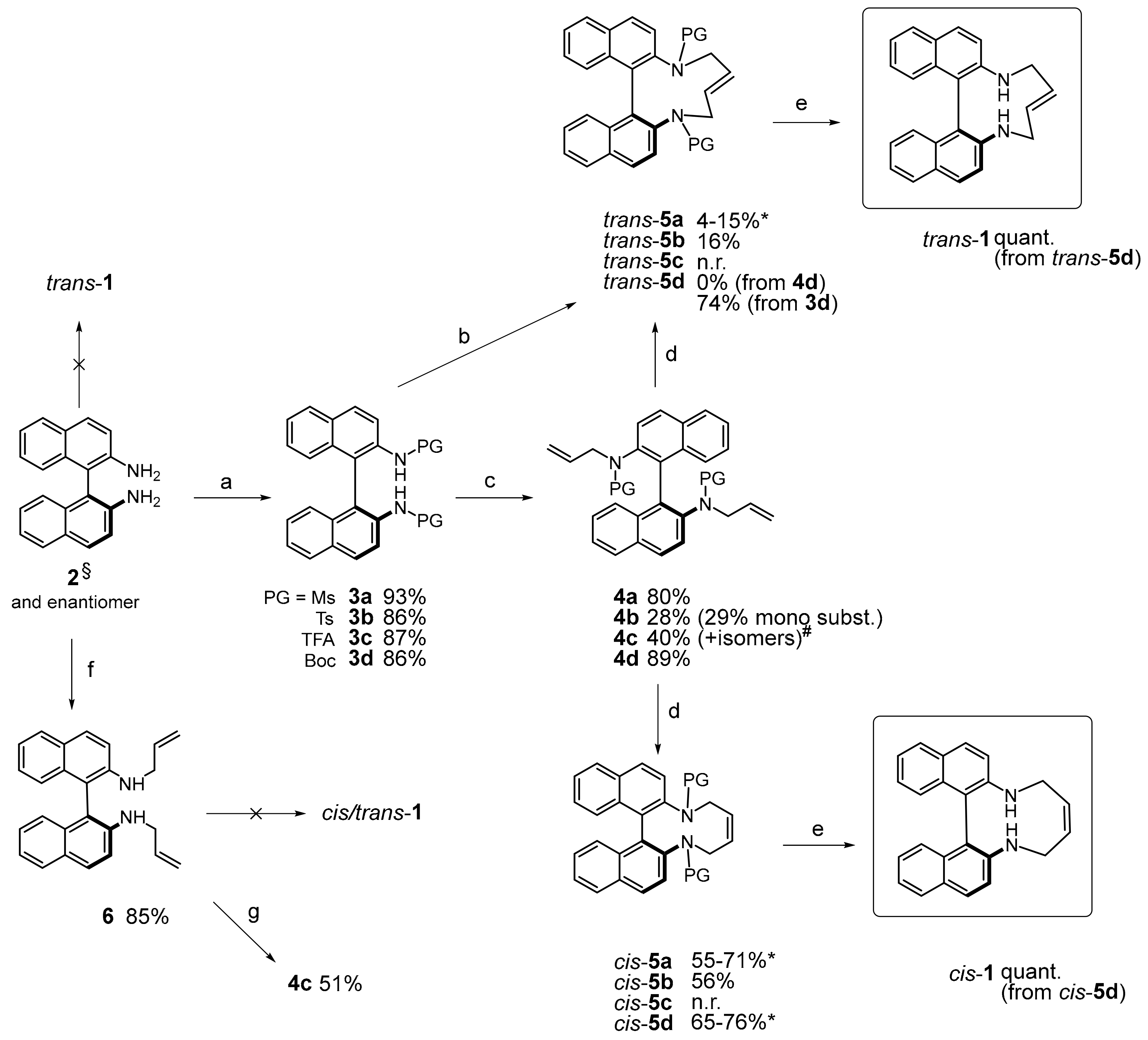
3.2. Synthesis
(E)-11,12,15,16-Tetrahydrodinaphtho [2,1-b:1′,2′-d][1,6]diazecine (trans-1): A solution of trans-5d (67 mg, 0.12 mmol) in DCM (3 mL) was cooled to 0 °C and an excess of TFA (1.5 mL) was added. The mixture was stirred for 2 h and then kept at 4 °C overnight. The reaction was quenched by careful addition of saturated NaHCO3 solution (10 mL) and extracted with DCM. The organic layer was dried (Na2SO4) and the solvent was evaporated at reduced pressure, affording 40 mg (99%) of trans-1 as colorless crystals; m.p.: 204–207 °C. 1H-NMR δ = 7.99 (d, J = 8.8 Hz, 2H); 7.91 (d, J = 8.2 Hz, 2H); 7.48 (d, J = 8.8 Hz; 2H); 7.43 (ddd, J = 8.2, 6.5, 1.7 Hz, 2H); 7.31 (ddd, J = 8.4, 6.6, 1.3 Hz, 2H); 7.27 (dm, J = 8.4 Hz, 2H); 4.67–4.76 (m, 2H); 3.42 (dm, J = 13.1 Hz, 2H); 3.22 (dm, J = 13.1 Hz, 2H); ~2.8 (br.s, 2H). 13C-NMR δ = 144.5 (C); 133.6 (C); 130.8 (C); 129.6 (CH); 128.7 (C); 128.3 (CH); 127.8 (CH); 127.3 (CH); 126.6 (CH); 125.0 (CH); 124.9 (CH); 52.2 (CH2). HRMS: calcd for C24H21N2 [M + H]+: 337.1698; found: 337.1696.
(Z)-11,12,15,16-Tetrahydrodinaphtho[2,1-b:1′,2′-d][1,6]diazecine (cis-1): The same procedure was applied as given for trans-1; yield: 34 mg (99%, colorless crystals, 0.1 mmol scale); m.p.: 184–185 °C. 1H-NMR δ = 7.90 (d, J = 8.7 Hz, 2H); 7.82 (br.d, J = 8.0 Hz, 2H); 7.35 (d, J = 8.8 Hz, 2H); 7.29 (ddd, J = 8.1, 6.6, 1.5 Hz, 2H); 7.21 (ddd, J = 8.5, 6.6, 1.4 Hz, 2H); 7.17 (dm, J = 8.5 Hz, 2H); 5.92–6.00 (m, 2H); 3.73–3.91 (m, 4H); 3.30 (br.s, 2H). 13C-NMR δ = 145.8 (C); 134.0 (C); 132.7 (CH); 129.7 (CH); 129.3 (C); 128.1 (CH); 126.7 (CH); 125.4 (CH); 123.2 (CH); 118.5 (CH); 117.7 (C); 45.1 (CH2). HRMS: calcd for C24H21N2 [M + H]+: 337.1705; found: 337.1696.
