Abstract
Decarboxylative aldol reaction of aliphatic carboxylic acids is a useful method for C–C bond formation because carboxylic acids are an easily available class of compounds. In this study, we found that the decarboxylative aldol reaction of tertiary β-ketocarboxylic acids and trifluoropyruvates proceeded smoothly to yield the corresponding aldol products in high yields and with high diastereoselectivity in the presence of a tertiary amine catalyst. In this reaction, we efficiently constructed a quaternary carbon center and an adjacent trifluoromethylated carbon center. This protocol was also extended to an enantioselective reaction with a chiral amine catalyst, and the desired product was obtained with up to 73% enantioselectivity.
1. Introduction
Decarboxylative functionalization of aliphatic carboxylic acids is a highly useful method in synthetic organic chemistry [1,2,3,4,5,6,7,8,9] because the carboxyl group is a fundamental and easily available functional group. In particular, the decarboxylative aldol reaction of β-oxocarboxylic acids has been intensively studied in recent years [2,5,6,10,11] because these acids easily release carbon dioxide to serve as a formal enolate equivalent [12,13,14,15,16,17,18,19,20]. A number of decarboxylative aldol reactions with primary and secondary β-oxocarboxylic acids have been reported, including a catalytic enantioselective version [2,5,6,10,11,21,22,23]. Most of these reactions are thought to proceed via the aldol adduct of the β-oxocarboxylic acid and the acceptor, followed by decarboxylative protonation, to yield the desired product (Figure 1a). On the other hand, decarboxylative aldol reactions with tertiary carboxylic acids have rarely been reported. To the best of our knowledge, there is only one report on the decarboxylative aldol reaction with tertiary β-oxocarboxylic acids to form an all-carbon quaternary carbon center [24], and there is no report on the enantioselective version, although there are some examples of reactions with α,α-difluoro- or α-monofluorocarboxylic acids to form a fluorinated tetrasubstituted carbon center [25,26,27,28]. These reactions proceed via an enol (or enolate) intermediate formed by decarboxylation of the β-oxocarboxylic acid, and the intermediate reacts with the aldol acceptor to give the desired product (Figure 1b). Therefore, the difficulty in the decarboxylative aldol reaction of tertiary carboxylic acids probably arises from the high steric hinderance of the trisubstituted enolate intermediate. Recently, our research group found that the decarboxylative halogenation of β-ketocarboxylic acids and 2-pyridylcarboxylic acids proceeds even with tertiary carboxylic acids as the substrate [29,30,31,32]. We hypothesized that this smooth decarboxylative functionalization of tertiary carboxylic acids was due to the high electrophilicity of the electrophilic halogenating agents, such as N-chlorosuccinimide (NCS), N-fluorobenzenesulfonimide (NFSI), and Selectfluor. Our previous results encouraged us to develop a decarboxylative aldol reaction of β-ketocarboxylic acids by employing a highly electrophilic aldol acceptor. Trifluoropyruvates are known to be good aldol acceptors with high electrophilicity [33,34,35,36,37,38], and the resulting aldol products, which have a trifluoromethylated stereogenic carbon center, are attractive building blocks for medicinally relevant compounds [39,40,41,42,43,44]. Therefore, we examined the decarboxylative aldol reaction of tertiary β-ketocarboxylic acids with trifluoropyruvates to form the corresponding aldol adduct (Figure 1c), and we disclose the results herein, including the application of this reaction to enantioselective synthesis.
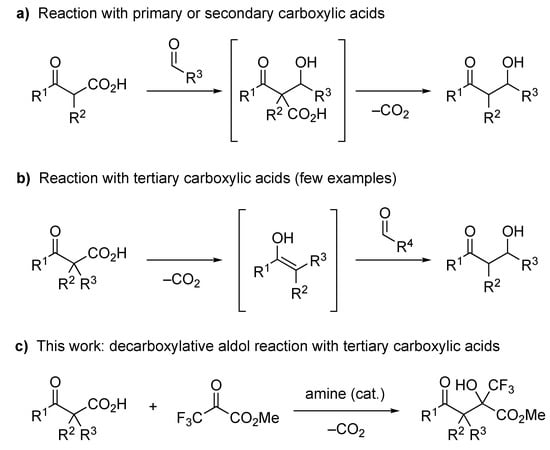
Figure 1.
Decarboxylative aldol reactions of β-oxocarboxylic acids: (a) Reaction with primary and secondary carboxylic acids; (b) Reaction with tertiary carboxylic acids; (c) This work.
2. Results and Discussion
First, we chose tetralone-derived β-ketocarboxylic acids 1a as the model substrate and then screened various amine catalysts for the decarboxylative aldol reaction with methyl trifluoropyruvate as the acceptor (Table 1). Tertiary amines showed superior catalytic activity than primary and secondary amines (entries 1–8). Bicyclic tertiary amines with bridgehead nitrogen(s) showed particularly high reactivity, diastereoselectivity (92:8–93:7 dr), and chemoselectivity between aldol product 2a and protonated product 3a (entries 7 and 8). Taking the reactivity, selectivity, and cost into account, we chose 1,4-diazabicyclo [2.2.2]octane (DABCO) as the best catalyst. Decreasing the catalyst loading to 15 mol% led to a satisfactory level of reactivity and selectivity (entry 9). Optimization of various reaction solvents revealed that toluene was the best choice for the model reaction (entries 9–13).

Table 1.
Optimization of the amine catalyst 1.
With the optimized reaction conditions in hand, next we focused on the substrate scope of the reaction. The results are summarized in Figure 2. The use of ethyl trifluoropyruvate gave the corresponding aldol product 2b in good yield and with high diastereoselectivity. Substituents on the benzoyl moiety or at the α-position of the carbonyl group had little effect on the reactivity and diastereoselectivity (2c and 2d). Indanone-derived β-ketocarboxylic acids 1e and 1f also afforded the corresponding products 2e and 2f in high yields and with high diastereoselectivity, whereas benzosuberone- and cyclohexanone-derived substrates 1g and 1h yielded poor diastereoselectivity of the respective products 2g and 2h. This reaction was also applicable to an acyclic substrate 1i. In addition, the reaction of α-fluoro-β-ketocarboxylic acids furnished the desired tertiary fluorides 2j and 2k in high yields, and with good diastereoselectivity.
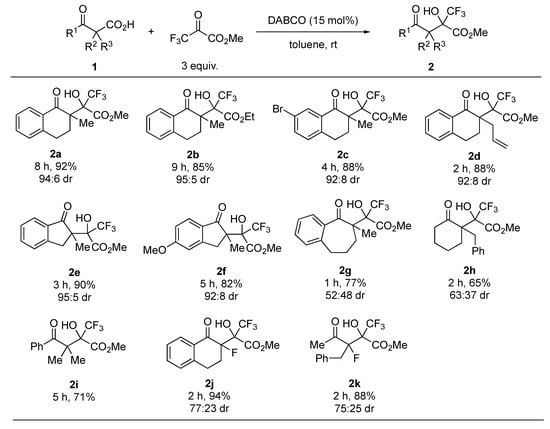
Figure 2.
Substrate scope 1. 1 All reactions were carried out at room temperature in toluene with 3 equiv. of trifluoropyruvates in the presence of 15 mol% DABCO. Isolated yields were described. Diastereomeric ratios were determined by 1H-NMR analyses.
