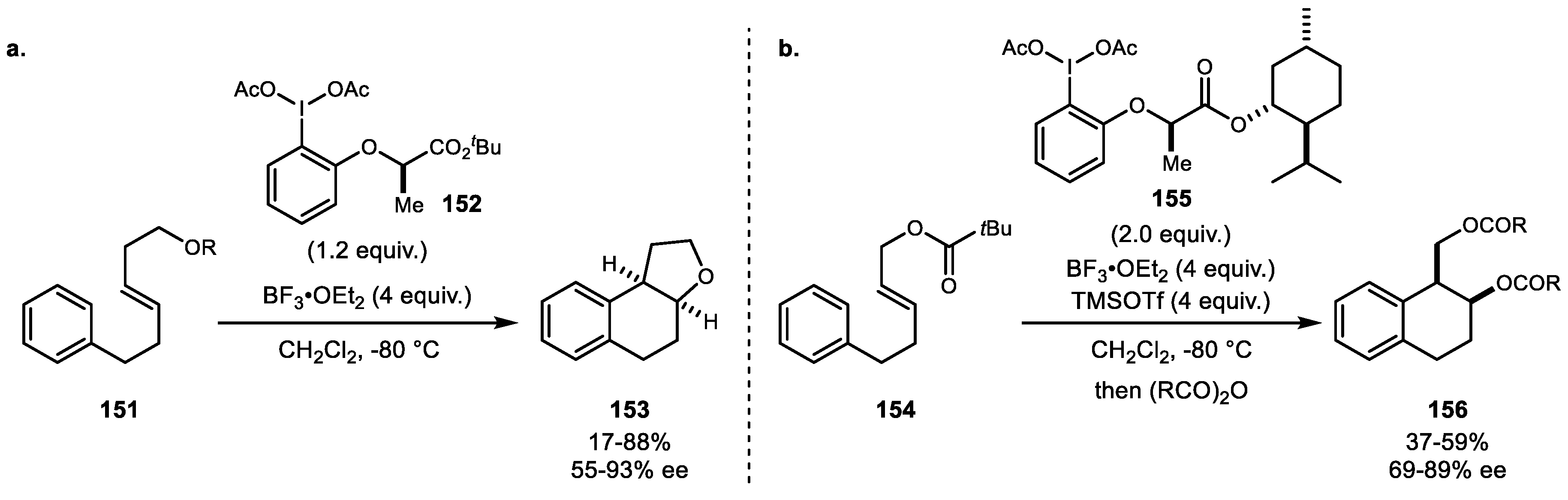
- Haubenreisser, S.; Wöste, T.H.; Martínez, C.; Ishihara, K.; Muñiz, K. Structurally Defined Molecular Hypervalent Iodine Catalysts for Intermolecular Enantioselective Reactions. Angew. Chem. Int. Ed. 2016, 55, 413–417. [Google Scholar] [CrossRef]
- Wöste, T.H.; Muñiz, K. Enantioselective Vicinal Diacetoxylation of Alkenes under Chiral Iodine(III) Catalysis. Synthesis 2016, 48, 816–827. [Google Scholar]
- Aertker, K.; Rama, R.J.; Opalach, J.; Muñiz, K. Vicinal Difunctionalization of Alkenes under Iodine(III) Catalysis involving Lewis Base Adducts. Adv. Synth. Catal. 2017, 359, 1290–1294. [Google Scholar] [CrossRef]
- Fujita, M.; Miura, K.; Sugimura, T. Enantioselective dioxytosylation of styrenes using lactate-based chiral hypervalent iodine(III). Beilstein J. Org. Chem. 2018, 14, 659–663. [Google Scholar] [CrossRef]
- Fujita, M.; Ookubo, Y.; Sugimura, T. Asymmetric cycloetherification based on a chiral auxiliary for 4-acyloxy-1-butene substrates during oxidation with iodosylbenzene via a 1,3-dioxan-2-yl cation. Tetrahedron Lett. 2009, 50, 1298–1300. [Google Scholar] [CrossRef]
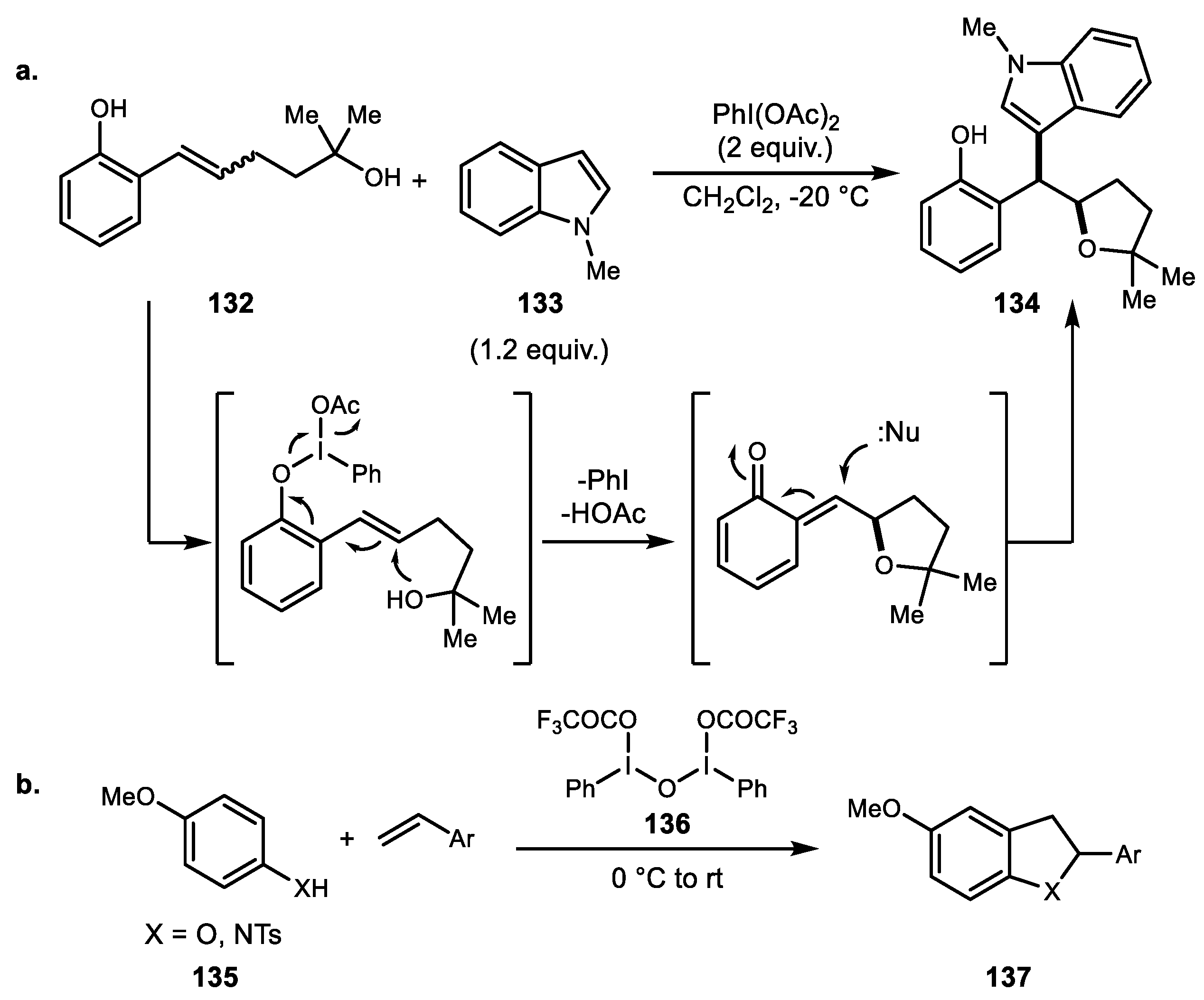