N,N′-([1,1′-Binaphthalene]-2,2′-diyl)dimethanesulfonamide (3a): To a solution of 2 (142 mg, 0.5 mmol) in pyridine (1 mL)/DCM (4 mL) was added mesylchloride (126 mg, 1.1 mmol) and the orange mixture was stirred at r.t. After 24 h, a second portion of mesylchloride was added (126 mg, 1.1 mmol) and stirring was continued. After complete conversion (TLC), the reaction was acidified (HCl, 1 M) and sufficiently extracted with DCM. The organic phase was dried (MgSO4) and the solvent removed under reduced pressure. The crude mixture was purified by MPLC (EtOAc (30→50%)/heptane) to yield 223 mg (quant.) of 3a as a mixture of tautomers; m.p.: 221–222 °C. 1H-NMR (C2-symmetric tautomer) δ = 8.10 (d, J = 8.9 Hz, 2H); 8.02 (d, J = 8.9 Hz, 2H); 7.95 (br.d, J = 8.2 Hz, 2H); 7.47 (ddd, J = 8.0, 6.8, 1.1 Hz, 2H); 7.31 (ddd, J = 8.4, 6.9, 1.3 Hz, 2H); 6.99 (br.d, J = 8.3 Hz, 2H); 6.02 (br.s, 2H); 2.97 (s, 6H). 13C-NMR δ = 134.4 (C); 132.5 (C); 131.5 (CH); 131.2 (C); 128.7 (CH); 128.2 (CH); 126.1 (CH); 124.5 (CH); 118.5 (C); 118.2 (CH); 41.0 (CH3). HRMS: calcd for C22H20NaN2O4S2 [M + Na]+: 463.0762; found 463.0762.
N,N′-([1,1′-Binaphthalene]-2,2′-diyl)bis(4-methylbenzenesulfonamide) (
3b): A similar procedure as given for
3a was applied, yielding 252 mg (85%, 0.5 mmol scale) of
3b as off-white solid. NMR spectra are in agreement with references [
28,
39].
N,N′-([1,1′-Binaphthalene]-2,2′-diyl)bis(2,2,2-trifluoroacetamide) (3c): To a solution of 1,1′-binaphthyl-2,2′-diamine 2 (569 mg, 2 mmol) in THF (30 mL) was added solid Na2CO3 (212 mg, 2 mmol) followed by dropwise addition of TFAA (1.27 mL, 9 mmol) in THF (30 mL). After 2 h the reaction was quenched with sat. NaHCO3 solution and extracted with EtOAc, washed with H2O and brine, dried (Na2SO4), and evaporated to give 826 mg (87%) of 3c; colorless crystals; m.p.: 195–196 °C. The product was pure enough for the next step. 1H-NMR: δ = 8.19 (d, J = 8.9 Hz, 2H); 8.14 (d, J = 8.9 Hz, 2H); 8.01 (d, J = 8.2 Hz, 2H); 7.72 (s, 2H); 7.56 (ddd, J = 8.2, 6.8, 1.2 Hz, 2H); 7.38 (ddd, J = 8.4, 6.8, 1.3 Hz, 2H); 7.13 (dm, J = 8.6 Hz, 2H). 13C-NMR δ = 132.3 (C); 131.8 (C); 131.7 (C); 130.9 (CH); 128.7 (CH); 128.2 (CH); 127.0 (CH); 124.7 (CH); 124.0 (C); 121.8 (CH); 115.3 (CF3, JCF ~280 Hz). HRMS: calcd for C24H15F6N2O2 [M + H]+: 477.1038; found 477.1041.
N,N′-([1,1′-Binaphthalene]-2,2′-diyl)bis(N-allylmethanesulfonamide) (4a): Bis(N-mesylate) 3a (220 mg, 0.5 mmol) was suspended in acetonitrile and degassed. To this was added allylbromide (420 mg, 3.5 mmol) and K2CO3 (350 mg, 2.5 mmol), and the mixture was stirred at 85 °C for 48 h. Extractive work-up with EtOAc/water left crude diallylated product which was purified by column chromatography (EtOAc (20→30%)/heptane) to yield 208 mg (80%) of 4a; m.p.: 197–199 °C. 1H-NMR: δ = 7.99 (d, J = 8.8 Hz, 2H); 7.92 (br.d, J = 8.2 Hz, 2H); 7.65 (d, J = 8.9 Hz, 2H); 7.49 (ddd, J = 8.0, 6.8, 1.0 Hz, 2H); 7.26 (ddd, J = 8.4, 6.9, 1.3 Hz, 2H); 7.08 (d, J = 8.4 Hz, 2H); 5.67 (br.s, 2H); 4.96–5.08 (m, 4H); 3.73–4.05 (br.m, 4H); 2.54 (br.s, 6H). 13C-NMR δ = 138.1 (br.C); 133.8 (C); 132.6 (C); 132.3 (C); 129.2 (CH); 128.2 (br.CH); 127.9 (CH); 127.7 (br.CH); 126.7 (CH); 126.6 (CH); 119.3 (CH2); 54.4 (br.CH2); 41.7 (br.CH3). HRMS: calcd for C28H28NaN2O4S2 [M + Na]+: 543.1388; found 543.1394.
N,N′-([1,1′-Binaphthalene]-2,2′-diyl)bis(N-allyl-4-methylbenzenesulfonamide) (4b): A similar procedure as given for the synthesis of 4a was applied using an excess of allylbromide (8 equ.) and 48 h reflux to afford 92 mg (28%, 0.5 mmol scale) of 4b (along with 92 mg, 29% of mono-allylated product); m.p.: 125–128 °C. 1H-NMR: δ = 7.94 (d, J = 8.8 Hz, 2H); 7.87 (d, J = 8.0 Hz, 2H); 7.61 (br.d, J = 8.8 Hz, 2H); 7.43 (ddd, J = 8.0, 6.9, 1.1 Hz, 2H); 7.14–7.29 (br.m, 4H); 7.18 (ddd, J = 8.2, 6.7, 1.1 Hz, 2H); 7.06 (br.d, J = 8.4 Hz, 2H); 6.96–7.08 (br.m, 4H); 5.68 (br.s, 2H); 4.76–4.91 (br.m, 4H); 4.03–4.21 (br.m, 2H); 3.73–3.93 (br.m, 2H); 2.35 (s, 6H). 13C-NMR δ = 143.0 (br.C); 137.3 (br.C); 134.4 (C); 134.0 (C); 133.6 (br.CH); 132.6 (C); 129.1 (CH); 128.8 (CH); 128.7 (br.CH); 127.8 (CH); 127.5 (br.CH); 126.6 (CH); 126.2 (CH); 118.7 (CH2); 21.5 (CH3). HRMS: calcd for C40H36NaN2O4S2 [M + Na]+: 695.2014; found 695.2026.
N,N′-([1,1′-binaphthalene]-2,2′-diyl)bis(N-allyl-2,2,2-trifluoroacetamide) (
4c): (
Method A) Bis(trifluoroacetamide)
3c (238 mg, 0.5 mmol) was dissolved in MeCN (10 mL) and degassed. To this was added K
2CO
3 (346 mg, 2.5 mM) and allylbromide (423 mg, 303 µL, 3.5 mM) and the mixture was stirred at reflux for 20 h. The reaction was worked up with DCM (50 mL)/water (20 mL). The organic phase was washed with water and brine and dried (MgSO
4). After removal of solvents the crude material was subjected to MPLC (EtOAc (5→20%)/heptane) afforded 109 mg (95% purity, 40% yield) of
4c as mixture of rotamers. Due to complexity of the
1H and
13C-NMR spectra, no signal assignment was possible (see
Supplementary Materials).
19F-NMR: δ = −66.43 (s); −66.53 (q,
J = 6.0 Hz); −68.43 (q,
J = 6.0 Hz); −68.65 (s). HRMS: calcd for C
30H
22NaF
6N
2O
2 [M + Na]
+: 579.1483; found 579.1467.