Next, we attempted to apply our method to catalytic enantioselective synthesis. First, we employed chiral primary amine C1 for the reaction of 1a with methyl trifluoropyruvate in toluene, because C1 showed high enantioselectivity in the decarboxylative chlorination of 1a in our previous study. To our surprise, the present reaction only yielded the desired aldol adduct in a moderate yield, with no enantioselectivity (Table 2, entry 1). The probable reason for this result was that the primary amine moiety of C1 reacted with the highly reactive keto carbonyl group of methyl trifluoropyruvate to form an imine or a hemiaminal, thereby causing C1 to lose its catalytic activity. Considering that quinuclidine and DABCO showed high catalytic activity (Table 1), we used cinchona alkaloids and their derivatives in the reaction of 1a and methyl trifluoropyruvate. As expected, all the tested catalysts showed high activity, with chemo- and diastereo-selectivity (Table 2, entries 2–9). Among simple cinchona alkaloids C2–C5, cinchonine C4 showed the best enantioselectivity of 56% ee (entries 2–5). Introducing benzoyl and thiourea moieties at the hydroxy group of cinchonine did not improve the enantioselectivity (entries 6 and 7). The use of dimeric cinchona alkaloids C8 and C9 also led to decreased enantioselectivity (entries 8 and 9). Therefore, we selected cinchonine as the best catalyst at present. Optimization of solvents revealed that the reaction showed higher enantioselectivity (73% ee, entry 12) in acetonitrile.
Table 2.
Screening of chiral amine catalyst 1.
Finally, we performed enantioselective decarboxylative aldol reactions with several β-ketocarboxylic acids (Figure 3). The use of ethyl trifluoropyruvate caused a slight decrease in both the diastereoselectivity and enantioselectivity (see 2b). The reaction of 7-bromotetralone-derived β-ketocarboxylic acid 1c with methyl trifluoropyruvate gave the corresponding product with high diastereoselectivity and good enantioselectivity. Introducing allyl and fluoro groups at the α-position of the keto carbonyl group of the substrate significantly decreased the enantioselectivity (2d and 2i). The use of indanone- and benzosuberone-derived substrates 1e and 1g, as well as acyclic substrate 1i, resulted in poor enantioselectivity.
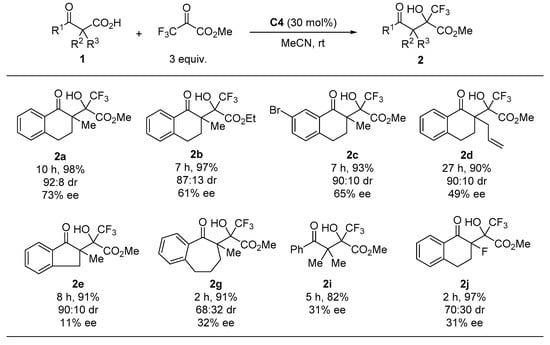
Figure 3.
Enantioselective decarboxylative aldol reaction of 1 1. 1 All reactions were carried out at room temperature in acetonitrile with 3 equiv. of trifluoropyruvates in the presence of 30 mol% C4. Isolated yields were described. Diastereomeric ratios were determined by 1H-NMR analyses. Enantiomeric excess was determined by chiral HPLC analysis.
In conclusion, we revealed that the decarboxylative aldol reaction of tertiary β-ketocarboxylic acids with trifluoropyruvates proceeded smoothly in the presence of a tertiary amine catalyst to afford the corresponding aldol product with high diastereoselectivity. We also examined a catalytic enantioselective reaction examined using cinchonine as the catalyst to obtain the product with moderate-to-good enantioselectivity (up to 73% ee). This was the first example of the enantioselective decarboxylative aldol reaction of tertiary β-ketocarboxylic acids to form an all-carbon quaternary stereogenic center. Since the reaction led to the simultaneous formation of a trifluoromethylated chiral carbon and an adjacent quaternary chiral carbon, it would be useful for the preparation of medicinally relevant compounds.
3. Materials and Methods
3.1. General
All non-aqueous reactions were carried out in dried glassware under an argon atmosphere and stirred using magnetic stir-plates. Thin-layer chromatography analyses were performed using pre-coated silica gel plates with a fluorescent indicator (F254) (Merck Millipore, Darmstadt, Germany). Visualization was accomplished using ultraviolet (UV) light (254 nm), phosphomolybdic acid, or p-anisaldehyde. Flash column chromatography was performed using silica gel 60 (mesh size 40–100) supplied by Kanto Chemical Co., Inc. (Tokyo, Japan). 1H, 13C, and 19F nuclear magnetic resonance (NMR) spectra were recorded on a JNM-ECS400 (400 MHz 1H, 100 MHz 13C, 376 MHz 19F) or a JNM-ECX500 (500 MHz 1H, 126 MHz 13C, 470 MHz 19F) instrument (JEOL Ltd., Tokyo, Japan). Chemical shift values (δ) are reported in ppm (tetramethylsilane δ 0.00 ppm for 1H; hexafluorobenzene δ −162.2 ppm for 19F; residual chloroform δ 77.0 ppm for 13C). Direct analyses in real time (DART) mass (positive mode) analyses were performed on a JMS-T100TD time-of-flight mass spectrometer (JEOL Ltd., Tokyo, Japan). Optical rotations were measured on a P-1030 digital polarimeter (JASCO Co., Ltd., Tokyo, Japan). Analytical high-performance liquid chromatography (HPLC) was performed on a PU1586 instrument with a MD-2018 plus diode array detector (JASCO Co., Ltd., Tokyo, Japan) using a chiral column under the conditions described below. The enantiomeric purity of the compounds was determined by HPLC analysis using chiral stationary phase columns. The 1H, 13C, and 19F-NMR spectra of compounds 1c and 2 and HPLC data of compounds 2 are available in the Supplementary Material.
3.2. Materials
Commercial grade reagents and solvents were used without further purification unless otherwise noted. Anhydrous acetonitrile was purchased from Sigma-Aldrich (St. Louis, MO, USA). Anhydrous toluene, dichloromethane, tetrahydrofurane (THF) were purchased from Kanto Chemical Co., Inc. (Tokyo, Japan) and they were used after purification via a Glass Contour solvent dispensing system (Pure Process Technology, Nashua, NH, USA). α,α-dialkyl-β-ketocarboxylic acid 1a, 1d, 1e, 1h [29], 1f, 1g [30], 1i [45], 1j, 1k [31], catalyst C6 [46], and C7 [47] were prepared according to literature procedures.
3.3. Synthesis of α,α-dialkyl-β-ketocarboxylic Acid 1c
Potassium hydroxide (22.0 mmol) was added to a solution of methyl 7-bromo-2-methyl-1-oxo-1,2,3,4-tetrahydronaphthalene-2-carboxylate (660 mg, 2.2 mmol) in MeOH, H2O, and CH2Cl2 (6 mL, 3mL, 0.6 mL), and the mixture was stirred at ambient temperature. After the reaction completed, the reaction mixture was diluted with H2O and washed with ethyl acetate. The resulting aqueous layer was acidified with 1.2N HCl until the solution became pH 1.0, and then extracted with CH2Cl2. The combined organic layer was dried over Na2SO4 and concentrated. The title compound was obtained as a colorless powder (430 mg, 68% yield).
