
Transfer of a Multiclass Method for over 60 Antibiotics in Food from High Resolution to Low Resolution Mass Spectrometry


 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Optimization of LC-MS/MS Conditions
2.2. Method Validation
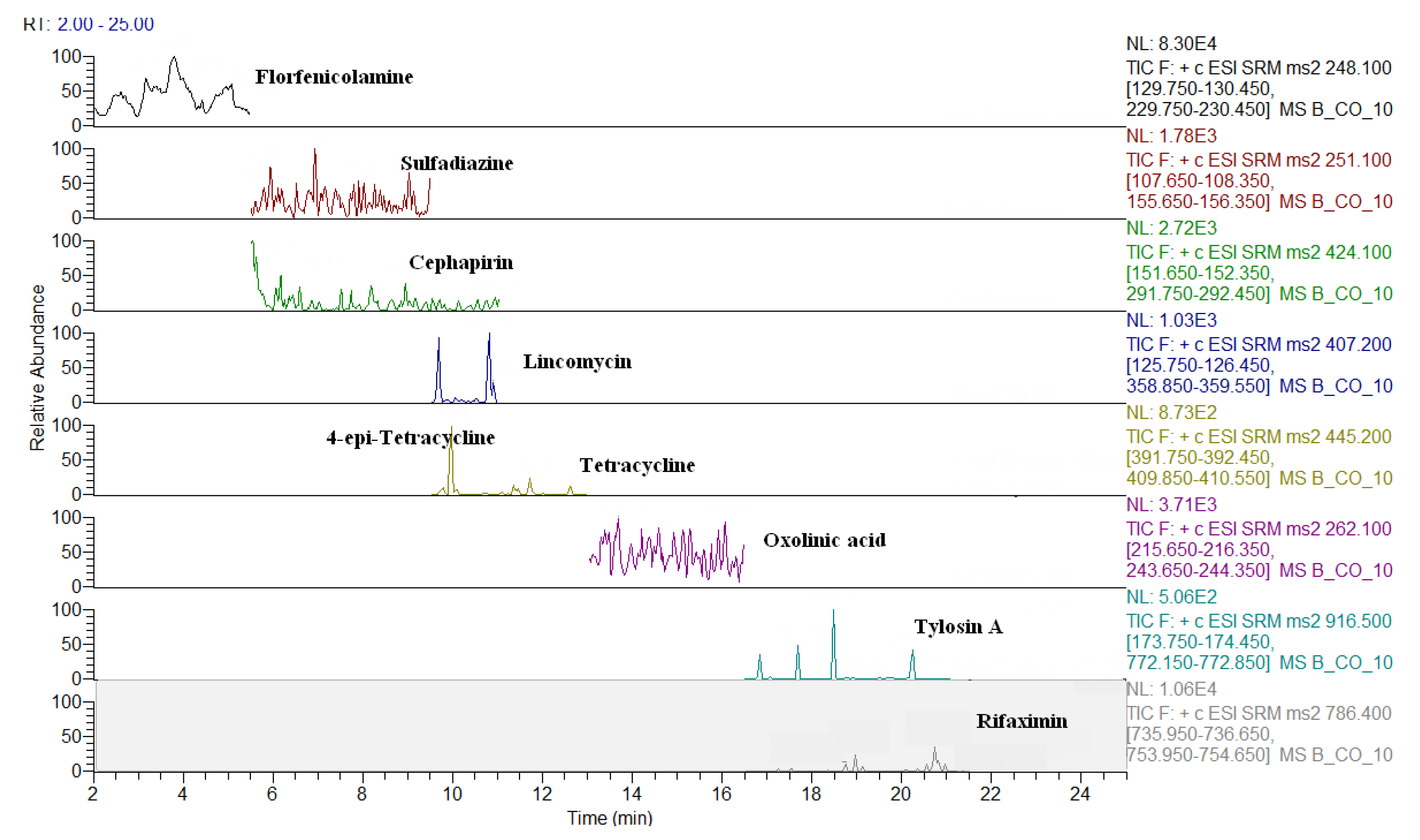
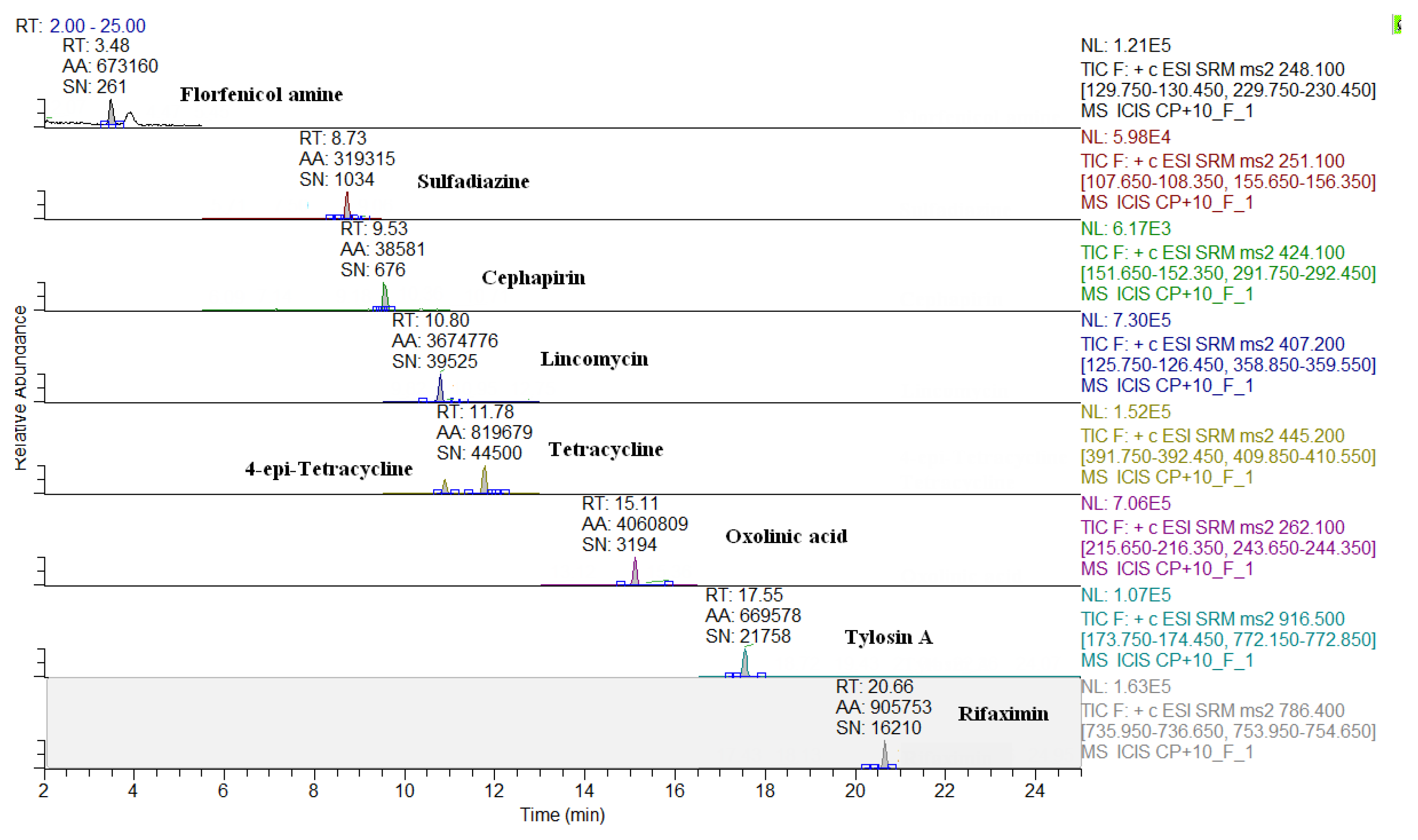
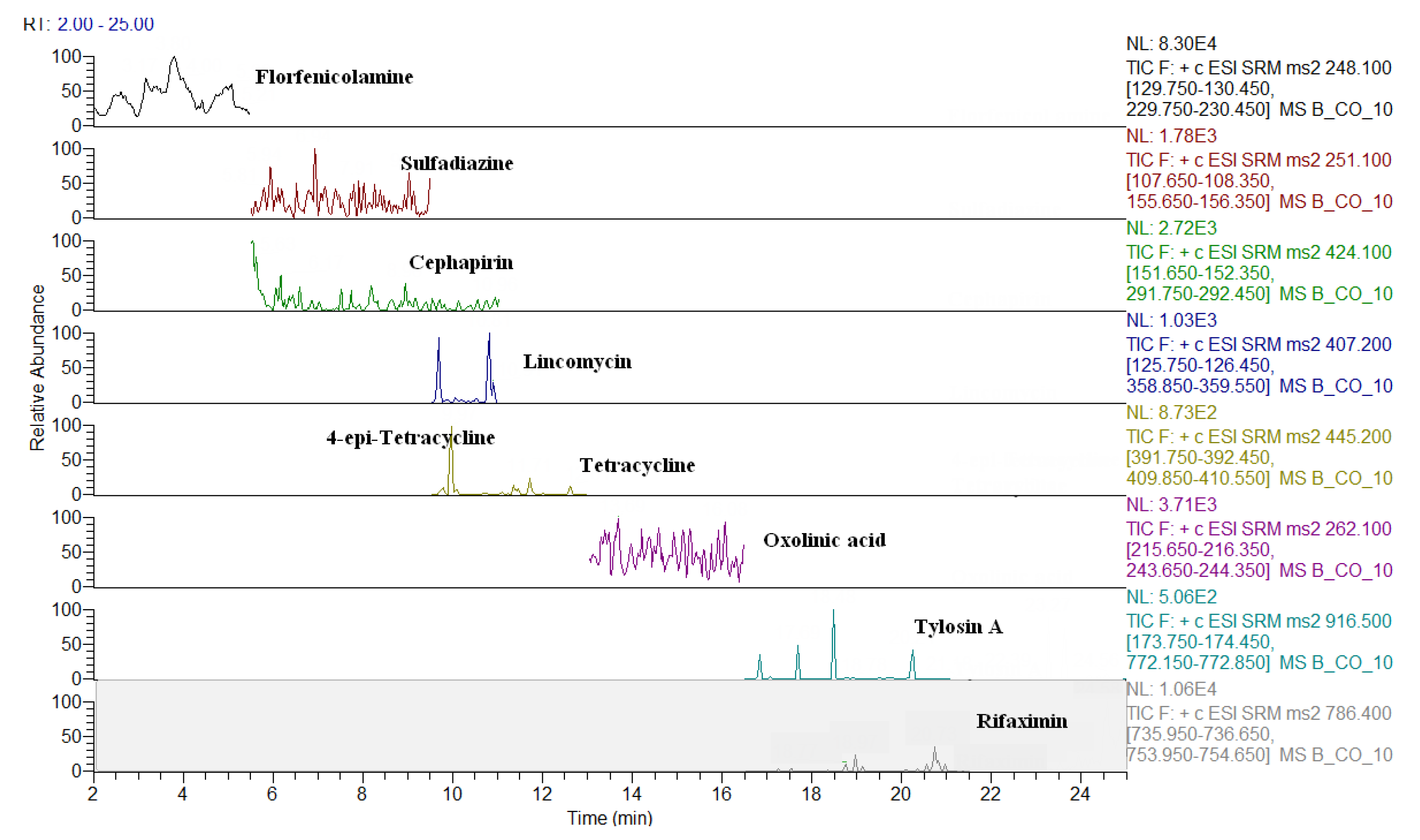