(Method B) To a solution of diallyldiamine 6 (142 mg, 0.5 mmol), Et3N (101 mg, 139 µL, 1 mmol) and DIMAP (122 mg, 1 mmol) in DCM (5 mL) was added trifluoroacetic anhydride (420 mg, 282 µL, 2 mmol) at r.t. and the solution was stirred for 24 h. Extractive work-up with DCM (30 mL)/water (20 mL) and MPLC (see above) afforded of 4c (140 mg, 50%, colorless crystals, m.p.: 175–176 °C).
Di-tert-butyl [1,1′-binaphthalene]-2,2′-diylbis(allylcarbamate) (
4d): A stirred suspension of Boc protected 2,2′-diamino-1,1′-binaphthyl
3d [
29] (242 mg, 0.5 mmol) in DMF (5 mL) was mixed at 0 °C with NaH (60 mg, 1.5 mmol, 60% in mineral oil) and then warmed up to r.t. during 30 min. The mixture was cooled to 0 °C again and treated with allylbromide (173 µL, 2 mmol) After stirring for 16 h at r.t. the reaction was diluted with EtOAc, washed with water and brine, dried (MgSO
4), and concentrated under reduced pressure. Purification by MPLC (EtOAc (10→30%)/heptane) afforded 270 mg (93% purity, 89% yield, colorless foam) of
4d as mixture of rotamers.
1H-NMR δ = 7.88 (d,
J = 7.9 Hz, ~2H); 7.87 (d,
J = 8.5 Hz, ~2H); 7.42 (ps.t,
J = 7.6 Hz, ~2H); 7.33 (d,
J = 8.9 Hz, ~1H); 7.17 (ps.t,
J = 7.5 Hz, ~1H); 6.85 (d,
J = 8.5 Hz, ~1H); 5.59–5.72 (m, ~1H); 4.79 (d,
J = 10.1 Hz, ~1H); 4.54 (d,
J = 17.1 Hz, ~1H); 3.99 (dd,
J = 15.4, 4.0 Hz, ~1H); 2.91 (dd,
J = 15.4, 7.8 Hz, ~1H); 1.44 (br.s, >9H). In addition several unresolved multiplets were observed between 2.8 and 8.0 ppm. HRMS: calcd for C
36H
40N
2NaO
4 [M + Na]
+: 587.2886; found: 587.2893.
Repetition of allylation of 3d in THF with a reaction time of 2 h at r.t. afforded 112 mg (71%, 0.3 mmol scale) of mono-allylated product, tert-butyl allyl(2′-((tert-butoxycarbonyl)amino)-[1,1′-binaphthalen]-2-yl)carbamate. 1H-NMR δ = 6.57–8.30 (several br.m, ~12H); 5.40–5.90 (m, 1H); 4.45–4.92 (m, 2H); 2.80–4.20 (m, 2H); 1.37; 1.28; 1.25 (3× br.s, ~18H). HRMS: calcd for C33H36N2NaO4 [M + Na]+: 547.2573; found: 547.2576.
(Z)- and (E)-11,16-Bis(methylsulfonyl)-11,12,15,16-tetrahydrodinaphtho[2,1-b:1′,2′-d][1,6]diazecine, (cis- and trans-5a) (Typical procedure): To a solution of 4a (52 mg, 0.1 mmol) in DCM (7 mL) was added at 40 °C Grubbs II catalyst (8.5 mg, 10 mol%) in DCM (3 mL) during 6 h by syringe pump. After 24 h the solvent was removed and the product mixture separated by chromatography (30→50% EtOAc/PE) to yield trans-5a (2 mg, 4%), cis-5a (35 mg, 71%), and a side product with shifted double bond cis-5a′ (9 mg, 18%). Repetition with Grubbs I catalysts afforded trans-5a (15%), cis-5a (55%), and 4a (2%).
trans-5a: Colorless crystals, m.p.: 248–255 °C, dec. 1H-NMR δ = 8.06 (d, J = 8.7 Hz, 2H); 7.92 (br.d, J = 8.2 Hz, 2H); 7.63 (br.d, J = 8.6 Hz, 2H), 7.62 (d, J = 8.6 Hz, 2H); 7.53 (ddd, J = 8.1, 6.9, 1.3 Hz, 2H); 7.38 (ddd, J = 8.3, 6.8, 1.3 Hz, 2H); 4.78–4.88 (m, 2H); 3.96–4.04 (m, 2H); 3.54–3.65 (m, 2H); 2.04 (s, 6H). 13C-NMR δ = 137.5 (C); 135.1 (C); 133.9 (C); 132.7 (C); 130.1 (CH); 129.5 (CH);129.1 (CH); 128.5 (CH); 127.5 (CH); 127.1 (CH); 126.5 (CH); 53.8 (CH2); 40.2 (CH3). HRMS (EI) calcd for C26H24N2O4S2 [M]+: 492.1178; found: 492.1171.
cis-5a: Colorless crystals, m.p.: 125–129 °C. 1H-NMR δ = 8.00 (d, J = 8.8 Hz, 2H); 7.90 (d, J = 8.2 Hz); 7.55 (d, J = 8.7 Hz, 2H); 7.47–7.53 (m, 2H); 7.29–7.35 (m, 4H); 5.80–5.88 (m, 2H); 4.16–4.23 (m, 4H); 1.88 (s, 6H). 13C-NMR δ = 137.8 (C); 135.0 (C); 133.5 (C); 132.4 (C); 129.9 (CH); 129.7 (CH); 128.4 (CH); 128.2 (CH); 127.7 (CH); 127.0 (CH); 126.7 (CH); 46.8 (CH2); 40.7 (CH3). HRMS (EI) calcd for C26H24N2O4S2 [M]+: 492.1178; found: 492.1168.
(Z)- and (E)-11,16-Ditosyl-11,12,15,16-tetrahydrodinaphtho[2,1-b:1′,2′-d][1,6]diazecine (cis- and trans-5b): A similar procedure as given for 5a was applied yielding a mixture of cis- and trans-5b, which was only in part separable affording trans-5b (~16%, enriched sample) and cis-5b (36 mg, 56%, 0.1 mmol scale) as a colorless foam.
trans-5b: 1H-NMR δ = 8.13 (d, J = 8.8 Hz, 2H); 8.04 (d, J = 8.1 Hz, 2H); 7.88 (d, J = 8.8 Hz, 2H); 7.81 (d, J = 8.4 Hz, 2H); 7.58 (ddd, J = 8.1, 6.8, 1.1 Hz, 2H); 7.39 (ddd, J = 8.3, 6.8, 1.3 Hz, 2H); 6.89 (d, J = 7.8 Hz, 4H); 6.70 (d, J = 8.2 Hz, 4H); 4.53–4.63 (m, 2H); 3.57–3.64 (m, 2H); 3.33–3.44 (m, 2H); 2.29 (s, 6H).
cis-5b: 1H-NMR δ = 7.98 (d, J = 8.8 Hz, 2H); 7.93 (br.d, J = 8.2 Hz, 2H); 7.50 (ddd, J = 8.1, 6.8, 1.1 Hz, 2H); 7.43 (d, J = 8.8 Hz, 2H); 7.38 (br.d, J = 8.4 Hz, 2H); 7.28 (ddd, J = 8.1, 6.7, 1.2 Hz, 2H); 6.94 (br.s, 8H); 5.42 (m, 2H); 3.92 (m, 4H); 2.31 (s, 6H). 13C-NMR δ = 143.5 (C); 137.4 (C); 135.9 (C); 135.6 (C); 134.1 (C); 132.6 (C); 129.41 (CH); 129.38 (CH); 129.3 (CH); 129.1 (CH); 128.1 (CH); 127.3 (CH); 126.7 (CH); 126.2 (2×CH), 47.2 (CH2); 21.4 (CH3). HRMS calcd for C38H33N2O4S2 [M + H]+: 645.1882; found: 645.1887.