1H-NMR (500 MHz) δ 8.18 (d, J = 2.3 Hz, 1H), 7.61 (d, J = 8.0, 2.3 Hz, 1H), 7.14 (d, J = 8.0 Hz, 1H), 3.07–3.00 (m, 1H), 2.96–2.90 (m, 1H), 2.57–2.52 (m, 1H), 2.19–2.14 (m, 1H), 1.54 (m, 3H); 13C-NMR (126 MHz) δ195.8, 142.0, 136.7, 132.6, 131.0, 130.6, 121.0, 32.8, 25.3, 20.8; HRMS (DART): [M + NH4]+ calcd. for C12H15Br1N1O3, 300.02353; found, 300.02335.
3.4. General Procedure for the Decarboxylative Aldol Reaction
α,α-Dialkyl-β-ketocarboxylic acid 1 was added to a stirred solution of DABCO (15 mol%) and trifluoropyruvates (3 equiv.) in toluene (0.1 M). Then, the mixture was stirred at ambient temperature. The reaction mixture was directly subjected to silica gel column chromatography purification to give aldol products. Some starting compounds 1 contained 2–10% of decarboxylated by-product and used as is in the aldol reaction, because they decomposed spontaneously even at ambient temperature.
Methyl 3,3,3-trifluoro-2-hydroxy-2-(2-methyl-1-oxo-1,2,3,4-tetrahydronaphthalen-2-yl)propanoate (2a)
Following general procedure, the reaction of 1a (0.53 mmol) was stirred for 7.5 h. The crude product was purified by flash column chromatography (hexane: ethyl acetate = 10: 1) to provide 2a (92% yield 152.0 mg, 94:6 d.r.) as a colorless oil. The diastereomeric ratio of 2a was determined to be 94:6 by the analysis of 1H-NMR.
1H-NMR (500 MHz) δ 7.98 (dd, J = 8.0, 1.2 Hz, 1H), 7.52 (dt, J = 7.6, 1.2 Hz, 1H), 7.32 (t, J = 7.6 Hz, 1H), 7.26 (d, J = 8.0 Hz, 1H), 5.02 (s, 1H), 3.76 (s, 3H), 3.22–3.15 (m, 1H), 3.01–2.88 (m, 2H), 2.23–2.18 (m, 1H), 1.55 (s, 3H); 13C-NMR (126 MHz) δ 201.6, 169.8, 142.5, 134.2, 130.2, 128.6, 128.2, 126.9, 123.4 (q, J = 287.9 Hz), 80.6 (q, J = 27.6 Hz), 53.6, 51.5, 30.5, 25.3, 16.7; 19F-NMR (470 MHz) δ –71.3; HRMS (DART): [M + NH4]+ calcd. for, C15H19F3N1O4, 334.1266; found, 334.1267.
Ethyl 3,3,3-trifluoro-2-hydroxy-2-(2-methyl-1-oxo-1,2,3,4-tetrahydronaphthalen-2-yl)propanoate (2b)
Following general procedure, the reaction of 1a (0.49 mmol) was stirred for 9 h. The crude product was purified by flash column chromatography (hexane: ethyl acetate = 10: 1) to provide 2b (85% yield 136.6 mg, 95:5 d.r.) as a colorless oil. The diastereomeric ratio of 2b was determined to be 95:5 by the analysis of 1H-NMR.
1H-NMR (500 MHz) δ 7.99 (dd, J = 8.0, 1.2 Hz, 1H), 7.52 (dt, J = 7.6, 1.2 Hz, 1H), 7.32 (t, J = 7.6 Hz, 1H), 7.25 (d, J = 8.0 Hz, 1H), 4.95 (s, 1H), 4.23 (q, J = 7.3 Hz, 2H), 3.22–3.15 (m, 1H), 3.00–2.89 (m, 2H), 2.22–2.18 (m, 1H), 1.55 (s, 3H), 1.17 (t, J = 7.3 Hz, 3H); 13C-NMR (126 MHz) δ 201.2, 169.2, 142.5, 134.1, 130.3, 128.6, 128.2, 126.9, 123.5 (q, J = 291.5 Hz), 80.6 (q, J = 27.6 Hz), 63.1, 51.4, 30.6, 25.4, 16.7, 13.7; 19F-NMR (470 MHz) δ –71.3; HRMS (DART): [M + H]+ calcd. for, C16H18F3O4, 331.1157; found, 331.1156.
Methyl 2-(7-bromo-2-methyl-1-oxo-1,2,3,4-tetrahydronaphthalen-2-yl)-3,3,3-trifluoro-2-hydroxypropanoate (2c)
Following general procedure, the reaction of 1c (0.18 mmol) was stirred for 4 h. The crude product was purified by flash column chromatography (hexane: ethyl acetate = 10: 1) to provide 2c (88% yield 59.8 mg, 92:8 d.r.) as a colorless oil. The diastereomeric ratio of 2c was determined to be 92:8 by the analysis of 1H-NMR.
1H-NMR (500 MHz) δ 8.01 (d, J = 1.9 Hz, 1H), 7.62 (dd, J = 8.0, 1.9 Hz, 1H), 7.15 (d, J = 8.0 Hz, 1H), 4.83 (s, 1H), 3.79 (s, 3H), 3.14–3.06 (m, 1H), 2.97–2.86 (m, 2H), 2.22–2.18 (m, 1H), 1.53 (s, 3H); 13C-NMR (126 MHz) δ 200.1, 169.7, 141.1, 136.9, 131.7, 130.9, 130.3, 123.3 (q, J = 289.1 Hz), 120.9, 80.4 (q, J = 27.6 Hz), 53.8, 51.4, 30.3, 24.9, 16.6; 19F-NMR (470 MHz) δ −71.4; HRMS (DART): [M + H]+ calcd. for, C15H15BrF3O4, 395.0106; found, 395.0105.
Methyl 2-(2-allyl-1-oxo-1,2,3,4-tetrahydronaphthalen-2-yl)-3,3,3-trifluoro-2-hydroxypropanoate (2d)
Following general procedure, the reaction of 1d (0.32 mmol) including 10% of decarboxylative product was stirred for 2 h. The crude product was purified by flash column chromatography (hexane: ethyl acetate = 10: 1) to provide 2d (88% yield 154.7 mg, 92:8 d.r.) as a pale yellow oil. The diastereomeric ratio of 2d was determined to be 92:8 by the analysis of 1H-NMR.
1H-NMR (500 MHz) δ 7.97–7.95 (m, 1H), 7.51 (m, 1H), 7.31 (t, J = 7.6 Hz, 1H), 7.23 (d, J = 7.6 Hz, 1H), 5.85–5.77 (m, 1H), 5.00 (s, 1H), 4.98–4.93 (m, 2H), 3.72 (s, 3H), 3.25–3.18 (m, 1H), 2.99–2.93 (m, 2H), 2.84–2.78 (m, 1H), 2.70–2.65 (m, 1H), 2.34–2.31 (m, 1H); 13C-NMR (126 MHz) δ 200.2, 169.9, 142.8, 134.5, 134.2, 132.0, 128.7, 128.1, 127.0, 123.4 (q, J = 287.9 Hz), 118.4, 81.5 (q, J = 27.6 Hz), 54.6, 53.7, 36.6, 29.0 (d, J = 3.6 Hz), 25.4; 19F-NMR (470 MHz) δ −70.4; HRMS (DART): [M + H]+ calcd. for, C17H18F3O4, 343.1157; found, 343.1159.