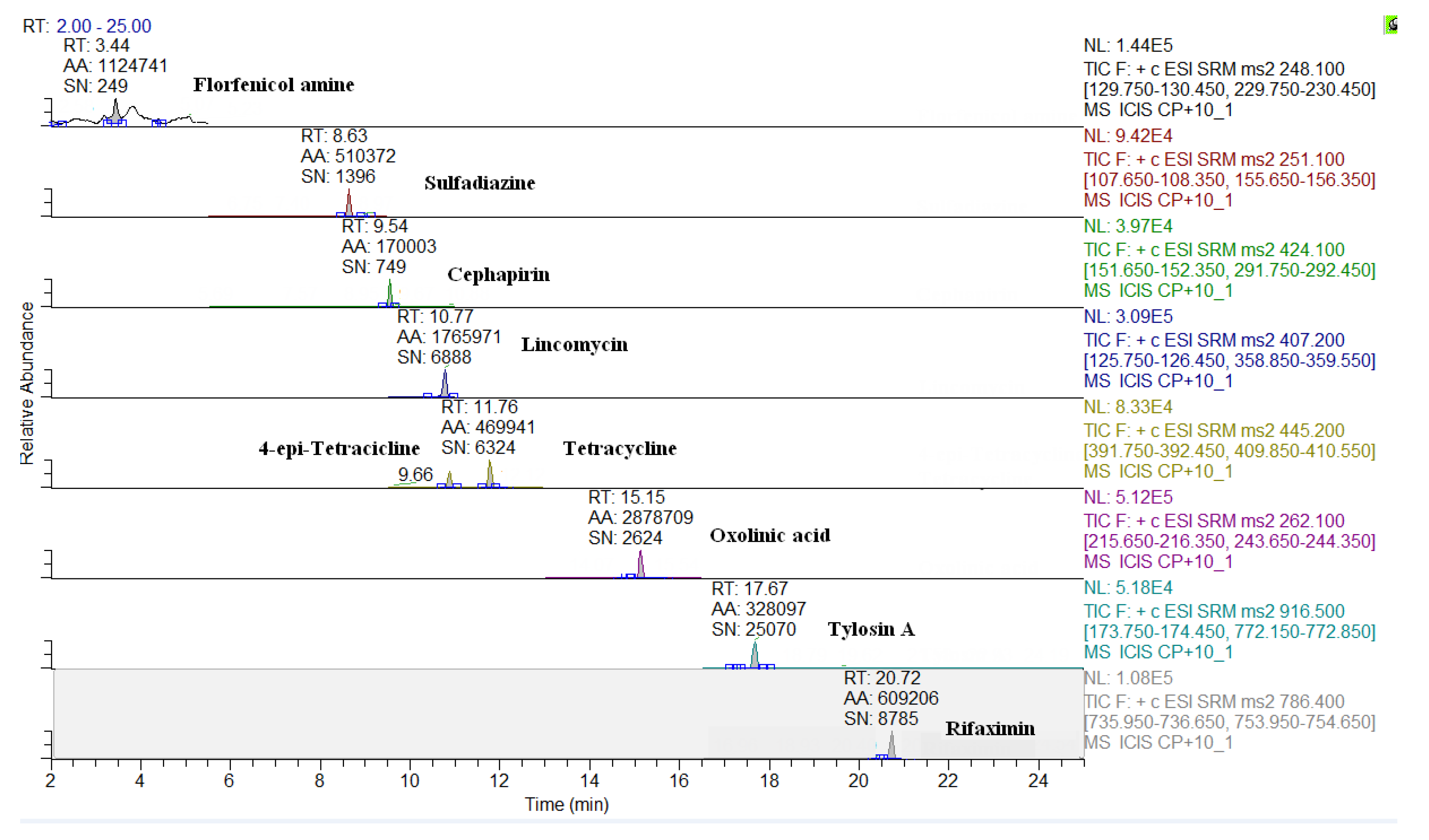
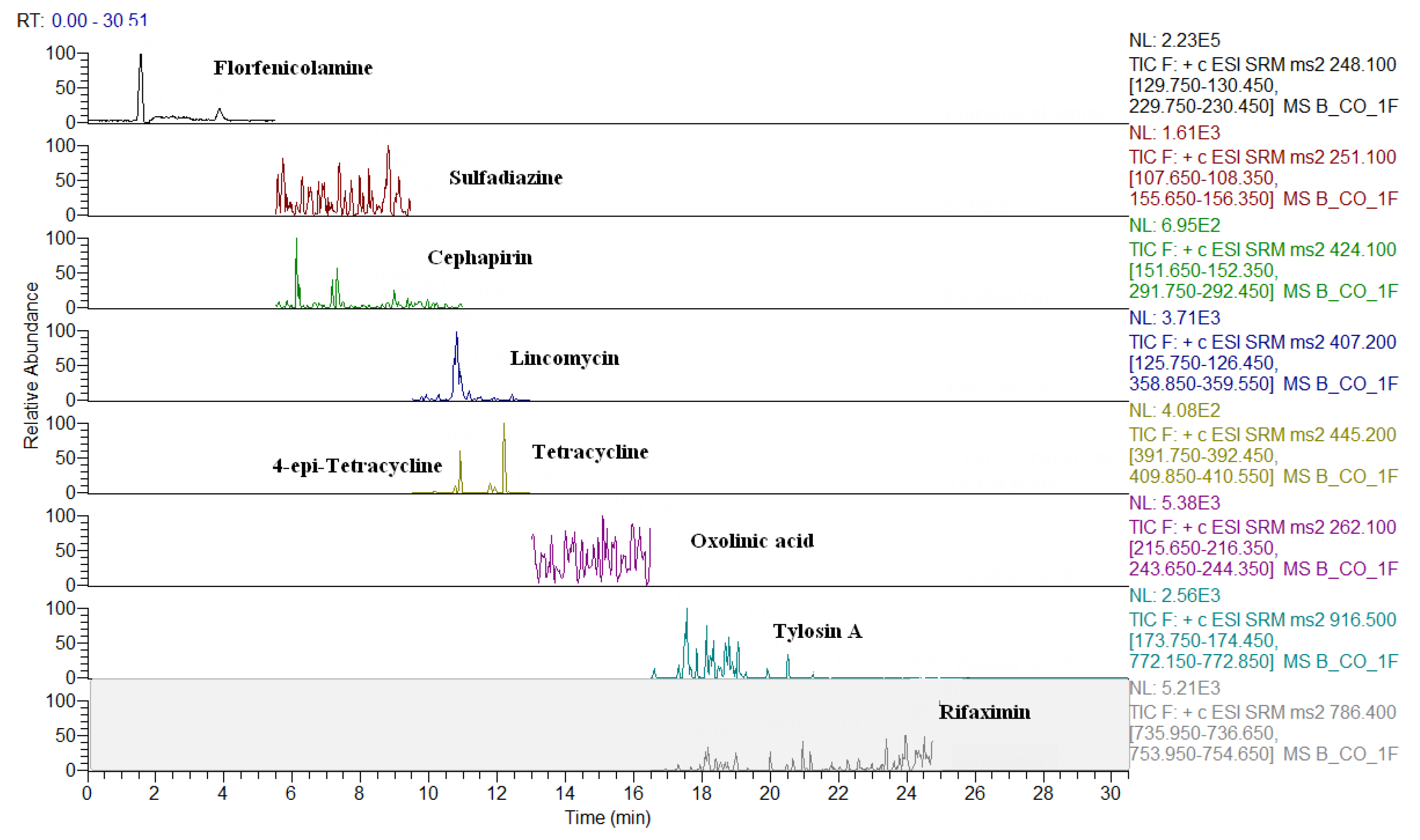
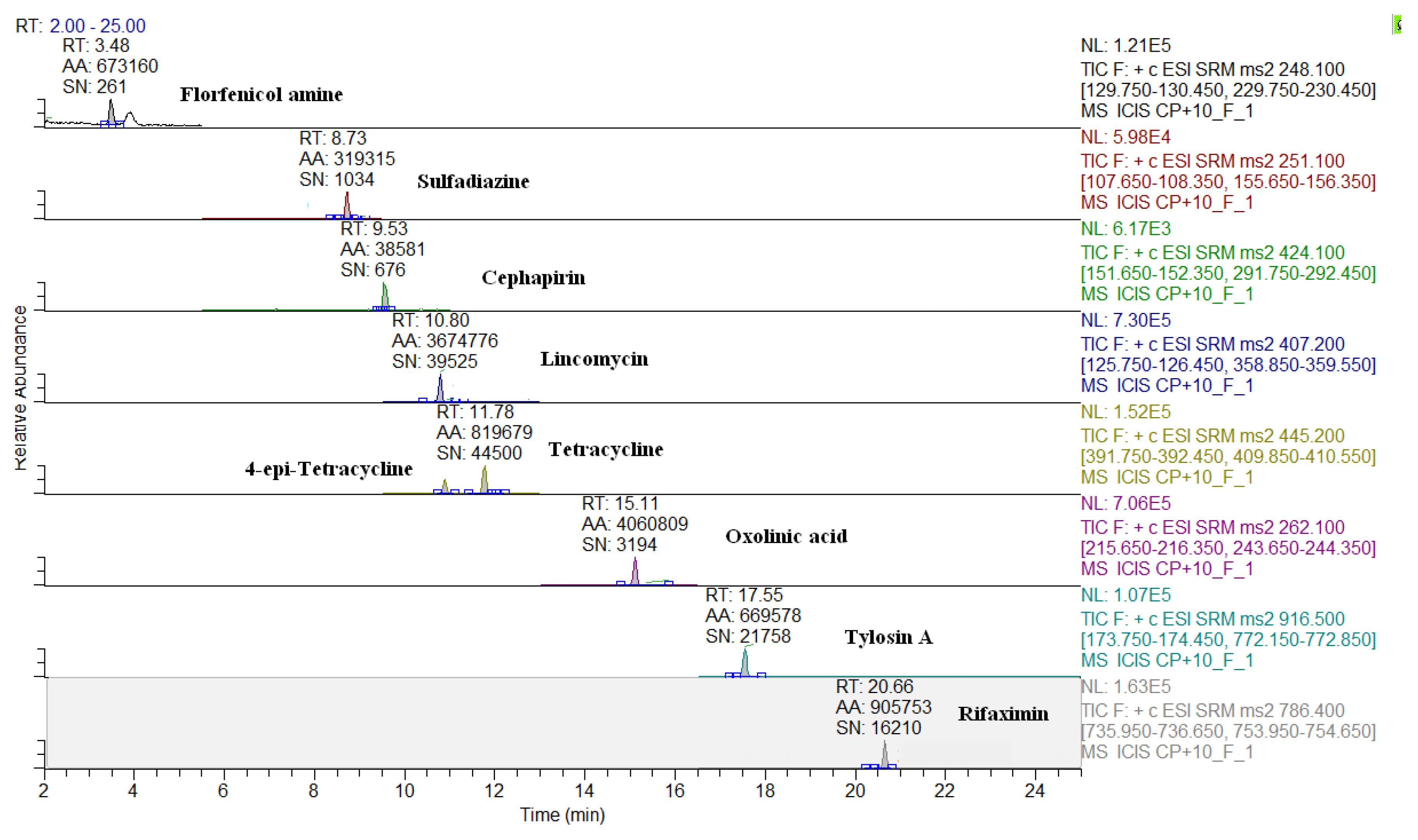
2.3. Comparison of LC-QqQ and LC-Q-Orbitrap Methods
3. Experimental
3.1. Chemical and Reagents
3.2. Standard Solutions
3.3. LC-MS-MS Conditions
3.4. Sample Preparation
3.5. Method Validation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- European Communities Commission Regulation (EU) No 37/2010 of 22 December 2009 on pharmacologically active substances and their classification regarding maximum residue limits in foodstuffs of animal origin. Off. J. Eur. Communities 2010, L15, 1–72. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32010R0037 (accessed on 30 May 2019).