- Zhang, K.; Wang, H.; Zheng, J.; Yu, L.; Ding, H. Hypervalent iodine mediated alkene difunctionalization of vinylphenols: Diastereoselective synthesis of substituted indoles and indolizines. Chem. Commun. 2015, 51, 6399–6402. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.H.; Pathak, T.P.; Zhang, Y.; Sigman, M.S. Palladium-Catalyzed Enantioselective Addition of Two Distinct Nucleophiles across Alkenes Capable of Quinone Methide Formation. J. Am. Chem. Soc. 2009, 131, 17074–17075. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.H.; Webb, J.D.; Sigman, M.S. Advancing the Mechanistic Understanding of an Enantioselective Palladium-Catalyzed Alkene Difunctionalization Reaction. J. Am. Chem. Soc. 2010, 132, 17471–17482. [Google Scholar] [CrossRef] [PubMed]
- Dohi, T.; Toyoda, Y.; Nakae, T.; Koseki, D.; Kubo, H.; Kamitanaka, T.; Kita, Y. Phenol and aniline oxidative coupling with alkenes by using hypervalent iodine dimer for the rapid access to dihydrobenzofurans and indolines. Heterocylces 2015, 90, 631–644. [Google Scholar]
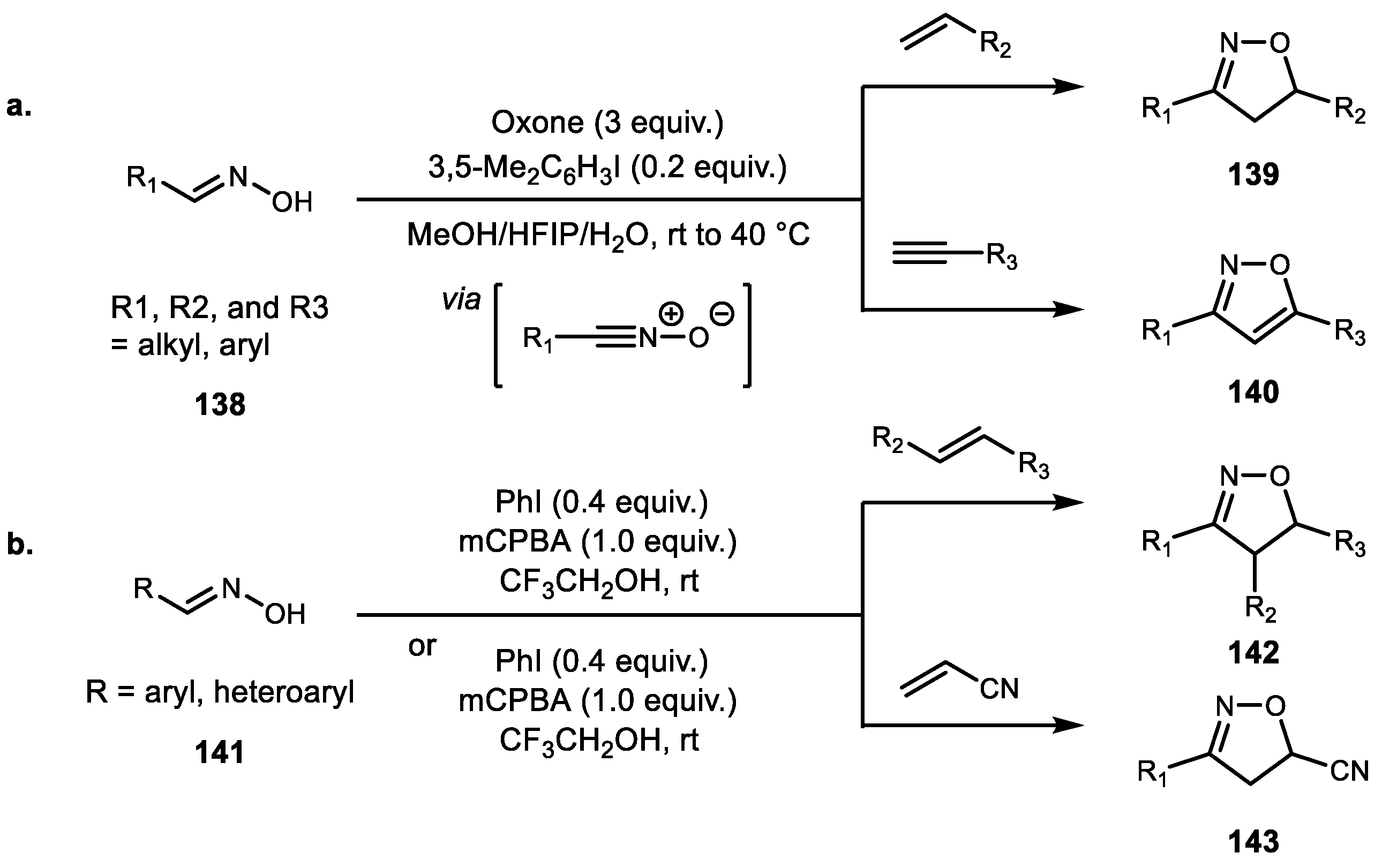