Methyl 3,3,3-trifluoro-2-hydroxy-2-(2-methyl-1-oxo-2,3-dihydro-1H-inden-2-yl)propanoate (2e)
Following general procedure, the reaction of 1e (0.38 mmol) including 9% of decarboxylative product was stirred for 3 h. The crude product was purified by flash column chromatography (hexane: ethyl acetate = 20: 3) to provide 2e (90% yield 108.8 mg, 95:5 d.r.) as a colorless oil. The diastereomeric ratio of 2e was determined to be 95:5 by the analysis of 1H-NMR.
1H-NMR (500 MHz) δ 7.77 (d, J = 7.6 Hz, 1H), 7.67 (m, 1H), 7.47 (d, J = 8.0 Hz, 1 H), 7.42 (t, J = 7.5 Hz, 1H), 5.32 (s, 1H), 3.84 (d, J = 17.6 Hz, 1H), 3.59 (s, 3H), 2.97 (d, J = 17.6 Hz, 1H), 1.47 (s, 3H); 13C-NMR (126 MHz) δ 208.3, 168.5, 152.4, 136.1, 133.5, 127.9, 126.5, 124.8, 123.0 (q, J = 287.9 Hz), 80.4 (q, J = 28.8 Hz), 53.3, 51.6, 38.3, 20.6; 19F-NMR (470 MHz) δ −72.0; HRMS (DART): [M + H]+ calcd. for, C14H14F3O4, 303.0844; found, 303.0846.
Methyl 3,3,3-trifluoro-2-hydroxy-2-(5-methoxy-2-methyl-1-oxo-2,3-dihydro-1H-inden-2-yl)propanoate (2f)
Following general procedure, the reaction of 1f (0.29 mmol) including 6% of decarboxylative product was stirred for 5 h. The crude product was purified by flash column chromatography (hexane: ethyl acetate = 10: 1) to provide 2f (82% yield 79.7 mg, 92:8 d.r.) as a colorless oil. The diastereomeric ratio of 2f was determined to be 92:8 by the analysis of 1H-NMR.
1H-NMR (500 MHz) δ 7.70 (d, J = 8.4 Hz, 1H), 6.94 (dd, J = 8.4, 2.9 Hz, 1H), 6.88 (d, J = 1.9 Hz, 1H), 5.63 (s, 1H), 3.90 (s, 3H), 3.81 (d, J = 17.8, 1H), 3.56 (s, 3H), 2.93 (d, J = 17.8 Hz, 1H), 1.47 (d, J = 1.2 Hz, 3H); 13C-NMR (126 MHz) δ 206.7, 168.5, 166.4, 155.9, 126.6, 126.4, 123.1 (q, J = 287.9 Hz), 116.3, 109.4, 80.6 (q, J = 28.8 Hz), 55.7, 53.2, 51.4, 38.3, 20.7; 19F-NMR (470 MHz) δ −72.0; HRMS (DART): [M + H]+ calcd. for, C15H16F3O5, 333.0950; found, 333.0951.
Methyl 3,3,3-trifluoro-2-hydroxy-2-(6-methyl-5-oxo-6,7,8,9-tetrahydro-5H-benzo[7]annulen-6-yl)propanoate (2g)
Following general procedure, the reaction of 1g (0.40 mmol) including 9% of decarboxylative product was stirred for 1 h. The crude product was purified by flash column chromatography (hexane: ethyl acetate = 10: 1) to provide 2g (77% yield 101.7 mg, 52:48 d.r.) as a colorless oil. The diastereomeric ratio of 2g was determined to be 52:48 by the analysis of 1H-NMR.
Major diastereomer; 1H-NMR (500 MHz) δ 7.40–7.32 (m, 1H), 7.28–7.21 (m, 2H), 7.13–7.11 (m, 1H), 4.49 (s, 1H), 3.90 (s, 3H), 2.87–2.85 (m, 2H), 2.61–2.57 (m, 1H), 2.06–1.92 (m, 3H), 1.42 (s, 3H); 13C-NMR (126 MHz) δ 209.8, 170.2, 140.2, 137.1, 130.7, 128.5, 128.0, 126.5, 123.8 (q, J = 289.1 Hz), 79.9 (q, J = 27.6 Hz), 57.8, 53.9, 34.0, 32.3, 23.2, 18.7; 19F-NMR (470 MHz) δ; –70.1; Minor diastereomer; 1H-NMR (500 MHz) δ 7.40–7.32 (m, 1H), 7.28–7.21 (m, 2H), 7.13–7.11 (m, 1H), 4.52 (s, 1H), 3.99 (s, 3H), 2.96–2.90 (m, 1H), 2.77–2.71 (m, 1H), 2.28–2.26 (m, 1H), 2.06–1.92 (m, 3H), 1.42 (s, 3H); 13C-NMR (126 MHz) δ 212.0, 170.3, 140.2, 137.3, 131.3, 128.4, 127.7, 126.5, 123.8 (q, J = 289.1 Hz), 81.4 (q, J = 27.6 Hz), 56.2, 54.3, 32.2, 30.4, 22.8, 19.5; 19F-NMR (470 MHz) δ −70.5; HRMS (DART): [M + H]+ calcd. for, C16H18F3O4, 331.1157; found, 331.1158.
Methyl 2-(1-benzyl-2-oxocyclohexyl)-3,3,3-trifluoro-2-hydroxypropanoate (2h)
Following general procedure, the reaction of 1h (0.88 mmol) including 2% of decarboxylative product was stirred for 2 h. The crude product was purified by flash column chromatography (hexane: ethyl acetate = 20: 3) to provide 2h (65% yield 179.2 mg, 63:37 d.r.) as a colorless oil. The diastereomeric ratio of 2h was determined to be 63:37 by the analysis of 1H-NMR.
Major diastereomer; 1H-NMR (500 MHz) δ 7.27–7.08 (m, 5H), 3.84 (s, 3H), 3.64 (d, J = 14.1 Hz, 1 H), 3.03 (d, J = 14.1 Hz, 1H), 2.59–2.41 (m, 1H), 2.38–2.27 (m,3H), 2.20–2.12 (m, 1H), 1.84–1.23 (m, 4H); 13C-NMR (126 MHz) δ 215.1, 170.0, 137.2, 130.8, 128.3, 126.9, 123.3 (q, J = 289.1 Hz), 82.0 (q, J = 27.6 Hz), 58.4, 53.7, 42.3, 40.8, 33.2, 23.8, 21.4; 19F-NMR (470 MHz) δ –69.8; Minor diastereomer; 1H-NMR (500 MHz) δ 7.27–7.08 (m, 5H), 4.02 (s, 3H), 3.75 (d, J = 13.2 Hz, 1H), 2.93 (d, J = 14.1 Hz, 1H), 2.59–2.41 (m, 1H), 2.38–2.27 (m,3H), 2.20–2.12 (m, 1H), 1.84–1.23 (m, 4H); 13C-NMR (126 MHz) δ 214.1, 169.9, 136.7, 130.7, 128.5, 126.9, 123.3 (q, J = 289.1 Hz), 81.0 (q, J = 27.6 Hz), 59.4, 54.2, 42.8, 41.9, 30.7, 22.0, 21.5; 19F-NMR (470 MHz) δ −69.5; HRMS (DART): [M + NH4]+ calcd. for, C17H23F3N1O4, 362.1579; found, 362.1578.