- Masiá, A.; Suarez-Varela, M.M.; Llopis-Gonzalez, A.; Picó, Y. Determination of pesticides and veterinary drug residues in food by liquid chromatography-mass spectrometry: A review. Anal. Chim. Acta 2016, 936, 40–61. [Google Scholar] [CrossRef] [PubMed]
- Mainero Rocca, L.; Gentili, A.; Pérez-Fernández, V.; Tomai, P. Veterinary drugs residues: A review of the latest analytical research on sample preparation and LC-MS based methods. Food Addit. Contam. - Part A Chem. Anal. Control. Expo. Risk Assess. 2017, 34, 766–784. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.; Saluti, G.; Moretti, S.; Diamanti, I.; Giusepponi, D.; Galarini, R. Multiclass methods for the analysis of antibiotic residues in milk by liquid chromatography coupled to mass spectrometry: A review. Food Addit. Contam. - Part A Chem. Anal. Control. Expo. Risk Assess. 2018, 35, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Aguilera-Luiz, M.M.; Vidal, J.L.M.; Romero-González, R.; Frenich, A.G. Multi-residue determination of veterinary drugs in milk by ultra-high-pressure liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2008, 1205, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Chico, J.; Rúbies, A.; Centrich, F.; Companyó, R.; Prat, M.D.; Granados, M. High-throughput multiclass method for antibiotic residue analysis by liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2008, 1213, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, A.; Butcher, P.; Maden, K.; Widmer, M. Quantitative multiresidue method for about 100 veterinary drugs in different meat matrices by sub 2-microm particulate high-performance liquid chromatography coupled to time of flight mass spectrometry. J. Chromatogr. A 2008, 1194, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Stolker, A.A.M.; Rutgers, P.; Oosterink, E.; Lasaroms, J.J.P.; Peters, R.J.B.; Van Rhijn, J.A.; Nielen, M.W.F. Comprehensive screening and quantification of veterinary drugs in milk using UPLC-ToF-MS. Anal. Bioanal. Chem. 2008, 391, 2309–2322. [Google Scholar] [CrossRef]
- Peters, R.J.B.B.; Bolck, Y.J.C.C.; Rutgers, P.; Stolker, A.A.M.M.; Nielen, M.W.F.F. Multi-residue screening of veterinary drugs in egg, fish and meat using high-resolution liquid chromatography accurate mass time-of-flight mass spectrometry. J. Chromatogr. A 2009, 1216, 8206–8216. [Google Scholar] [CrossRef]
- Stubbings, G.; Bigwood, T. The development and validation of a multiclass liquid chromatography tandem mass spectrometry (LC-MS/MS) procedure for the determination of veterinary drug residues in animal tissue using a QuEChERS (QUick, Easy, CHeap, Effective, Rugged and Safe) approac. Anal. Chim. Acta 2009, 637, 68–78. [Google Scholar] [CrossRef]
- Gaugain-Juhel, M.; Delépine, B.; Gautier, S.; Fourmond, M.P.; Gaudin, V.; Hurtaud-Pessel, D.; Verdon, E.; Sanders, P. Validation of a liquid chromatography-tandem mass spectrometry screening method to monitor 58 antibiotics in milk: A qualitative approach. Food Addit. Contam. - Part A Chem. Anal. Control. Expo. Risk Assess. 2009, 26, 1459–1471. [Google Scholar] [CrossRef] [PubMed]
- Martínez Vidal, J.L.; Frenich, A.G.; Aguilera-Luiz, M.M.; Romero-González, R. Development of fast screening methods for the analysis of veterinary drug residues in milk by liquid chromatography-triple quadrupole mass spectrometry. Analytical and Bioanalytical Chemistry. 2010, 397, 2777–2790. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, A.; Butcher, P.; Maden, K.; Walker, S.; Widmer, M. Development of an improved high resolution mass spectrometry based multi-residue method for veterinary drugs in various food matrices. Anal. Chim. Acta 2011, 700, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Hurtaud-Pessel, D.; Jagadeshwar-Reddy, T.; Verdon, E. Development of a new screening method for the detection of antibiotic residues in muscle tissues using liquid chromatography and high resolution mass spectrometry with a LC-LTQ-Orbitrap instrument. Food Addit. Contam. - Part A Chem. Anal. Control. Expo. Risk Assess. 2011, 28, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Leung, D. The challenges of developing a generic extraction procedure to analyze multi-class veterinary drug residues in milk and honey using ultra-high pressure liquid chromatography quadrupole time-of-flight mass spectrometry. Drug Test. Anal. 2012, 1, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Bittencourt, M.S.; Martins, M.T.; de Albuquerque, F.G.S.; Barreto, F.; Hoff, R. High-throughput multiclass screening method for antibiotic residue analysis in meat using liquid chromatography-tandem mass spectrometry: A novel minimum sample preparation procedure. Food Addit. Contam. - Part A Chem. Anal. Control. Expo. Risk Assess. 2012, 29, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.T.; Melo, J.; Barreto, F.; Barcellos Hoff, R.; Jank, L.; Soares Bittencourt, M.; Bazzan Arsand, J.; Scherman Schapoval, E.E. A simple, fast and cheap non-SPE screening method for antibacterial residue analysis in milk and liver using liquid chromatography-tandem mass spectrometry. Talanta 2014, 129, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, A.; Butcher, P.; Maden, K.; Walker, S.; Widmer, M. Multi-residue quantification of veterinary drugs in milk with a novel extraction and cleanup technique: Salting out supported liquid extraction (SOSLE). Anal. Chim. Acta 2014, 820, 56–68. [Google Scholar] [CrossRef]
- Freitas, A.; Barbosa, J.; Ramos, F. Multidetection of antibiotics in liver tissue by ultra-high-pressure-liquid-chromatography-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2015, 976-977, 49–54. [Google Scholar] [CrossRef]
- Martins, M.T.; Barreto, F.; Hoff, R.B.; Jank, L.; Arsand, J.B.; Feijó, T.C.; Schapoval, E.E.S. Determination of quinolones and fluoroquinolones, tetracyclines and sulfonamides in bovine, swine and poultry liver using LC-MS/MS. Food Addit. Contam. - Part A Chem. Anal. Control. Expo. Risk Assess. 2015, 32, 333–341. [Google Scholar] [CrossRef]
- Wang, J.; Leung, D.; Chow, W.; Chang, J.; Wong, J.W. Development and Validation of a Multiclass Method for Analysis of Veterinary Drug Residues in Milk Using Ultrahigh Performance Liquid Chromatography Electrospray Ionization Quadrupole Orbitrap Mass Spectrometry. J. Agric. Food Chem. 2015, 63, 9175–9187. [Google Scholar] [CrossRef] [PubMed]
- Dasenaki, M.E.; Thomaidis, N.S. Multi-residue determination of 115 veterinary drugs and pharmaceutical residues in milk powder, butter, fish tissue and eggs using liquid chromatography-tandem mass spectrometry. Anal. Chim. Acta 2015, 880, 103–121. [Google Scholar] [CrossRef] [PubMed]
- Dasenaki, M.E.; Michali, C.S.; Thomaidis, N.S. Analysis of 76 veterinary pharmaceuticals from 13 classes including aminoglycosides in bovine muscle by hydrophilic interaction liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2016, 1452, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Moretti, S.; Dusi, G.; Giusepponi, D.; Pellicciotti, S.; Rossi, R.; Saluti, G.; Cruciani, G.; Galarini, R. Screening and confirmatory method for multiclass determination of 62 antibiotics in meat. J. Chromatogr. A 2016, 1429, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Moretti, S.; Cruciani, G.; Romanelli, S.; Rossi, R.; Saluti, G.; Galarini, R. Multiclass method for the determination of 62 antibiotics in milk. J. Mass Spectrom. 2016, 51, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Anumol, T.; Lehotay, S.J.; Stevens, J.; Zweigenbaum, J. Comparison of veterinary drug residue results in animal tissues by ultrahigh-performance liquid chromatography coupled to triple quadrupole or quadrupole–time-of-flight tandem mass spectrometry after different sample preparation methods, including use of. Anal. Bioanal. Chem. 2017, 409, 2639–2653. [Google Scholar] [CrossRef] [PubMed]
- Lehotay, S.J.; Lightfield, A.R. Simultaneous analysis of aminoglycosides with many other classes of drug residues in bovine tissues by ultrahigh-performance liquid chromatography–tandem mass spectrometry using an ion-pairing reagent added to final extracts. Anal. Bioanal. Chem. 2018, 2018. 410, 1095–1109. [Google Scholar] [CrossRef]
- Gaspar, A.F.; Santos, L.; Rosa, J.; Leston, S.; Barbosa, J.; Vila Pouca, A.S.; Freitas, A.; Ramos, F. Development and validation of a multi-residue and multi-class screening method of 44 antibiotics in salmon (Salmo salar) using ultra-high-performance liquid chromatography/time-of-flight mass spectrometry: Application to farmed salmon. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2019, 1118–1119, 78–84. [Google Scholar] [CrossRef]
- Romero-González, R.; Aguilera-Luiz, M.M.; Plaza-Bolaños, P.; Frenich, A.G.; Vidal, J.L.M. Food contaminant analysis at high resolution mass spectrometry: Application for the determination of veterinary drugs in milk. J. Chromatogr. A 2011, 1218, 9353–9365. [Google Scholar] [CrossRef]
- Commission Decision (2002/657/EC) of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off. J. Eur. Communities 2002, L221, 8–36.