- Yoshimura, A.; Middleton, K.R.; Todora, A.D.; Kastern, B.J.; Koski, S.R.; Maskaev, A.V.; Zhdankin, V.V. Hypervalent Iodine Catalyzed Generation of Nitrile Oxides from Oximes and their Cycloaddition with Alkenes or Alkynes. Org. Lett. 2013, 15, 4010–4013. [Google Scholar] [CrossRef] [PubMed]
- Xiang, C.; Li, T.; Yan, J. Hypervalent Iodine–Catalyzed Cycloaddition of Nitrile Oxides to Alkenes. Synth. Commun. 2014, 44, 682–688. [Google Scholar] [CrossRef]
- Kumar, R.; Kumar, M.; Prakash, O. A Simple and Efficient Method for the Synthesis of Novel Pyrazolyl Isoxazoline Derivatives Using Hypervalent Iodine (III) Reagent. Heteroat. Chem. 2016, 27, 228–234. [Google Scholar] [CrossRef]
- Nocquet-Thibault, S.; Retailleau, P.; Cariou, K.; Dodd, R.H. Iodine(III)-Mediated Umpolung of Bromide Salts for the Ethoxybromination of Enamides. Org. Lett. 2013, 15, 1842–1845. [Google Scholar] [CrossRef]
- Nocquet-Thibault, S.; Minard, C.; Retailleau, P.; Cariou, K.; Dodd, R.H. Iodine(III)-mediated ethoxychlorination of enamides with iron(III) chloride. Tetrahedron 2014, 70, 6769–6775. [Google Scholar] [CrossRef]
- Nocquet-Thibault, S.; Rayar, A.; Retailleau, P.; Cariou, K.; Dodd, R.H. Iodine(III)-Mediated Diazidation and Azido-oxyamination of Enamides. Chem. Eur. J. 2015, 21, 14205–14210. [Google Scholar] [CrossRef] [PubMed]