Methyl 2-hydroxy-3,3-dimethyl-4-oxo-4-phenyl-2-(trifluoromethyl)butanoate (2i)
Following general procedure, the reaction of 1i (0.52 mmol) was stirred for 4.5 h. The crude product was purified by flash column chromatography (hexane: ethyl acetate = 6: 1) to provide 2i (71% yield 112.5 mg) as a colorless oil.
1H-NMR (500 MHz) δ 7.52–7.38 (m, 5H), 4.57 (s, 1H), 3.96 (s, 3H), 1.60 (dd, J = 33.8, 1.0 Hz, 6H); 13C-NMR (126 MHz) δ 209.1, 170.2, 138.4, 128.0, 127.1, 123.7 (q, J = 287.9 Hz), 80.1 (q, J = 27.6 Hz), 55.1, 54.1, 23.5 (d, J = 2.4 Hz), 22.8 (d, J = 2.4 Hz); 19F-NMR (470 MHz) δ −70.6; HRMS (DART): [M + NH4]+ calcd. for, C14H19F3N1O4, 322.1266; found, 322.1266.
Methyl 3,3,3-trifluoro-2-(2-fluoro-1-oxo-1,2,3,4-tetrahydronaphthalen-2-yl)-2-hydroxypropanoate (2j)
Following general procedure, the reaction of 1j (0.48 mmol) was stirred for 2 h. The crude product was purified by flash column chromatography (hexane: ethyl acetate = 10: 1) to provide 2j (94% yield 144.2 mg, 77:23 d.r.) as a colorless oil. The diastereomeric ratio of 2j was determined to be 77:23 by the analysis of 1H-NMR.
Major diastereomer; 1H-NMR (500 MHz) δ 7.98 (dd, J = 7.6, 1.2 Hz), 7.57 (dt, J = 7.6, 1.2 Hz), 7.36 (t, J = 7.6 Hz), 7.29 (d, J = 7.6 Hz), 4.63 (s, 3H), 3.36–3.30 (m, 1H), 3.01 (dt, J = 17.2, 4.4 Hz, 1H), 2.95–2.88 (m, 1H), 2.78–2.71 (m, 1H); 13C-NMR (126 MHz) δ 191.2 (d, J = 20.4 Hz), 167.4, 143.4, 134.9, 130.3, 128.6, 128.6, 127.2, 122.3 (q, J = 287.9 Hz), 93.4 (d, J = 192.0 Hz), 79.6–78.9 (m), 54.3, 30.5 (dd, J = 22.8, 2.4 Hz), 24.5 (d, J = 7.2 Hz); 19F-NMR (470 MHz) δ −72.7, −172.1; Minor diastereomer; 1H-NMR (500 MHz), 7.99 (dd, J = 8.0, 0.8 Hz, 1H), 7.55 (dt, J = 7.6, 1.2 Hz, 1H), 7.35 (t, J = 7.6 Hz, 1H), 7.29 (8.0 Hz, 1H), 4.36 (s, 1H), 4.04 (s, 3H), 3.29–3.20 (m, 1H), 3.05–3.00 (m, 1H), 2.79–2.65 (m, 1H); 13C-NMR (126 MHz), 189.0 (d, J = 19.2 Hz), 169.4, 143.7, 139.4–139.1 (m), 136.8–136.6 (m), 134.8, 130.3, 128.7, 128.5, 127.2, 122.9 (q, J = 288.5 Hz), 93.6 (d, J = 196.5 Hz), 76.4–75.9 (m), 54.8, 29.4 (d, J = 22.0 Hz), 24.0 (d, J = 4.8 Hz); 19F-NMR (470 MHz); −72.2, −167.4; HRMS (DART): [M + NH4]+ calcd. for, C14H16F4N1O4, 338.1016; found, 338.1014.
Methyl 3-benzyl-3-fluoro-2-hydroxy-4-oxo-2-(trifluoromethyl)pentanoate (2k)
Following general procedure, the reaction of 1k (0.72 mmol) including 2% of decarboxylative product was stirred for 2 h. The crude product was purified by flash column chromatography (hexane: ethyl acetate = 10: 1) to provide 2k (88% yield, 203.8 mg, 75:25 d.r.) as a colorless oil. The diastereomeric ratio of 2k was determined to be 75:25 by the analysis of 1H-NMR.
Major diastereomer; 1H-NMR (500 MHz) δ 7.30–7.25 (m, 3H), 7.15–7.13 (m, 2H), 4.54 (s, 1H), 3.92 (s, 3H), 3.51–3.28 (m, 2H), 1.70 (d, J = 5.7 Hz, 3H); 13C-NMR (126 MHz) δ 208.0 (d, J = 30.0 Hz), 167.6, 132.5, 130.9, 128.4, 127.4, 121.1 (dq, J = 287.3, 2.4 Hz), 100.6 (d, J = 203.3 Hz), 80.1–79.1 (m), 54.4, 38.6 (d, J = 20.4 Hz), 28.0; 19F-NMR (470 MHz) δ −71.8 (d, J = 14.7 Hz), –166.9 (d, J = 44.0 Hz); Minor diastereomer; 1H-NMR (500 MHz) δ 7.30–7.25 (m, 3H), 7.15–7.13 (m, 2H), 4.47 (s, 1H), 4.05 (s, 3H), 3.58–3.36 (m, 2H), 1.70 (d, J = 5.7 Hz, 3H); 13C-NMR (126 MHz) δ 208.6 (d, J = 31.2 Hz), 166.5 (d, J = 6.0 Hz), 130.9, 132.5, 128.5, 127.5, 122.4 (q, J = 287.9 Hz), 101.5 (d, J = 226.3 Hz), 80.1–79.1 (m), 54.9, 39.1 (d, J = 22.8 Hz), 28.3; 19F-NMR (470 MHz) δ −71.9 (d, J = 14.7 Hz), −168.7 (d, J = 44.0 Hz); HRMS (DART): [M + NH4]+ calcd. for, C14H18F4N1O4, 340.1172; found, 340.1173.
3.5. General Procedure of the Enantioselective Decarboxylative Aldol Reaction
α,α-Dialkyl-β-ketocarboxylic acid 1 was added to a stirred solution of cinchonine (30 mol%) and trifluoropyruvates (3.0 equiv.) in toluene (0.1 M). Then, the reaction mixture was stirred at ambient temperature. The mixture was purified by flash column chromatography on silica gel to give aldol products. When the reactions were performed, some starting compounds 1 contained 5–33% of decarboxylated by-product as an impurity, as noted in the description of each compound, because some of 1 decomposed slowly while standing at ambient temperature.
Methyl 3,3,3-trifluoro-2-hydroxy-2-(2-methyl-1-oxo-1,2,3,4-tetrahydronaphthalen-2-yl)propanoate (2a)
Following general procedure, the reaction of 1a (0.30 mmol) was stirred for 10 h. The crude product was purified by flash column chromatography (hexane: ethyl acetate = 10: 1) to provide 2a (98% yield 151.9 mg, 92:8 d.r., 73% e.e. (major), 42% e.e. (minor)) as a colorless oil. The diastereomeric ratio of 2a was determined to be 92:8 by the analysis of 1H-NMR.