Di-tert-butyl(E)-12,15-dihydrodinaphtho[2,1-b:1′,2′-d][1,6]diazecine-11,16-dicarboxylate (trans-5d): To a suspension of 3d (484 mg, 1 mmol) in DMF (10 mL) was added NaH (120 mg, 3 mmol, 60% in mineral oil) at 0 °C with stirring and after gas evolution ceased stirring was continued at r.t. for 30 min. The turbid mixture was again cooled to 0 °C, solid trans-1,4-dibromobut-2-ene (214 mg, 1 mmol) was added and reaction stirred at r.t. for 20 h. The mixture was diluted with EtOAc, washed with water and brine, dried and concentrated under reduced pressure. The crude product was purified by column chromatography (EtOAc(10→30%)/heptane) to give 399 mg (74%) of trans-5d as an off-white solid, m.p.: 242–243 °C. 1H-NMR (unresolved mixture of conformers) δ = 7.99 (br.d, J = 8.6 Hz, 2H); 7.86 (br.d, J = 8.2 Hz, 2H); 7.41–7.60 (br.m, 2H); 7.45 (br.pt, J = 7.4 Hz); 7.18–7.34 (br.m, 2H); 7.23 (ddd, J = 8.4, 7.0, 1.1 Hz, 2H); 4.79–4.89 (br.m, 2H); 4.40–4.90 (br.m, 2H); 3.18–3.42 (br.m, 2H); 1.15–1.45 (br.m, 18H). HRMS calcd for C34H36N2Na2O4 [M + Na]+: 559.2573; found: 559.2567.
Di-tert-butyl(Z)-12,15-dihydrodinaphtho[2,1-b:1′,2′-d][1,6]diazecine-11,16-dicarboxylate (cis-5d): A similar procedure as given for 5a was applied affording exclusively cis-5d in 41 mg (76%, Grubbs I or Grubbs-Hoveyda II) or 35 mg (65%, Grubbs II) yield. Experiments were performed on a 0.1 mmol scale and 10 mol% of catalyst; colorless crystals, m.p.: 230–231 °C. 1H-NMR (unresolved mixture of conformers) δ = 7.86–7.98 (br.m, 2H); 7.84 (br.d, J = 7.7 Hz, 2H); 7.37–7.46 (br.m, 2H); 7.16–7.36 (br.m, 6H); 5.62–5.73 (br.m, 2H); 3.69–4.39 (br.m, 4H); 0.95–1.38 (br.m, 18H). HRMS calcd for C34H36N2Na2O4 [M + Na]+: 559.2573; found: 559.2566.