- Dickson, L.C. Performance characterization of a quantitative liquid chromatography-tandem mass spectrometric method for 12 macrolide and lincosamide antibiotics in salmon, shrimp and tilapia. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 967, 203–210. [Google Scholar] [CrossRef] [PubMed]
- SANCO/2004/2726-rev 4-December 2008 Guidelines for the Implementation of Decision 2002/657/EC. European Commission Health & Consumer Protection Directorate-General. 2008. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/cs_vet-med-residues_cons_2004-2726rev4_en.pdf (accessed on 30 May 2019).
- SANTE/11813/2017. Guidance document on analytical quality control and validation procedures for pesticide residues analysis in food and feed. In European Commission Health & Consumer Protection Directorate-General. 2017. Available online: https://ec.europa.eu/food/sites/food/files/plant/docs/pesticides_mrl_guidelines_wrkdoc_2017-11813.pdfwebsite (accessed on 30 May 2019).
- Barvick, V.J.; Wllison, S.R.L. Part (d): Protocol for uncertainty evaluation from validation data. In VAM Project 3.2.1. Development and Harmonisation of Measurement Uncertainty Principles; LGC: Teddington, UK, 2000. [Google Scholar]
- European Communities Commission Regulation (EU) No 37/2010 of 22 December 2009 on pharmacologically active substances and their classification regarding maximum residue limits in foodstuffs of animal origin. Consolidated test. 3 March 2019. Available online: https://eur-lex.europa.eu/collection/eu-law/consleg.html (accessed on 30 May 2019).
- Galarini, R.; Moretti, S.; Saluti, G. Quality Assurance and Validation General Considerations and Trends. In Chromatographic Analysis of the Environment Mass Spectrometry Based Approaches, Fourth Edition; Leo, M.L., Nollet, D.A.L., Eds.; CRC Press: Boca Raton, FL, USA, 2017; ISBN 9781315316208. [Google Scholar]
- Grujic, S.; Vasiljevic, T.; Lausevic, M.; Ast, T. Study on the formation of an amoxicillin adduct with methanol using electrospray ion trap tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2008, 22, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Mastovska, K.; Lightfield, A.R. Streamlining methodology for the multiresidue analysis of beta-lactam antibiotics in bovine kidney using liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2008, 1202, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Asteggiante, L.G.; Nunez, A.; Lehotay, S.J.; Lightfield, A.R. Structural characterization of product ions by electrospray ionization and quadrupole time-of-flight mass spectrometry to support regulatory analysis of veterinary drug residues in foods. Rapid Commun. Mass Spectrom. 2014, 28, 1061–1081. [Google Scholar] [CrossRef] [PubMed]
- Niessen, W.M.A.; Correa, R.A.C. Interpretation of MS-MS Mass Spectra of Drugs and Pesticides; John Wilewy and Sons: Hoboken, NJ, USA, 2017. [Google Scholar]
- Kaufmann, A.; Walker, S. Extension of the Q Orbitrap intrascan dynamic range by using a dedicated customized scan. Rapid. Commun. Mass Spectrom. 2016, 30, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- General requirements for the competence of testing and calibration laboratories, Second ed.; International Standard Organization: Geneva, Switzerland, 2005.
- Berendsen, B.J.A.A.; Elbers, I.J.W.W.; Stolker, A.A.M.M. Determination of the stability of antibiotics in matrix and reference solutions using a straightforward procedure applying mass spectrometric detection. Food Addit. Contam. - Part A Chem. Anal. Control. Expo. Risk Assess. 2011, 28, 1657–1666. [Google Scholar] [CrossRef]
- Gaugain, M.; Chotard, M.P.; Verdon, E. Stability study for 53 antibiotics in solution and in fortified biological matrixes by LC/MS/MS. J. AOAC Int. 2013, 96, 471–480. [Google Scholar] [CrossRef]
Sample Availability: Not available. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N° of Veterinary Drugs | Matrix | Equipment | Reference | Laboratory/Centre a | |
|---|---|---|---|---|---|
| 1 | 18 | Milk | LC-QqQ | Aguilera-Luiz et al. 2008 [5] | Almeria University (Spain) |
| 2 | 39 | Chicken muscle | LC-QqQ | Chico et al. 2008 [6] | Barcelona University (Spain) |
| 3 | >100 | Muscle | LC-TOF | Kaufmann et al. 2008 [7] | OFCA-Zurich (Switerland) |
| 4 | >100 | Milk | LC-TOF | Stolker et al. 2008 [8] | RIKILT (The Netherlands) |
| 5 | Ca 100 | Meat and other food | LC-TOF | Peters et al. 2009 [9] | RIKILT (The Netherlands) |
| 6 | Ca 26 | Animal tissues | LC-QqQ | Stubbings et al. 2009 [10] | FERA (UK) |
| 7 | 58 | Milk | LC-QqQ | Gaugain-Juhel et al. 2009 [11] | EURL (France) |
| 8 | 21 | Milk | LC-QqQ | Martinez-Vidal et al. 2010 [12] | Almeria University (Spain) |
| 9 | 30 | Milk | LC-Orbitrap, LC-Q-TOF, LC-QqQ | Romero-González et al. 2011 [5] | Almeria University (Spain) |