[α]D28 = −38.1 (c 0.5, CHCl3) (diastereomeric mixture); Major diastereomer; The enantiomeric purity of the title compound was determined by HPLC analysis (DAICEL CHIRALPAK IC-3, hexane: 2-propanol = 200: 1, flow rate = 1.0 mL/min, retention time; 37.8 min (minor enantiomer) and 44.9 min (major enantiomer)), Minor diastereomer; The enantiomeric purity of the title compound was determined by HPLC analysis (DAICEL CHIRALPAK IC-3, hexane: 2-propanol = 200: 1, flow rate = 1.0 mL/min, retention time; 22.6 min (major enantiomer), 29.4 min (minor enantiomer)).
Ethyl 3,3,3-trifluoro-2-hydroxy-2-(2-methyl-1-oxo-1,2,3,4-tetrahydronaphthalen-2-yl)propanoate (2b)
Following general procedure, the reaction of 1b (0.25 mmol) was stirred for 7 h. The crude product was purified by flash column chromatography (hexane: ethyl acetate = 10: 1) to provide 2b (97% yield 76.5 mg, 87:13 d.r., 61% e.e. (major), 36% e.e. (minor)) as a colorless oil. The diastereomeric ratio of 2b was determined to be 87:13 by the analysis of 1H-NMR.
[α]D28 = −26.5 (c 0.72, CHCl3) (diastereomeric mixture); Major diastereomer; The enantiomeric purity of the title compound was determined by HPLC analysis (DAICEL CHIRALPAK ID and ID-3, hexane: 2-propanol = 100: 1, flow rate = 1.0 mL/min, retention time; 52.7 min (minor enantiomer) and 57.1 (major enantiomer)), Minor diastereomer; The enantiomeric purity of the title compound was determined by HPLC analysis (DAICEL CHIRALPAK ID and ID-3, hexane: 2-propanol = 100: 1, flow rate = 1.0 mL/min, retention time; 32.7 min (major enantiomer), 45.6 min (minor enantiomer)).
Methyl 2-(7-bromo-2-methyl-1-oxo-1,2,3,4-tetrahydronaphthalen-2-yl)-3,3,3-trifluoro-2-hydroxypropanoate (2c)
Following general procedure, the reaction of 1c (0.35 mmol) was stirred for 7 h. The crude product was purified by flash column chromatography (hexane: ethyl acetate = 10: 1) to provide 2c (93% yield 124.0 mg, 90:10 d.r., 65% e.e. (major), 32% e.e. (minor)) as a colorless oil. The diastereomeric ratio of 2c was determined to be 90:10 by the analysis of 1H-NMR.
[α]D28 = −22.7 (c 1.6, CHCl3) (diastereomeric mixture); Major diastereomer; The enantiomeric purity of the title compound was determined by HPLC analyses (DAICEL CHIRALPAK ID-3, hexane: 2-propanol = 50: 1, flow rate = 1.0 mL/min, retention time; 19.9 min (minor enantiomer) and 23.5 min (major enantiomer), Minor diastereomer; The enantiomeric purity of the title compound was determined by HPLC analysis (DAICEL CHIRALPAK ID-3, hexane: 2-propanol = 50: 1, flow rate = 1.0 mL/min, retention time;13.5 min (major enantiomer), 17.7 min (major enantiomer)).
Methyl 2-(2-allyl-1-oxo-1,2,3,4-tetrahydronaphthalen-2-yl)-3,3,3-trifluoro-2-hydroxypropanoate (2d)
Following general procedure, the reaction of 1d (0.32 mmol) including 5% of decarboxylative product was stirred for 27 h. The crude product was purified by flash column chromatography (hexane: ethyl acetate = 10: 1) to provide 2d (90% yield 91.5 mg, 90:10 d.r., 49% e.e. (major), 9% e.e. (minor)) as a pale yellow oil. The diastereomeric ratio of 2d was determined to be 90:10 by the analysis of 1H-NMR.
[α]D28 = −32.3 (c 0.77, CHCl3) (diastereomeric mixture); Major diastereomer; The enantiomeric purity of the title compound was determined by HPLC analysis (DAICEL CHIRALPAK IE-3, hexane: 2-propanol = 200: 1, flow rate = 1.0 mL/min, retention time; 35.5 min (major enantiomer) and 40.2 min (minor enantiomer)), Major diastereomer; The enantiomeric purity of the title compound was determined by HPLC analysis (DAICEL CHIRALPAK IE-3, hexane: 2-propanol = 200: 1, flow rate = 1.0 mL/min, retention time; 22.0 min (major enantiomer), 30.2 min (minor enantiomer)).
Methyl 3,3,3-trifluoro-2-hydroxy-2-(2-methyl-1-oxo-2,3-dihydro-1H-inden-2-yl)propanoate (2e)
Following general procedure, the reaction of 1e (0.38 mmol) including 5% of decarboxylative product was stirred for 3 h. The crude product was purified by flash column chromatography (hexane: ethyl acetate = 20: 3) to provide 2e (91% yield 76.4 mg, 90:10 d.r., 11% e.e. (major), 26% e.e. (minor)) as a colorless oil. The diastereomeric ratio of 2e was determined to be 90:10 by the analysis of 1H-NMR.
Major diastereomer; The enantiomeric purity of the title compound was determined by HPLC analysis (DAICEL CHIRALPAK IE-3, hexane: 2-propanol = 200: 1, flow rate = 1.0 mL/min, retention time; 52.7 min (minor enantiomer) and 68.5 min (major enantiomer)), Minor diastereomer; The enantiomeric purity of the title compound was determined by HPLC analysis (DAICEL CHIRALPAK IE-3, hexane: 2-propanol = 200: 1, flow rate = 1.0 mL/min, retention time; 34.0 min (minor diastereomer, minor enantiomer), 43.2 min (minor diastereomer, major enantiomer)).
Methyl 3,3,3-trifluoro-2-hydroxy-2-(6-methyl-5-oxo-6,7,8,9-tetrahydro-5H-benzo[7]annulen-6-yl)propanoate (2g)
Following general procedure, the reaction of 1g (0.20 mmol) including 33% of decarboxylative product was stirred for 2 h. The crude product was purified by flash column chromatography (hexane: ethyl acetate = 10: 1) to provide 2g (91% yield 66.1 mg, 68:32 d.r., 32% e.e. (major), 16% e.e. (minor)) as a colorless oil. The diastereomeric ratio of 2g was determined to be 52:48 by the analysis of 1H-NMR.
Major diastereomer; The enantiomeric purity of the title compound was determined by HPLC analysis (DAICEL CHIRALPAK IB-3, hexane: 2-propanol = 200: 1, flow rate = 1.0 mL/min, retention time; 20.8 min (major enantiomer) and 33.0 min (minor enantiomer)), Minor diastereomer; The enantiomeric purity of the title compound was determined by HPLC analysis (DAICEL CHIRALPAK IB-3, hexane: 2-propanol = 200: 1, flow rate = 1.0 mL/min, retention time; 12.9 min (major enantiomer), 15.7 min (minor enantiomer)).
Methyl 2-hydroxy-3,3-dimethyl-4-oxo-4-phenyl-2-(trifluoromethyl)butanoate (2i)
Following general procedure, the reaction of 1i (0.39 mmol) was stirred for 4.5 h. The crude product was purified by flash column chromatography (hexane: ethyl acetate = 6: 1) to provide 2i (82% yield 97.3 mg, 31% e.e.) as a colorless oil.