N2,N2′-Diallyl-[1,1′-binaphthalene]-2,2′-diamine (6): To a solution of 2 (1.421 g, 5 mmol) in benzene (5 mL) was added allylalcohol (0.850 mL, 12.5 mmol) and dried molsieve (1 g, 4 Å) and the mixture was degassed. Subsequently, Ti(i-OPr)4, (710 mg, 740 µL, 2.5 mmol), PPh3 (105 mg, 0.4 mmol), and Pd(OAc)2 (22.5 mg, 0.1 mmol) was added and the reaction was stirred under Ar at 50 °C. The conversion was monitored by TLC. After extractive work-up with DCM/water, drying (MgSO4), and evaporation, the crude product was purified by chromatography in EtOAc (5→20%)/heptane to afford 1.55 g (85%) of 6 as a slightly brown crystaline solid; m.p.: 95–99 °C. 1H-NMR δ = 7.87 (d, J = 9.0 Hz, 2H); 7.78 (dm, J = 7.7 Hz, 2H); 7.21 (d, J = 9.1 Hz, 2H); 7.14–7.22 (m, 4H); 6.99 (dm, J = 7.9 Hz, 2H); 5.77 (ddm, J = 17.3, 10.3 Hz, 2H); 5.12 (dm, J = 17.3 Hz, 2H); 5.02 (dm, J = 10.3 Hz, 2H); 3.92 (br.s, 2H); 3.77–3.86 (br.m, 4H). 13C-NMR δ = 144.2 (C); 135.7 (CH); 133.9 (C); 129.5 (CH); 128.1 (CH); 127.7 (C); 126.7 (CH); 123.9 (CH); 122.0 (CH); 115.6 (CH2); 114.2 (CH); 112.0 (C); 46.1 (CH2). HRMS calcd for C26H25N2 [M + H]+: 365.2018; found: 365.2011.
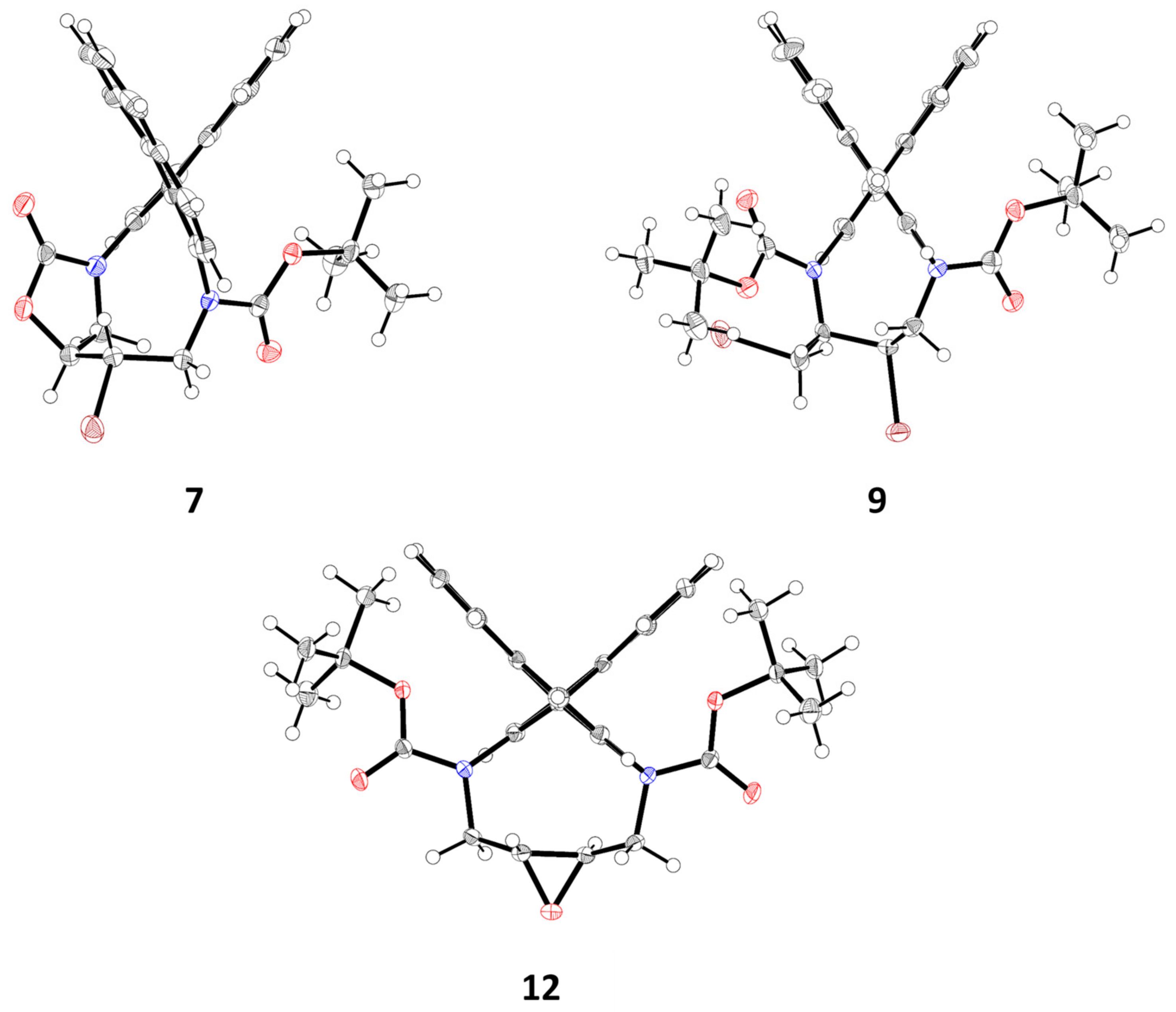
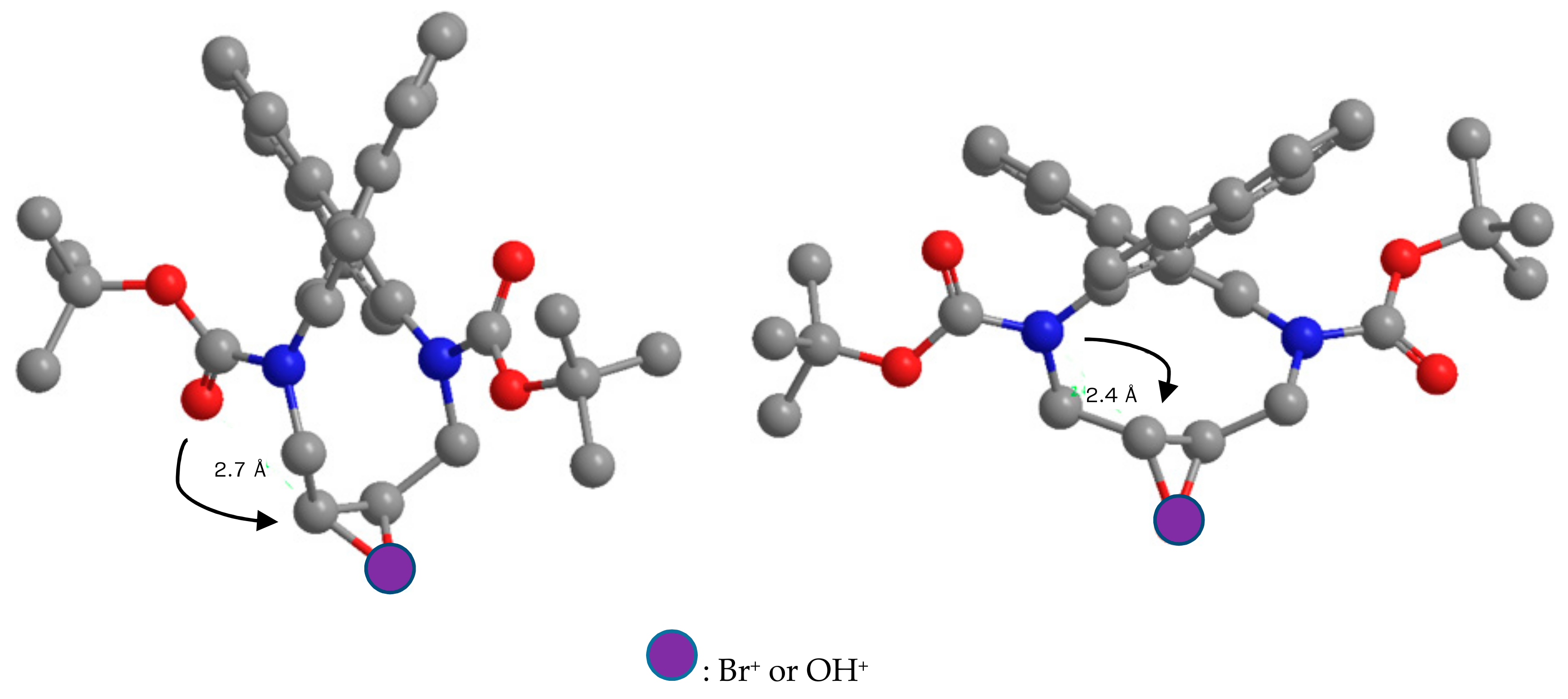
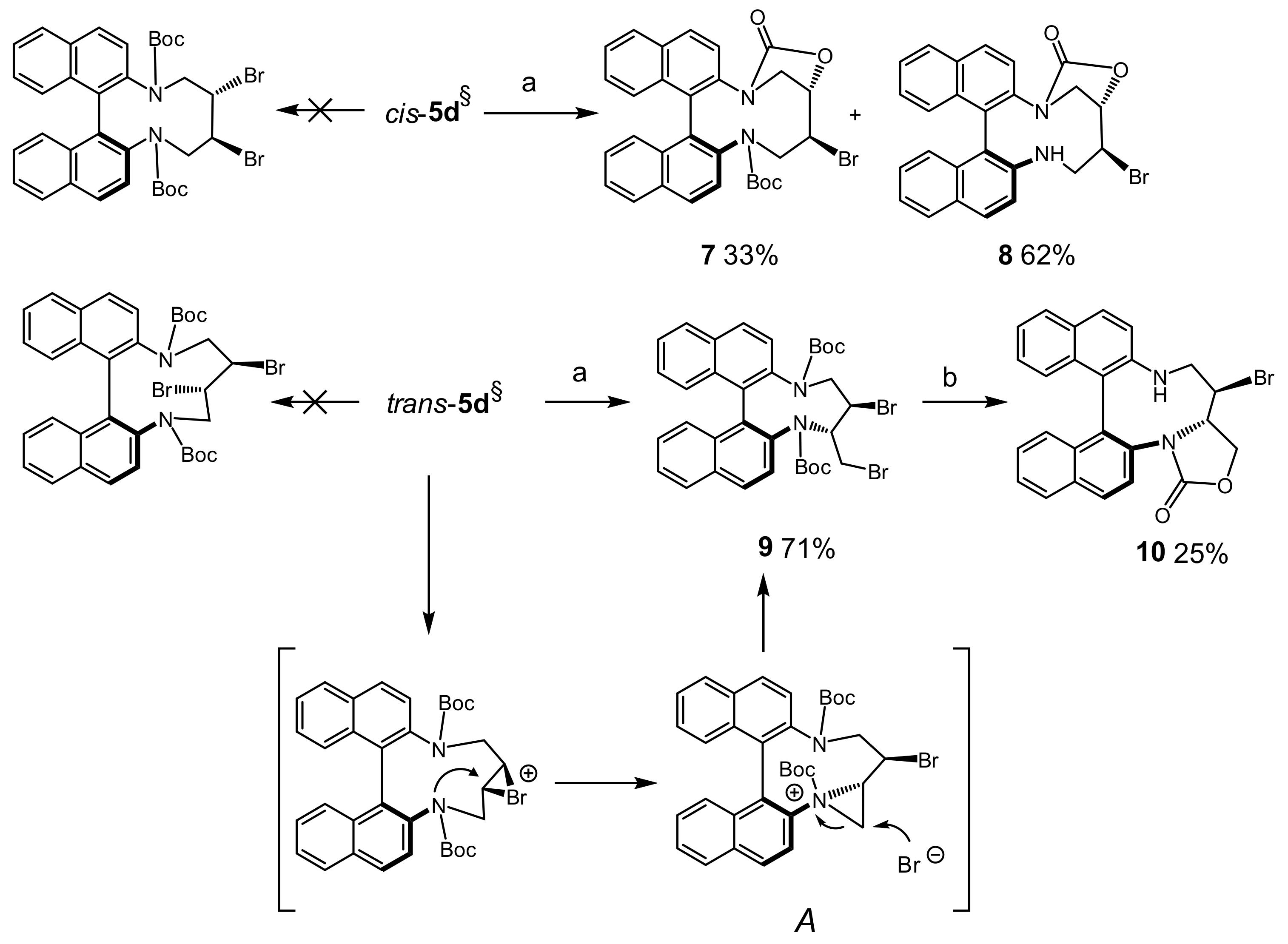
tert-Butyl (10S*,11S*)-11-bromo-8-oxo-11,12-dihydro-8H-7,10-methanodinaphtho[2,1-d:1′,2′-f][1]oxa[3,8]di-azacycloundecine-13(10H)-carboxylate (7) and (10S*,11S*)-11-Bromo-10,11,12,13-tetrahydro-8H-7,10-methanodinaphtho[2,1-d:1′,2′-f][1]oxa[3,8]diazacycloundecin-8-one (8): Diazecine cis-5d (54 mg, 0.1 mmol) was added to DCM (2 mL) and the solution was cooled to 0 °C. Bromine (26 mg, 0.16 mmol) dissolved in DCM (1 mL) was added dropwise. After 16 h at r.t. the pale yellow reaction was diluted with DCM (10 mL) and stirred with NaHSO3 solution (10%, 3 mL). The organic phase was separated, dried, and evaporated. MPLC (EtOAc(20→40%)/heptane) afforded fractions containing 7 (18 mg, 33%, colorless crystals, m.p.: 180–185 °C, dec.) and 8 (28 mg, 62%, colorless crystals, m.p.: 254–256 °C, dec.).