| 10 | >100 | Meat and other food | LC-Orbitrap | Kaufmann et al. 2011 [13] | OFCA-Zurich (Switzerland) |
| 11 | >60 | Meat | LC-LTQ-Orbitrap | Hurtaud-Pessel et al. 2011 [14] | EURL (France) |
| 12 | 59 | Milk and honey | LC-Q-TOF | Wang et al. 2012 [15] | CFIA-Calgary (Canada) |
| 13 | 21 | Meat | LC-QqQ | Bittencourt et al. 2012 [16] | LANAGRO (Brazil) |
| 14 | 24 | Milk and liver | LC-QqQ | Martins et al. 2014 [17] | LANAGRO (Brazil) |
| 15 | >100 | Milk | LC-Q-Orbitrap | Kaufmann et al. 2014 [18] | OFCA-Zurich (Switzerland) |
| 16 | 39 | Liver | LC-QqQ | Freitas et al. 2015 [19] | INIAV (Portugal) |
| 17 | 23 | Liver | LC-QqQ | Martins et al. 2015 [20] | LANAGRO (Brazil) |
| 18 | >100 | Milk | LC-Q-Orbitrap | Wang et al. 2015 [21] | CFIA-Calgary (Canada) |
| 19 | >100 | Various food | LC-Q-TOF | Dasenaki et al. 2015 [22] | University of Athens (Greece) |
| 20 | 76 | Bovine muscle | LC-QqQ | Dasenaki et al. 2016 [23] | University of Athens (Greece) |
| 21 | 62 | Animal muscle | LC-Q-Orbitrap | Moretti et al. 2016 [24] | IZSUM (Italy) |
| 22 | 62 | Milk | LC-Q-Orbitrap | Moretti et al. 2016 [25] | IZSUM (Italy) |
| 23 | >120 | Animal tissues | LC-QqQ/LC-Q-TOF | Anumol et al. 2017 [26] | USDA (USA) |
| 24 | 174 | Bovine tissues | LC-QqQ | Lehotay et al. 2018 [27] | USDA (USA) |
| 25 | 44 | Salmon | LC-Q-TOF | Gaspar et al. 2019 [28] | INIAV (Portugal) |
| N° | Analyte | Retention Time (min) | Adduct (m/z) | Precursor Ion (m/z) | Product Ions (m/z) | Collision Energy (eV) |
|---|---|---|---|---|---|---|
| 1 | Sulfaguanidine | 2.85 | [M + H]+ | 215.1 | 92.0 | 15 |
| 156.0 | 20 | |||||
| 2 | Florfenicolamine | 3.20 | [M + H]+ | 248.1 | 230.1 | 10 |
| 130.1 | 30 | |||||
| 3 | Sulfanilamide | 3.30 | [M + H − NH3]+ | 156.0 | 92.0 | 12 |
| 108.1 | 10 | |||||
| Sulfanilamide-13C6 | 3.30 | [M + H − NH3]+ | 162.0 | 98.1 | 13 | |
| 114.1 | 13 | |||||
| 4 | Desacetylcephapyrin | 6.80 | [M + H]+ | 382.1 | 152.0 | 30 |
| 226.0 | 20 | |||||
| 5 | Amoxicillin | 8.30 | [M + H]+ | 366.1 | 349.1 | 10 |
| 114.0 | 20 | |||||
| 6 | Sulfadiazine | 8.50 | [M + H]+ | 251.1 | 108.0 | 26 |
| 156.0 | 15 | |||||
| 7 | Sulfathiazole | 9.20 | [M + H]+ | 256.0 | 92.1 | 28 |
| 156.0 | 15 | |||||
| 8 | Cephapyrin | 9.45 | [M + H]+ | 424.1 | 292.1 | 20 |
| 152.0 | 30 | |||||
| 9 | Sulfapyridine | 9.50 | [M + H]+ | 250.1 | 108.0 | 26 |
| 156.0 | 17 | |||||
| 10 | Tildipirosin | 9.90 | [M+2H]++ | 367.7 | 281.2 | 20 |
| 98.1 | 18 | |||||
| 11 | Sulfamerazine | 9.90 | [M + H]+ | 265.1 | 108.0 | 27 |
| 156.0 | 17 | |||||
| 12 | Cefquinome | 10.00 | [M + 2H]++ | 265.1 | 134.2 | 20 |
| 199.1 | 20 | |||||
| 13 | Cefacetrile | 10.15 | [M + Na]+ | 362.0 | 258.0 | 10 |
| 302.0 | 10 | |||||
| 14 | Cefalonium | 10.50 | [M + H]+ | 459.1 | 337.0 | 10 |
| 152.0 | 20 | |||||
| 15 | Lincomycin | 10.50 | [M + H]+ | 407.2 | 126.1 | 30 |
| 359.2 | 10 | |||||
| 16 | Tulathromycin marker | 10.60 | [M + 2H]++ | 289.0 | 158.3 | 17 |
| 420.5 | 17 | |||||
| 17 | Thiamphenicol | 10.60 | [M + H]+ | 356.0 | 308.0 | 20 |
| 229.0 | 20 | |||||
| 18 | Epitetracycline | 10.60 | [M + H]+ | 445.2 | 410.2 | 20 |
| 392.1 | 30 | |||||
| 19 | Trimethoprim | 10.70 | [M + H]+ | 291.1 | 261.1 | 30 |
| 230.1 | 30 | |||||
| 20 | Marbofloxacin | 10.80 | [M + H]+ | 363.1 | 276.1 | 14 |
| 320.1 | 14 | |||||
| 21 | Sulfamethazine | 11.10 | [M + H]+ | 279.1 | 92.1 | 31 |
| 124.1 | 28 | |||||
| Sulfamethazine-13C6 | 11.10 | [M + H]+ | 285.1 | 186.1 | 17 | |
| 22 | Epioxytetracycline | 11.35 | [M + H]+ | 461.2 | 426.1 | 20 |
| 337.1 | 30 | |||||
| 23 | Norfloxacina | 11.50 | [M + H]+ | 320.1 | 231.2 | 39 |
| 282.1 | 29 | |||||
| 24 | Tetracycline | 11.50 | [M + H]+ | 445.2 | 410.2 | 20 |
| 392.1 | 30 | |||||
| 25 | Cefalexin | 11.70 | [M + H]+ | 348.1 | 158.0 | 10 |
| 174.1 | 20 | |||||
| 26 | Oxytetracycline | 11.80 | [M + H]+ | 461.2 | 426.1 | 20 |
| 337.1 | 30 | |||||
| 27 | Ciprofloxacin | 11.80 | [M + H]+ | 332.1 | 245.1 | 23 |
| 288.1 | 17 | |||||
| 28 | Enrofloxacin | 11.90 | [M + H]+ | 360.2 | 245.0 | 26 |
| 316.1 | 19 | |||||
| Enrofloxacin -d5 | 11.90 | [M + H]+ | 365.2 | 321.4 | 18 | |
| 29 | Tulathromycin | 11.90 | [M + 2H]++ | 404.0 | 158.1 | 20 |
| 116.1 | 20 | |||||
| 30 | Danofloxacin | 11.95 | [M + H]+ | 358.2 | 283.1 | 24 |
| 340.1 | 22 | |||||
| 31 | Cefazolin | 12.00 | [M + H]+ | 455.0 | 323.1 | 10 |
| 156.0 | 20 | |||||
| 32 | Sulfamethoxazole | 12.10 | [M + H]+ | 254.1 | 108.1 | 28 |
| 156.0 | 16 | |||||
| 33 | Difloxacin | 12.30 | [M + H]+ | 400.1 | 299.1 | 28 |
| 356.1 | 18 | |||||
| 34 | Ampicillin | 12.30 | [M + H]+ | 350.1 | 106.1 | 20 |
| 160.0 | 20 | |||||
| 35 | Sulfamonomethoxine | 12.30 | [M + H]+ | 281.1 | 108.1 | 28 |
| 156.0 | 16 | |||||