[α]D28 = +7.6 (c 1.2, CHCl3); The enantiomeric purity of the title compound was determined by HPLC analysis (DAICEL CHIRALPAK IA-3, hexane: 2-propanol = 200: 1, flow rate = 1.0 mL/min, retention time; 24.8 min (major enantiomer), 29.5 min (minor enantiomer)).
Methyl 3,3,3-trifluoro-2-(2-fluoro-1-oxo-1,2,3,4-tetrahydronaphthalen-2-yl)-2-hydroxypropanoate (2j)
Following general procedure, the reaction of 1j (0.48 mmol) was stirred for 2 h. The crude product was purified by flash column chromatography (hexane: ethyl acetate = 10: 1) to provide 2j (97% yield 148.0 mg, 70:30 d.r., 30% e.e. (major), 31% e.e. (minor)) as a colorless oil. The diastereomeric ratio of 2j was determined to be 70:30 by the analysis of 1H-NMR.
Major diastereomer; [α]D28 = +21.1 (c 0.9, CHCl3); The enantiomeric purity of the title compound was determined by HPLC analysis (DAICEL CHIRALPAK IC-3, hexane: 2-propanol = 10: 1, flow rate = 1.0 mL/min, retention time; 11.1 min (major enantiomer), 17.2 min (minor enantiomer)); Minor diastereomer; [α]D28 = +6.3 (c 1.0, CHCl3); The enantiomeric purity of the title compound was determined by HPLC analysis (DAICEL CHIRALPAK IC-3, hexane: 2-propanol = 10: 1, flow rate = 1.0 mL/min, retention time; 5.7 min (major enantiomer), 6.2 min (minor enantiomer)).
Supplementary Materials
The following are available online, 1H, 13C, and 19F-NMR spectra of compounds 2 and HPLC data of compound 2.
Author Contributions
K.S. conceived and designed the project; R.K. and S.H. performed the experiments and analyzed the data while discussing with S.I. and K.S.; R.K. and K.S. wrote the paper.
Funding
This study was supported by the Grants-in-Aid for Scientific Research (B) (18H01974) and Daiko Foundation.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rodríguez, N.; Goossen, L.J. Decarboxylative coupling reactions: A modern strategy for C-C-bond formation. Chem. Soc. Rev. 2011, 40, 5030–5048. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Tan, C.-H. Catalytic decarboxylative reactions: Biomimetic approaches inspired by polyketide biosynthesis. Synthesis 2011, 13, 2044–2053. [Google Scholar] [CrossRef]
- Dzik, W.I.; Lange, P.P.; Gooßen, L.J. Carboxylates as sources of carbon nucleophiles and electrophiles: Comparison of decarboxylative and decarbonylative pathways. Chem. Sci. 2012, 3, 2671–2678. [Google Scholar] [CrossRef]
- Cornella, J.; Larrosa, I. Decarboxylative carbon-carbon bond-forming transformations of (Hetero)aromatic carboxylic acids. Synthesis 2012, 44, 653–676. [Google Scholar] [CrossRef]
- Wang, Z.-L. Recent advances in catalytic asymmetric decarboxylative addition reactions. Adv. Synth. Catal. 2013, 355, 2745–2755. [Google Scholar] [CrossRef]
- Nakamura, S. Catalytic enantioselective decarboxylative reactions using organocatalysts. Org. Biomol. Chem. 2014, 12, 394–405. [Google Scholar] [CrossRef]
- Xuan, J.; Zhang, Z.-G.; Xiao, W.J. Visible-light-induced decarboxylative functionalization of carboxylic acids and their derivatives. Angew. Chem. Int. Ed. 2015, 54, 15632–15641. [Google Scholar] [CrossRef]
- Liu, P.; Zhang, G.; Sun, P. Transition metal-free decarboxylative alkylation reactions. Org. Biomol. Chem. 2016, 14, 10763–10777. [Google Scholar] [CrossRef]
- Wei, Y.; Hu, P.; Zhang, M.; Su, W. Metal-catalyzed decarboxylative C-H functionalization. Chem. Rev. 2017, 117, 8864–8907. [Google Scholar] [CrossRef]
- Bernardi, L.; Fochi, M.; Franchini, M.; Ricci, A. Bioinspired organocatalytic asymmetric reactions. Org. Biomol. Chem. 2012, 10, 2911–2922. [Google Scholar] [CrossRef]
- Bae, H.Y. Malonic acid half thioesters (MAHTs) as efficient enolate precursors in biomimetic catalysis. Synlett 2015, 26, 705–706. [Google Scholar] [CrossRef][Green Version]
- Pedersen, K.J. The ketonic decomposition of beta-keto carboxylic acids. J. Am. Chem. Soc. 1929, 51, 2098–2107. [Google Scholar] [CrossRef]
- Pedersen, K.J. The decomposition of α-nitorocarboxylic acids. J. Phys. Chem. 1933, 38, 559–571. [Google Scholar] [CrossRef]
- Pedersen, K.J. Amine catalysis of the ketonic decomposition of α, α-dimethylacetoacetic acid. J. Am. Chem. Soc. 1938, 60, 595–601. [Google Scholar] [CrossRef]
- Westheimer, F.H.; Jones, W.A. The effect of solvent on some reaction rates. J. Am. Chem. Soc. 1941, 63, 3283–3286. [Google Scholar] [CrossRef]
- Brown, B.B.R.; Phil, D. The mechanism of thermal decarboxylation. Quartery Rev. Chem. Soc. 1950, 5, 131–146. [Google Scholar] [CrossRef]
- Swain, C.G.; Bader, R.F.W.; Esteve, R.M.; Griffine, R.N. Use of substituent effects on isotope effects to distinguish between proton and hydride transfers. part II. mechanism of decarboxylation of β-keto acids in benzene. J. Am. Chem. Soc. 1961, 83, 1951–1954. [Google Scholar] [CrossRef]
- Brower, K.R.; Gay, B.; Konkol, T.L. The volume of activation in unimolecular decomposition reactions. Decarboxylation and demercuration. J. Am. Chem. Soc. 1966, 88, 1681–1685. [Google Scholar] [CrossRef]
- Straub, T.S.; Bender, M.L.; Straub, T.S. Cycloamyloses as enzyme models. The decarboxylation of benzoylacetic acids. J. Am. Chem. Soc. 1972, 94, 8881–8888. [Google Scholar] [CrossRef]
- Louge, M.W.; Pollack, R.M.; Vitullo, V.P. The nature of the transition state for the decarboxylation of β-keto acids. J. Am. Chem. Soc. 1975, 97, 6868–6869. [Google Scholar] [CrossRef]
- Lalic, G.; Aloise, A.D.; Shair, M.D. An Exceptionally mild catalytic thioester aldol reaction inspired by polyketide biosynthesis. J. Am. Chem. Soc. 2003, 125, 2852–2853. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, S.; Benaglia, M.; Cozzi, F. Cu(II)-catalyzed enantioselective aldol condensation between malonic acid hemithioesters and aldehydes. Tetrahedron Lett. 2004, 45, 1747–1749. [Google Scholar] [CrossRef]