7: 1H-NMR δ = 8.02 (d, J = 8.7 Hz, 1H); 7.96 (d, J = 8.5 Hz, 1H); 7.88 (br.d, J = 8.3 Hz); 7.48 (d, J = 8.8 Hz, 1H); 7.43–7.48 (m, 2H); 7.42 (d, J = 8.7 Hz, 1H); 7.18–7.27 (m, 3H); 7.01 (dm, J = 8.7 Hz, 1H); 5.01 (dd, J = 6.4, 3.6 Hz, 1H); 4.72 (dd, J = 12.7, 15.0 Hz, 1H); 4.29 (dd, J = 9.8, 6.4 Hz, 1H); 4.18 (dd, J = 15.0, 3.2 Hz, 1H); 4.09 (d, J = 9.9 Hz, 1H); 3.79 (dps.t, J = 12.7, 3.4 Hz, 1H); 0.71 (s, 9H). 13C-NMR δ = 153.4 (C); 134.23 (C); 134.17 (C); 133.7 (C); 133.1 (C); 132.4 (C); 132.2 (C); 132.1 (C); 131.9 (C); 130.3 (CH); 130.1 (CH); 128.5 (CH); 128.0 (CH); 127.9 (CH); 127.7 (CH); 126.6 (CH); 126.5 (CH); 126.4 (CH); 126.2 (CH); 123.3 (CH); 120.9 (CH); 82.0 (CH); 81.8 (C); 52.8 (CH2); 50.5 (CH2); 49.0 (CH); 27.4 (CH3). HRMS: calcd for C30H27BrN2NaO4 [M + Na]+: 583.1031; found: 583.1024.
8:
1H-NMR (mixture of conformers) δ = 8.03 (d,
J = 8.5 Hz, 0.4H); 8.02 (d,
J = 8.4 Hz, 0.6H); 7.92–7.96 (m, 1.4H); 7.90 (d,
J = 8.8 Hz, 0.6H); 7.50–7.55 (m, 1H); 7.47 (d,
J = 8.5 Hz, 0.6H); 7.46 (d,
J = 8.7 Hz, 0.4H); 7.16-7.30 (m, 5H); 6.99 (dm,
J = 8.5 Hz, 0.6H); 6.85 (dm,
J = 9.1 Hz, 0.4H); 5.10–5.14 (m, 1H); 4.47–4.56 (m, 2H); 4.27–4.36 (m, 1H); 3.81–3.89 (m, 2H); 3.42–3.51 (m, 1H).
13C-NMR (mixture of conformers [
40]) δ = 152.4 (C); 139.4 (C); 139.0 (C); 134.14 (C); 134.07 (C); 134.05 (C); 133.9 (C); 133.7 (C); 132.9 (C); 132.6 (C); 130.5 (CH
B); 130.4 (CH
A); 130.2 (CH
A); 130.1 (CH
B); 129.98 (CH
B); 129.95 (C); 129.6 (C); 129.5 (CH
B); 128.9 (C); 128.3 (CH
B); 128.22 (CH
A); 128.21 (CH
A); 127.43 (CH
B); 127.42 (CH
B); 127.36 (CH
B); 127.2 (CH
A); 127.1 (CH
B); 126.8 (CH
A,CH
B); 126.6 (CH
A); 125.6 (CH
A); 123.41 (CH
A); 123.38 (CH
B); 123.3 (CH
A); 117.01 (C); 116.99 (C); 115.5 (CH
B); 114.4 (CH
A); 79.1 (CH
A); 79.0 (CH
B); 50.4 (CH
A); 50.1 (CH
2A); 50.04 (CH
B); 50.00 (CH
2B); 48.4 (CH
2A); 48.3 (CH
2B). HRMS: calcd for C
25H
19BrN
2NaO
4 [M + Na]
+: 481.0528; found: 481.0516.