| 36 | Florfenicol | 12.40 | [M + H]+ | 358.0 | 241.0 | 20 |
| 340.0 | 10 | |||||
| Florfenicol -d3 | 12.40 | [M + H]+ | 361.0 | 241.0 | 16 | |
| 37 | Cefoperazone | 12.60 | [M + H]+ | 646.1 | 530.3 | 10 |
| 143.1 | 30 | |||||
| 38 | Sarafloxacin | 12.60 | [M + H]+ | 386.1 | 342.1 | 18 |
| 299.1 | 26 | |||||
| 39 | Epichlortetracycline | 12.85 | [M + H]+ | 479.1 | 444.1 | 20 |
| 154.0 | 30 | |||||
| 40 | Neospiramycin | 13.40 | [M + 2H]++ | 350.2 | 160.1 | 10 |
| 174.1 | 20 | |||||
| 41 | Chlortetracycline | 13.80 | [M + H]+ | 479.1 | 441.1 | 20 |
| 154.0 | 30 | |||||
| 42 | Spiramycin | 14.05 | [M + 2H]++ | 422.3 | 702.4 | 10 |
| 174.1 | 20 | |||||
| Spiramycin -d3 | 14.05 | [M + 2H]++ | 423.8 | 174.0 | 20 | |
| 43 | Sulfadimethoxine | 14.40 | [M + H]+ | 311.1 | 108.1 | 29 |
| 156.0 | 21 | |||||
| 44 | Sulfaquinoxaline | 14.80 | [M + H]+ | 301.1 | 92.1 | 30 |
| 156.0 | 21 | |||||
| 45 | Oxolinic Acid | 15.00 | [M + H]+ | 262.1 | 216.0 | 29 |
| 244.0 | 18 | |||||
| 46 | Ceftiofur | 15.10 | [M + H]+ | 524.0 | 241.0 | 20 |
| 126.0 | 30 | |||||
| Ceftiofur-d3 | 15.10 | [M + H]+ | 527.0 | 244.1 | 15 | |
| Metacycline | 15.15 | [M + H]+ | 443.1 | 426.2 | 16 | |
| 47 | Gamithromycin | 15.40 | [M + H]+ | 777.5 | 158.1 | 39 |
| 619.7 | 32 | |||||
| 48 | Tilmicosin | 15.70 | [M + 2H]++ | 435.3 | 695.5 | 20 |
| 174.1 | 30 | |||||
| 49 | Doxycycline | 15.50 | [M + H]+ | 445.2 | 428.1 | 10 |
| 321.1 | 35 | |||||
| 50 | Nalidixic Acida | 16.80 | [M + H]+ | 233.1 | 159.0 | 30 |
| 187.0 | 25 | |||||
| 51 | Tiamulin | 17.10 | [M + H]+ | 494.3 | 192.1 | 20 |
| 119.0 | 30 | |||||
| 52 | Penicillin G | 17.15 | [M + Na]+ | 357.1 | 198.1 | 20 |
| 182.0 | 20 | |||||
| Penicillin G-d7 | 17.15 | [M + Na]+ | 364.0 | 205.2 | 13 | |
| 53 | Flumequine | 17.20 | [M + H]+ | 262.1 | 202.0 | 33 |
| 244.0 | 19 | |||||
| 54 | Tylosina A | 17.40 | [M + H]+ | 916.5 | 174.1 | 36 |
| 772.5 | 28 | |||||
| 55 | Erythromycin | 17.60 | [M + H]+ | 734.5 | 576.4 | 20 |
| 158.1 | 30 | |||||
| 56 | 3-O-Acetyltylosin | 17.75 | [M + H]+ | 958.5 | 174.0 | 36 |
| 772.5 | 28 | |||||
| 57 | Oxacillin | 18.20 | [M + Na]+ | 424.1 | 265.1 | 20 |
| 182.0 | 20 | |||||
| 58 | Penicillin V | 18.20 | [M + Na]+ | 373.1 | 182.0 | 20 |
| 214.0 | 20 | |||||
| 59 | Cloxacillin | 18.50 | [M + Na]+ | 458.1 | 299.0 | 20 |
| 182.0 | 20 | |||||
| 60 | Valnemulin | 19.10 | [M + H]+ | 565.4 | 263.1 | 20 |
| 72.1 | 30 | |||||
| 61 | Dicloxacillin | 19.20 | [M + Na]+ | 492.0 | 333.0 | 20 |
| 182.0 | 20 | |||||
| 62 | Nafcillin | 19.30 | [M + H]+ | 415.1 | 171.1 | 34 |
| 199.1 | 13 | |||||
| 63 | Tilvalosin | 19.45 | [M + H]+ | 1042.6 | 174.0 | 39 |
| 814.5 | 30 | |||||
| 64 | Rifaximin | 20.60 | [M + H]+ | 786.4 | 754.3 | 20 |
| 736.3 | 30 |
| Muscle | Milk | |||||||
|---|---|---|---|---|---|---|---|---|
| Analyte a,b | CVr,pooled (%) | CVRw, pooled (%) | Rec (%) | ME c (%) | CVr,pooled (%) | CVRw, pooled (%) | Rec (%) | ME c (%) |
| Sulfaguanidine | 5.8 | 10 | 83 | −14 | 5.0 | 14 | 61 | 253 |
| Florfenicol Amine | 3.2 | 6.1 | 85 | −22 | 2.5 | 3.8 | 94 | −13 |
| Sulfanilamide | 6.6 | 8.9 | 74 | −35 | 5.8 | 12 | 66 | −21 |
| Desacetylcephapirin | 7.6 | 7.3 | 76 | −1 | 5.9 | 6.0 | 91 | −21 |
| Amoxicillin | 4.5 | 6.3 | 64 | −1 | 5.8 | 5.8 | 89 | −10 |
| Sulfadiazine | 5.1 | 8.2 | 86 | −18 | 4.8 | 8.6 | 74 | −16 |
| Sulfathiazole | 5.2 | 8.0 | 83 | −6 | 5.2 | 8.3 | 71 | −15 |
| Cephapirin | 6.9 | 7.8 | 75 | 1 | 12 | 12 | 94 | −5 |
| Sulfapyridine | 4.6 | 7.2 | 85 | 13 | 5.8 | 9.5 | 67 | −13 |
| Tildipirosin | 6.2 | 10 | 72 | −8 | 6.0 | 20 | 87 | 5 |
| Cefquinome | 6.1 | 7.2 | 97 | −4 | 11 | 19 | 78 | −16 |
| Sulfamerazine | 3.8 | 5.9 | 89 | 11 | 6.7 | 8.7 | 70 | −13 |
| Cefacetrile | 12 | 13 | 80 | −26 | 22 | 27 | 92 | −5 |
| Cefalonium | 6.7 | 7.3 | 78 | 3 | 8.7 | 8.7 | 93 | −6 |
| Lincomycin | 4.3 | 7.2 | 83 | −12 | 2.3 | 4.4 | 92 | 30 |
| Epitetracycline | 6.2 | 9.2 | 66 | 22 | 4.7 | 7.6 | 96 | 5 |
| Trimethoprim | 3.4 | 7.8 | 90 | −3 | 2.7 | 4.9 | 94 | 4 |
| Thiamphenicol | 10 | 11 | 87 | −13 | 13 | 13 | 92 | −13 |
| Tulathromycin marker | 5.6 | 8.6 | 81 | −13 | 8.7 | 26 | 81 | −18 |
| Marbofloxacin | 6.1 | 7.0 | 87 | −27 | 4.3 | 4.9 | 96 | −11 |
| Sulfamethazine | 4.1 | 7.2 | 85 | −1 | 7.8 | 14 | 68 | −13 |
| Epioxytetracycline | 8.8 | 13 | 62 | 40 | 13 | 15 | 90 | −3 |
| Norfloxacin | - | - | - | - | 7.0 | 8.0 | 94 | 3 |
| Tetracycline | 6.3 | 9.4 | 71 | 27 | 4.5 | 5.0 | 91 | 40 |
| Cefalexin | 6.2 | 8.4 | 64 | 1 | 6.2 | 6.7 | 90 | −7 |
| Oxytetracycline | 6.7 | 8.1 | 63 | 10 | 4.4 | 5.2 | 90 | 1 |
| Ciprofloxacin | 7.5 | 8.4 | 81 | −25 | 6.1 | 7.5 | 95 | −10 |
| Enrofloxacin | 4.7 | 6.6 | 93 | −16 | 4.1 | 5.9 | 99 | −1 |
| Tulathromycin | 8.3 | 16 | 69 | 10 | 7.5 | 13 | 94 | 5 |
| Danofloxacin | 5.7 | 7.2 | 90 | −17 | 4.2 | 5.0 | 97 | 3 |