- Magdziak, D.; Lalic, G.; Lee, H.M.; Fortner, K.C.; Aloise, A.D.; Shair, M.D. Catalytic Enantioselective thioester aldol reactions that are compatible with protic functional groups. J. Am. Chem. Soc. 2005, 127, 7284–7285. [Google Scholar] [CrossRef] [PubMed]
- Baudoux, J.; Lefebvre, P.; Legay, R.; Lasne, M.C.; Rouden, J. Environmentally benign metal-free decarboxylative aldol and Mannich reactions. Green Chem. 2010, 12, 252–259. [Google Scholar] [CrossRef]
- Yuan, J.-W.; Liu, S.-N.; Mai, W.-P. Copper-catalysed difluoroalkylation of aromatic aldehydes via a decarboxylation/aldol reaction. Org. Biomol. Chem. 2017, 15, 7654–7659. [Google Scholar] [CrossRef]
- Huang, D.-K.; Lei, Z.-L.; Zhu, Y.-J.; Liu, Z.-J.; Hu, X.-J.; Mao, H.-F. An expedient synthesis of α, α-difluoro-β-hydroxy ketones via decarboxylative aldol reaction of α, α-difluoro-β-keto acids with aldehydes. Tetrahedron Lett. 2017, 58, 3394–3397. [Google Scholar] [CrossRef]
- Tarui, A.; Oduti, M.; Shinya, S.; Sato, K.; Omote, M. Decarboxylative aldol reaction of α, α-difluoro-β-ketocarboxylate salt: A facile method for generation of difluoroenolate. RSC Adv. 2018, 8, 20568–20575. [Google Scholar] [CrossRef]
- Li, Y.-L.; Wang, X.-L.; Xiao, D.; Liu, M.-Y.; Du, Y.; Deng, J. Organocatalytic biomimetic decarboxylative aldol reaction of fluorinated β-keto acids with unprotected isatins. Adv. Synth. Catal. 2018, 360, 4147–4152. [Google Scholar] [CrossRef]
- Shibatomi, K.; Kitahara, K.; Sasaki, N.; Kawasaki, Y.; Fujisawa, I.; Iwasa, S. Enantioselective decarboxylative chlorination of β-ketocarboxylic acids. Nat. Commun. 2017, 8, 15600. [Google Scholar] [CrossRef]
- Katada, M.; Kitahara, K.; Iwasa, S.; Shibatomi, K. Catalyst-free decarboxylative fluorination of tertiary β -keto carboxylic acids. Synlett 2018, 29, 2408–2411. [Google Scholar]
- Naruse, A.; Kitahara, K.; Iwasa, S.; Shibatomi, K. Synthesis of α-fluoroenones by elimination of α-chloro-α-fluoroketones. Asian J. Org. Chem. 2019, 8, 691–693. [Google Scholar] [CrossRef]
- Kawanishi, R.; Phongphane, L.; Iwasa, S.; Shibatomi, K. Decarboxylative fluorination of 2-pyridylacetates. Chem. Eur. J. 2019, 25, 7453–7456. [Google Scholar] [CrossRef] [PubMed]
- Gathergood, N.; Juhl, K.; Poulsen, T.B.; Thordrup, K.; Jørgensen, K.A. Direct catalytic asymmetric aldol reactions of pyruvates: Scope and mechanism. Org. Biomol. Chem. 2004, 2, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, W.; Poulsen, T.B.; Jørgensen, K.A. A versatile catalyst for asymmetric reactions of carbonyl groups working purely by activation through hydrogen bonding: Mukaiyama-aldol, hetero Diels-Alder and Friedel-Crafts reactions. Org. Biomol. Chem. 2005, 3, 3284–3289. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Shibata, N.; Inagaki, J.; Nakamura, S.; Toru, T.; Shiro, M. Cinchona-alkaloid-catalyzed enantioselective direct aldol-type reaction of oxindoles with ethyl trifluoropyruvate. Angew. Chem. Int. Ed. 2007, 46, 8666–8669. [Google Scholar] [CrossRef] [PubMed]
- Frings, M.; Atodiresei, I.; Runsink, J.; Raabe, G.; Bolm, C. Catalyzed vinylogous mukaiyama aldol reactions with controlled enantio-and diastereoselectivities. Chem. Eur. J. 2009, 15, 1566–1569. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Liu, X.; Zheng, K.; Li, W.; Hu, X.; Lin, L.; Feng, X. Catalytic asymmetric synthesis of 3-(α-hydroxy-β-carbonyl) oxindoles by a ScIII-catalyzed direct aldol-type reaction. Chem. Eur. J. 2010, 16, 3736–3742. [Google Scholar] [CrossRef]
- Dong, X.; Sun, J. Catalytic asymmetric α-aldol reaction of vinylogous N-heterocyclic carbene enolates: Formation of quaternary and labile tertiary stereocenters. Org. Lett. 2014, 16, 2450–2453. [Google Scholar] [CrossRef]
- Ma, J.-A.; Cahard, D.; Recherche, I.; Fine, O. Update 1 of asymmetric fluorination, trifluoromethylation, and perfluoroalkylation reactions. Chem. Rev. 2008, 108, PR1–PR43. [Google Scholar] [CrossRef]
- Nie, J.; Guo, H.; Cahard, D.; Ma, J.-A. Asymmetric construction of stereogenic carbon centers featuring a trifluoromethyl group from prochiral trifluoromethylated substrates. Chem. Rev. 2011, 111, 455–529. [Google Scholar] [CrossRef]
- Sorochinsky, A.E.; Soloshonok, V.A. Recent advances in the asymmetric synthesis of α-(Trifluoromethyl) containing α-amino acids. Synthesis 2012, 44, 1592–1602. [Google Scholar]
- Bizet, V.; Besset, T.; Ma, J.A.; Cahard, D. Recent progress in asymmetric fluorination and trifluoromethylation reactions. Curr. Top. Med. Chem. 2014, 14, 901–940. [Google Scholar] [CrossRef] [PubMed]
- Shibatomi, K. Catalytic enantioselective constructions of trifluoromethylated chiral stereogenic centers via the conjugate addition to CF3-alkenes: Overview of current progress. Curr. Org. Chem. 2015, 19, 1619–1637. [Google Scholar] [CrossRef]
- Mei, H.; Xie, C.; Han, J.; Soloshonok, V.A. Ntert -Butylsulfinyl-3 3 3-trifluoroacetaldimine: Versatile reagent for asymmetric synthesis of trifluoromethyl containing amines and amino acids of pharmaceutical. Eur. J. Org. Chem. 2016, 2016, 5917–5932. [Google Scholar] [CrossRef]
- Kluger, R.; Brandl, M. β-Deuterium secondary isotope effects in heterolytic decarboxylation reactions. manifestations of negative hyperconjugation. J. Org. Chem. 1986, 51, 3964–3968. [Google Scholar] [CrossRef]
- Dummer, N.F.; Jenkins, R.; Li, X.; Bawaked, S.M.; McMorn, P.; Burrows, A.; Kiely, C.J.; Wells, R.P.K.; Willock, D.J.; Hutchings, G.J. Inversion of enantioselectivity for the hydrogenation of ethyl pyruvate in the gas-phase over Pt/SiO2 modified with derivatives of hydroquinidine. J. Catal. 2006, 243, 165–170. [Google Scholar] [CrossRef]
- Yoneda, N.; Fujii, Y.; Matsumoto, A.; Asano, K.; Matsubara, S. Organocatalytic enantio and diastereoselective cycloetherification via dynamic kinetic resolution of chiral cyanohydrins. Nat. Commun. 2017, 8, 1397. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).