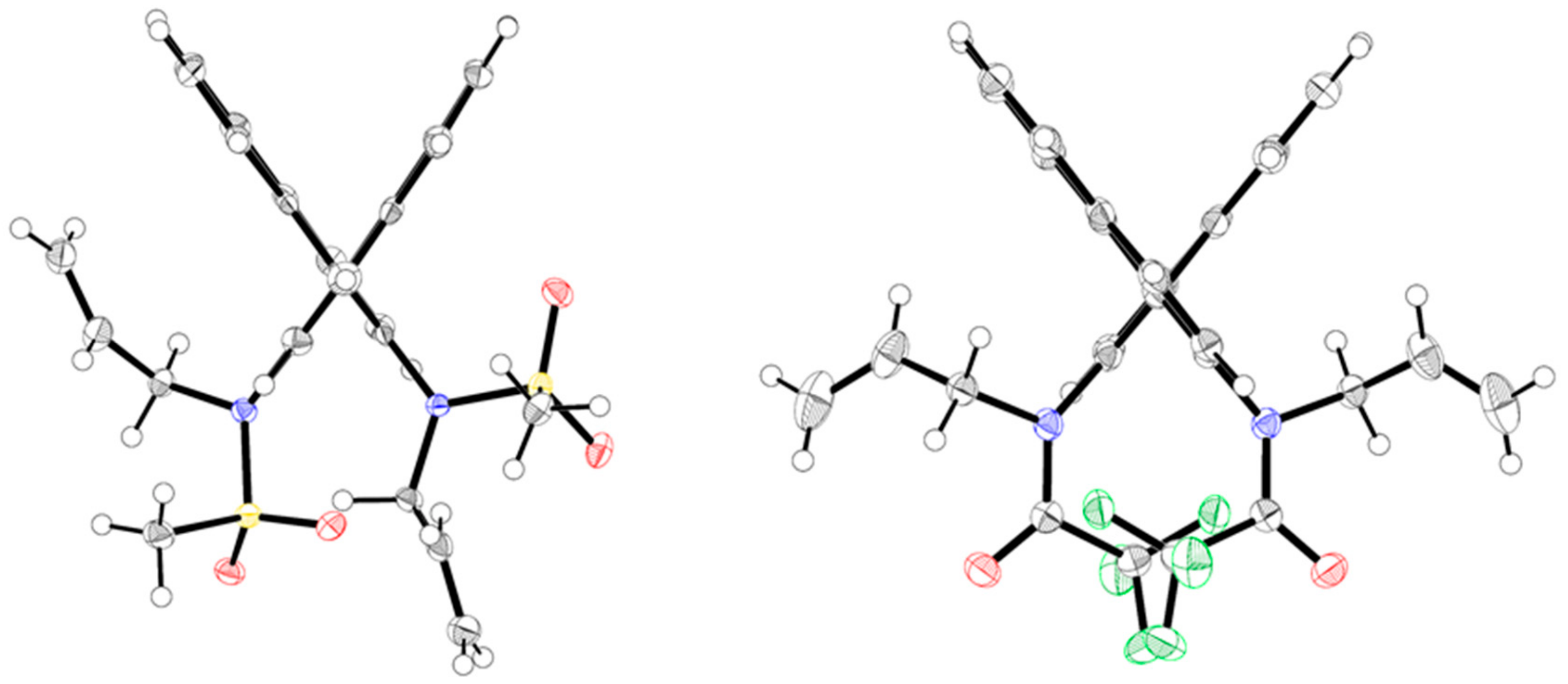
Di-tert-butyl (8S*,9R*)-9-bromo-8-(bromomethyl)-9,10-dihydro-7H-dinaphtho[2,1-f:1′,2′-h][1,5] diazonine-7,11(8H)-dicarboxylate (9): Diazecine trans-5d was treated with bromine in DCM similarly as described for cis-5d to afford 9 (49 mg, 71%, 0.1 mmol scale) after MPLC (EtOAc(5→20%)/heptane); m.p.: 180–183 °C, dec. 1H-NMR (mixture of conformers, THF solvate) δ = 7.63–8.03 (m, 5.13H); 6.85–7.52 (m, 6.90H); 5.10–5.19 (m, 0.42H); 4.49–4.79 (m, 2.60H); 3.90–3.95 (m, 0.49H); 3.73–3.77 (m, 2H, THF); 3.57–3.89 (m, 4.11H); 3.48 (dd, J = 14.4, 8.4 Hz, 0.48H); 1.81–1.90 (m, 2H, THF); 0.70–1.12 (6× s, 18.2H). HRMS: calcd for C34H3679Br81BrN2NaO4 [M + Na]+: 717.0919; found: 717.0923.
Attempted deprotection of9: Treatment of 9 with excess of TFA in DCM (1:1) afforded bromocarbamate 10 as white solid (12 mg, 25%, 0.1 mmol scale). 1H-NMR δ = 8.03 (dm, J = 8.9 Hz, 1H); 7.91 (dm, J = 8.3 Hz, 1H); 7.84 (dm, J = 8.9 Hz, 1H); 7.78 (dm, J = 8.9 Hz, 1H); 7.77 (d, J = 8.8 Hz, 1H); 7.47 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H); 7.23–7.27 (m, 2H); 7.14 (ddd, J = 8.4, 6.9, 1.4 Hz, 1H); 7.09 (d, J = 8.9 Hz, 1H); 7.01 (dm, J = 8.4 Hz, 1H); 6.84 (dm, J = 8.4 Hz, 1H); 4.80 (ddd, J = 10.9, 9.6, 8.3 Hz, 1H); 4.34 (dd, J = 8.8, 8.2 Hz, 1H); 4.08 (dt, J = 11.1, 2.1 Hz, 1H); 3.84 (dd, J = 9.4, 9.0 Hz, 1H); 3.53 (dd, J = 17.2, 2.0 Hz, 1H); 2.93 (dd, J = 17.1, 2.4 Hz, 1H). 13C-NMR δ = 156.3 (C); 142.8 (C); 134.6 (C); 133.6 (C); 133.4 (C); 132.5 (C); 130.2 (CH); 130.0 (C); 129.6 (CH); 128.6 (C); 128.0 (CH); 127.6 (CH); 127.5 (CH); 127.2 (CH); 126.6 (CH); 125.64 (CH); 125.55 (CH); 123.6 (CH); 123.5 (CH); 120.5 (CH); 114.0 (C); 69.5 (CH2); 60.0 (CH); 57.3 (CH); 48.0 (CH2). HRMS: calcd for C25H19BrN2NaO2 [M + Na]+: 483.0507; found: 483.0505.