- Shimogaki, M.; Fujita, M.; Sugimura, T. Metal-Free Enantioselective Oxidative Arylation of Alkenes: Hypervalent-Iodine-Promoted Oxidative C–C Bond Formation. Angew. Chem. Int. Ed. 2016, 55, 15797–15801. [Google Scholar] [CrossRef] [PubMed]
- Shimogaki, M.; Fujita, M.; Sugimura, T. Enantioselective C-C Bond Formation during the Oxidation of 5-Phenylpent-2-enyl Carboxylates with Hypervalent Iodine(III). J. Org. Chem. 2017, 82, 11836–11840. [Google Scholar] [CrossRef] [PubMed]
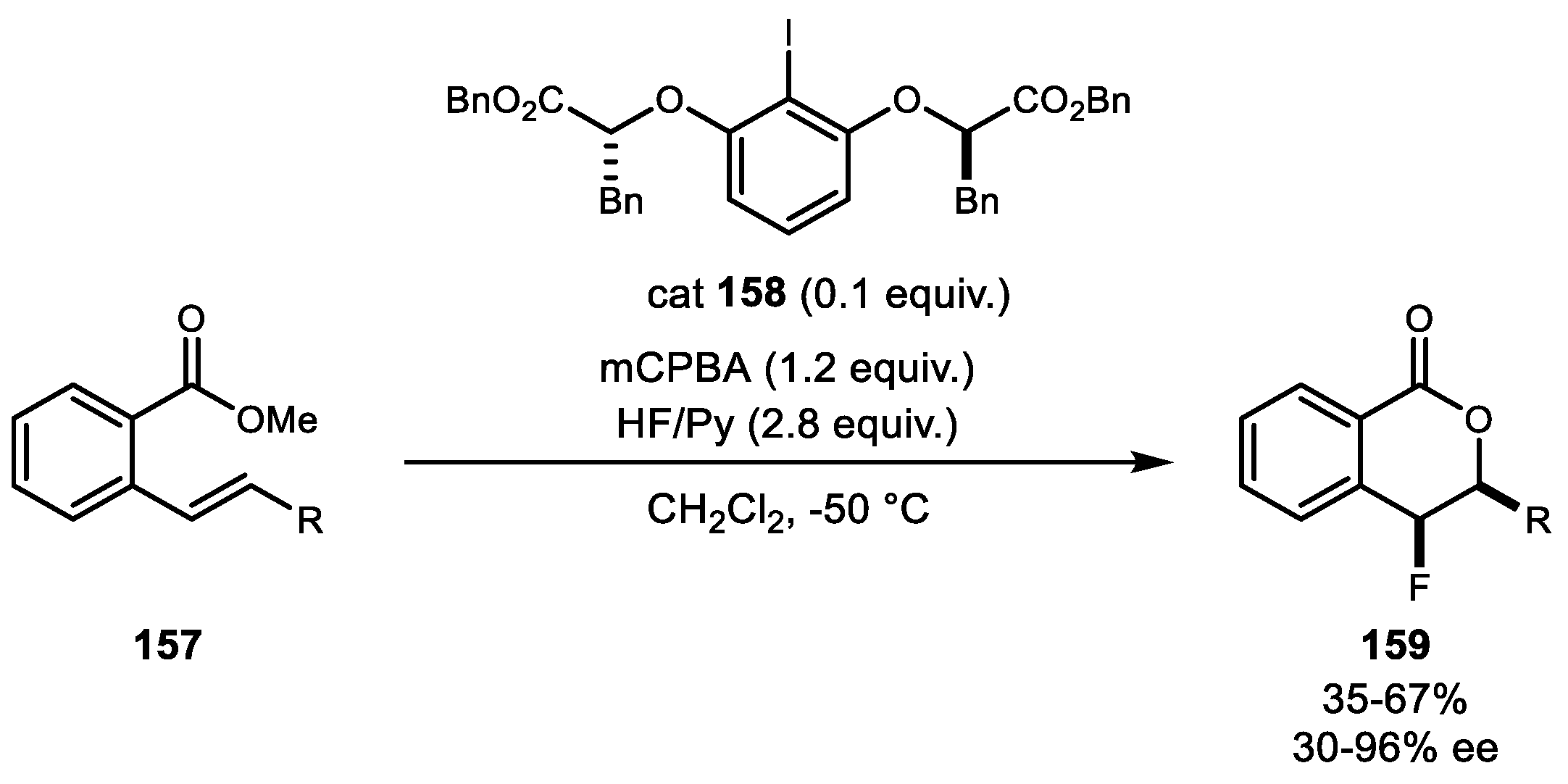
- Woerly, E.M.; Banik, S.M.; Jacobsen, E.N. Enantioselective, Catalytic Fluorolactonization Reactions with a Nucleophilic Fluoride Source. J. Am. Chem. Soc. 2016, 138, 13858–13861. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, S.; Yan, J. PhI Catalyzed Acetoxyselenylation and Formyloxyselenylation of Alkenes. Helv. Chim. Acta 2017, 100, e1600306. [Google Scholar] [CrossRef]
- Ngatimin, M.; Gartshore, C.J.; Kindler, J.P.; Naidu, S.; Lupton, D.W. The α-halogenation of α,β-unsaturated carbonyls and dihalogenation of alkenes using bisacetoxyiodobenzene/pyridine hydrohalides. Tetrahedron Lett. 2009, 50, 6008–6011. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Simmons, N.L.; Ying, Y.; Heretsch, P.M.; Chen, J.S. Enantioselective Dichlorination of Allylic Alcohols. J. Am. Chem. Soc. 2011, 133, 8134–8137. [Google Scholar] [CrossRef]
- Hara, S.; Nakahigashi, J.; Ishi-i, K.; Sawaguchi, M.; Sakai, H.; Fukuhara, T.; Yoneda, N. Difluorination of Alkenes with Iodotoluene Difluoride. Synlett 1998, 1998, 495–496. [Google Scholar] [CrossRef]
- Molnár, I.G.; Gilmour, R. Catalytic Difluorination of Olefins. J. Am. Chem. Soc. 2016, 138, 5004–5007. [Google Scholar] [CrossRef]
- Scheidt, F.; Schäfer, M.; Sarie, J.C.; Daniliuc, C.G.; Molloy, J.J.; Gilmour, R. Enantioselective, Catalytic Vicinal Difluorination of Alkenes. Angew. Chem. Int. Ed. 2018, 57, 16431–16435. [Google Scholar] [CrossRef]
- Banik, S.M.; Medley, J.W.; Jacobsen, E.N. Catalytic, Diastereoselective 1,2-Difluorination of Alkenes. J. Am. Chem. Soc. 2016, 138, 5000–5003. [Google Scholar] [CrossRef] [PubMed]
- Grayfer, T.D.; Retailleau, P.; Dodd, R.H.; Dubois, J.; Cariou, K. Chemodivergent, Tunable, and Selective Iodine(III)-Mediated Bromo-Functionalizations of Polyprenoids. Org. Lett. 2017, 19, 4766–4769. [Google Scholar] [CrossRef] [PubMed]