| Cefazolin | 8.4 | 9.0 | 83 | −8 | 14 | 14 | 94 | −11 |
| Sulfamethoxazole | 7.7 | 9.7 | 88 | −16 | 6.7 | 6.7 | 84 | −24 |
| Difloxacin | 5.0 | 7.4 | 93 | −15 | 3.3 | 4.2 | 96 | −13 |
| Ampicillin | 6.6 | 8.0 | 68 | −2 | 9.4 | 10 | 88 | −10 |
| Sulfamonomethoxine | 4.6 | 7.4 | 86 | 0 | 6.4 | 9.9 | 78 | −19 |
| Florfenicol | 9.8 | 16 | 90 | −17 | 13 | 13 | 94 | −28 |
| Cefoperazone | 6.8 | 8.4 | 85 | −13 | 16 | 18 | 103 | −25 |
| Sarafloxacin | 5.8 | 7.4 | 86 | −26 | 4.8 | 6.6 | 99 | −22 |
| Epichlorotetracycline | 9.3 | 13 | 71 | 65 | 7.4 | 11 | 100 | 22 |
| Neospiramycin | 7.8 | 16 | 67 | 12 | 7.3 | 12 | 86 | 13 |
| Chlortetracycline | 5.8 | 7.7 | 69 | 47 | 5.7 | 7.2 | 92 | 48 |
| Spiramycin | 8.4 | 17 | 74 | 20 | 4.5 | 7.6 | 91 | 21 |
| Sulfadimethoxine | 4.5 | 8.1 | 88 | −12 | 3.9 | 3.9 | 90 | −29 |
| Sulfaquinoxaline | 6.1 | 7.7 | 86 | −26 | 4.6 | 4.8 | 91 | −38 |
| Oxolinic Acid | 4.9 | 6.7 | 97 | 4 | 2.6 | 6.6 | 96 | −3 |
| Ceftiofur | 7.5 | 9.7 | 72 | −21 | 6.7 | 7.1 | 95 | −24 |
| Gamithromycin | 5.6 | 7.1 | 94 | 55 | 3.2 | 4.8 | 98 | 26 |
| Tilmicosin | 6.9 | 10 | 88 | 49 | 3.2 | 4.4 | 95 | 38 |
| Doxycycline | 7.0 | 9.5 | 69 | 1 | 6.6 | 7.4 | 95 | 10 |
| Nalidixic Acid | - | - | - | - | 3.6 | 6.0 | 94 | −3 |
| Penicillin G | 7.9 | 9.0 | 85 | −21 | 12 | 13 | 92 | 37 |
| Tiamulin | 9.3 | 13 | 88 | −21 | 2.5 | 4.1 | 98 | −5 |
| Flumequine | 4.3 | 7.3 | 95 | −3 | 2.9 | 4.5 | 95 | 52 |
| Tylosin A | 7.7 | 15 | 85 | 29 | 2.5 | 4.8 | 96 | 54 |
| Erythromycin A | 4.9 | 8.6 | 89 | −27 | 4.7 | 12 | 67 | −21 |
| 3-O-Acetyltylosin | 9.3 | 16 | 88 | 42 | 3.7 | 5.0 | 95 | 53 |
| Oxacillin | 6.0 | 11 | 83 | −29 | 8.0 | 8.5 | 100 | 1 |
| Penicillin V | 7.9 | 11 | 85 | −28 | 7.5 | 7.7 | 99 | 6 |
| Cloxacillin | 8.2 | 11 | 84 | −27 | 9.8 | 12 | 110 | −25 |
| Valnemulin | 19 | 31 | 75 | −26 | 2.7 | 5.4 | 108 | 55 |
| Dicloxacillin | 6.7 | 10 | 81 | −25 | 13 | 13 | 99 | −10 |
| Nafcillin | 5.0 | 8.2 | 84 | −7 | 5.0 | 5.4 | 99 | −19 |
| Tylvalosin | 9.0 | 19 | 93 | 50 | 5.4 | 6.9 | 101 | 90 |
| Rifaximin | 9.0 | 12 | 91 | 33 | 4.8 | 9.6 | 98 | 7 |
| Method | LC-Q-Orbitrap | LC-QqQ | ||||
|---|---|---|---|---|---|---|
| Sample Code/Year | Matrix | Analyte | Found Concentration (µg·kg−1) | Found Concentration (µg·kg−1) | Consensus Value (µg·kg−1) | Acceptability Range (µg·kg−1) |
| MI1432-A1/2014 a | Milk | Sulfamethazine | 144 | 96 | 103 | 57–150 |
| MI1432-A2/2014 a | Milk | Amoxicillin | 5 | ND | Not assigned | - |
| M1435-A1/2014 a | Pig muscle | Sulfamethazine | 88 | 75 | 69 | 36–102 |
| Pig muscle | Sulfadimethoxine | 32 | 23 | 27 | 12–42 | |
| M1433-A2/2014 a | Turkey muscle | Ciprofloxacin | 5.5 | 5 | 5.6 | 3.2–8.1 |
| Turkey muscle | Enrofloxacin | 173 | 152 | 160 | 92–227 | |
| MI1532-A1/2015 a | Milk | Amoxicillin | 16 | ND | 14 | 5.6–22 |
| MI1532-A2/2015 a | Milk | Sulfamethazine | 165 | 131 | 134 | 75–191 |
| MI1623-A1/2016 a | Milk | Flumequine | 91 | 111 | 88 | 47–129 |
| MI1623-A2/2016 a | Milk | Oxytetracycline | 93 | 55 | 91 | 49–132 |
| MI1715-A2/2017 a | Milk | Danofloxacin | 91 | 80 | 74 | 39–109 |
| 484 (material C)/2018 b | Bovine Muscle | Marbofloxacin | 178 | 193 | 170 | 100–240 |
| 484 (material C)/2018 b | Bovine muscle | Oxytetracycline | 89 | 79 | 106 | 59–152 |
| 334/2019 b | Bovine muscle | Ciprofloxacin | 10 | 10 | NAc | - |
| 334/2019 b | Bovine muscle | Enrofloxacin | 82 | 84 | NAc | - |
| 544/2019 b | Bovine muscle | Tylosin A | 54 | 87 | NAc | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giusepponi, D.; Paoletti, F.; Barola, C.; Moretti, S.; Saluti, G.; Ianni, F.; Sardella, R.; Galarini, R. Transfer of a Multiclass Method for over 60 Antibiotics in Food from High Resolution to Low Resolution Mass Spectrometry. Molecules 2019, 24, 2935. https://doi.org/10.3390/molecules24162935
Giusepponi D, Paoletti F, Barola C, Moretti S, Saluti G, Ianni F, Sardella R, Galarini R. Transfer of a Multiclass Method for over 60 Antibiotics in Food from High Resolution to Low Resolution Mass Spectrometry. Molecules. 2019; 24(16):2935. https://doi.org/10.3390/molecules24162935
Chicago/Turabian StyleGiusepponi, Danilo, Fabiola Paoletti, Carolina Barola, Simone Moretti, Giorgio Saluti, Federica Ianni, Roccaldo Sardella, and Roberta Galarini. 2019. "Transfer of a Multiclass Method for over 60 Antibiotics in Food from High Resolution to Low Resolution Mass Spectrometry" Molecules 24, no. 16: 2935. https://doi.org/10.3390/molecules24162935
APA StyleGiusepponi, D., Paoletti, F., Barola, C., Moretti, S., Saluti, G., Ianni, F., Sardella, R., & Galarini, R. (2019). Transfer of a Multiclass Method for over 60 Antibiotics in Food from High Resolution to Low Resolution Mass Spectrometry. Molecules, 24(16), 2935. https://doi.org/10.3390/molecules24162935