- Peilleron, L.; Grayfer, T.D.; Dubois, J.; Dodd, R.H.; Cariou, K. Iodine(III)-mediated halogenations of acyclic monoterpenoids. Beilstein J. Org. Chem. 2018, 14, 1103–1111. [Google Scholar] [CrossRef] [PubMed]


































© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.H.; Choi, S.; Hong, K.B. Alkene Difunctionalization Using Hypervalent Iodine Reagents: Progress and Developments in the Past Ten Years. Molecules 2019, 24, 2634. https://doi.org/10.3390/molecules24142634
Lee JH, Choi S, Hong KB. Alkene Difunctionalization Using Hypervalent Iodine Reagents: Progress and Developments in the Past Ten Years. Molecules. 2019; 24(14):2634. https://doi.org/10.3390/molecules24142634
Chicago/Turabian StyleLee, Ji Hoon, Sungwook Choi, and Ki Bum Hong. 2019. "Alkene Difunctionalization Using Hypervalent Iodine Reagents: Progress and Developments in the Past Ten Years" Molecules 24, no. 14: 2634. https://doi.org/10.3390/molecules24142634
APA StyleLee, J. H., Choi, S., & Hong, K. B. (2019). Alkene Difunctionalization Using Hypervalent Iodine Reagents: Progress and Developments in the Past Ten Years. Molecules, 24(14), 2634. https://doi.org/10.3390/molecules24142634