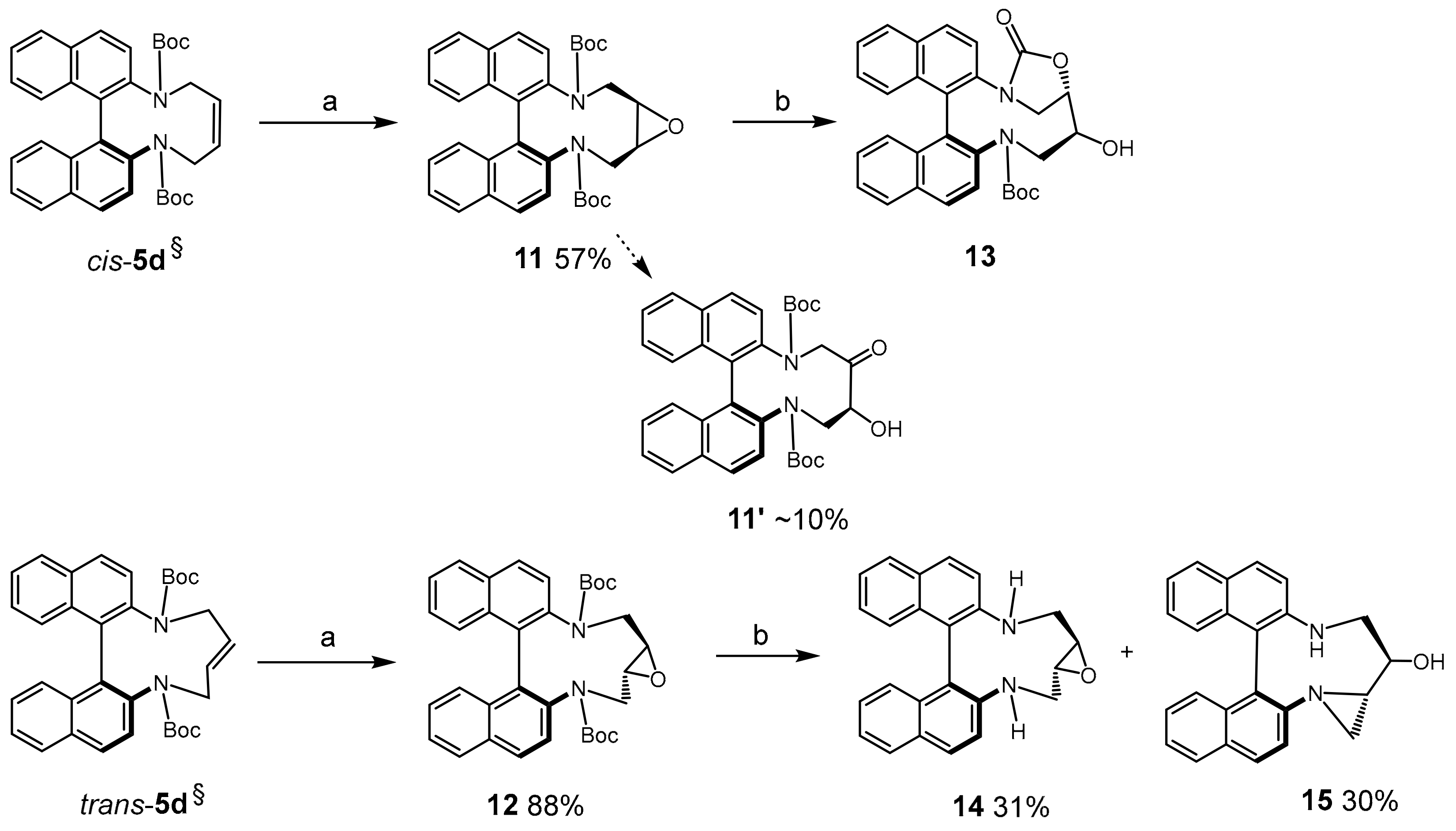
Di-tert-butyl(1aR*,17aS*)-1a,2,17,17a-tetrahydrodinaphtho[2,1-b:1′,2′-d]oxireno[2,3-h][1,6]diazecine- 3,16-dicarboxylate (11): To a solution of cis-5d (0.1 mmol, 54 mg) in DCM (4 mL) was added m-CPBA in portions (120 mg, 0.7 mmol) and the mixture was kept at r.t. overnight. To destroy excess of reagent, NaHSO3 (10%) was added and the organic phase was washed with Na2CO3 (2 M) and dried (Na2SO4). The crude material was purified by MPLC (EtOAc(20→50%)/heptane) to afford 11 as semisolid product (35 mg, 57%) and 11′ as a by-product (6 mg, 10%). 11: 1H-NMR δ = 7.77–8.07 (br.m, 4H); 7.16–7.55 (br.m, 8H); 4.03–4.71 (br.m, 2H); 2.89–3.14 (br.m, 2H); 2.45–2.89 (br.m, 2H); 1.00–1.42 (3× br.s, 18H). HRMS: calcd for C34H36N2NaO5 [M + Na]+: 575.2522; found: 575.2529. 11′: m.p.: 192–8 °C (dec.). 1H-NMR (DMSO-d6, 353K) δ = 8.13 (d, J = 8.9 Hz, 1H); 8.05 (d, J = 8.9 Hz, 1H); 7.98 (d, J = 8.3 Hz, 1H); 7.94 (d, J = 8.3 Hz, 1H); 7.84 (d, J = 8.9 Hz, 1H); 7.54 (d, J = 8.8 Hz, 1H); 7.46 (m, 2H); 7.21 (m, 2H); 7.01 (d, J = 8.6 Hz, 1H); 6.97 (d, J = 8.5 Hz, 1H); 4.79 (br.d, J = 6.3 Hz, 1H); 4.54 (d, J = 16.3 Hz, 1H); 4.24 (br.m, 1H); 3.99 (m, 2H); 3.91 (d, J = 16.3 Hz, 1H); 0.84 (s, ~9H); 0.80 (s, ~9H). 13C-NMR (DMSO-d6, 353K) δ = 204.4 (C); 152.5 (C); 138.1 (C); 136.0 (C); 132.8 (C); 132.7 (C); 131.9 (C); 131.4 (C); 131.04 (C); 130.99 (C); 129.4 (CH); 128.8 (CH); 128.0 (CH); 127.7 (CH); 127.0 (CH); 125.23 (CH); 125.16 (CH); 125.0 (CH); 124.2 (CH); 122.7 (CH); 80.4 (C); 79.6 (C); 68.7 (CH); 58.9 (CH2); 52.0 (CH2); 26.8 (CH3); 26.7 (CH3). HRMS: calcd for C34H36N2NaO6 [M + Na]+: 591.2471; found. 591.2466.
Di-tert-butyl (1aR*,17aR*)-1a,2,17,17a-tetrahydrodinaphtho[2,1-b:1′,2′-d]oxireno[2,3-h][1,6]diazecine-3,16-dicarboxylate (12): Epoxide 12 was accessed from trans-5d similarly as described for 11, with the exception that 3 equ. of m-CPBA were used; the reaction was complete after 6 h at r.t. Crystalline colorless material was obtained by slow evaporation from DCM/heptane solution; 49 mg (88% yield, 0.1 mmol scale). 1H-NMR δ = 7.14–8.12 (m, ~12H); 4.43–5.18 (br.m, 2H); 2.48 (br.m, 2H); 2.08 (dm, J = 9.3 Hz, 2H); 1.27 (br.s, ~18H). HRMS: calcd for C34H36N2NaO5 [M + Na]+: 575.2522; found: 575.2526.
Attempted deprotection of 11 and 12: To epoxide 11 (32 mg, 0.06 mmol) in DCM (2 mL) was added TFA (19 µL). After 22 h the reaction was neutralized (NaHCO3) and extracted. MPLC (MeOH(0→5%)/DCM) afforded 23 mg of 13 (80–90% purity). 1H-NMR δ = 7.98 (d, J = 8.8 Hz, 1H); 7.97 (d, J = 8.7 Hz, 1H); 7.88 (br.d, J = 8.1 Hz, 1H); 7.85 (br.d, J = 8.1 Hz, 1H); 7.46 (d, J = 8.7 Hz, 1H); 7.43 (ddd, J = 8.1, 6.7, 1.3 Hz, 1H); 7.40 (ddd, J = 8.0, 6.7, 1.1 Hz, 1H); 7.32 (d, J = 8.7 Hz, 1H); 7.21 (ddd, J = 8.6, 6.6, 1.3 Hz, 1H); 7.16 (dm, J = 8.7 Hz, 1H); 7.10 (ddd, J = 8.6, 6.8, 1.4 Hz, 1H); 6.85 (br.s, 1H); 6.83 (br.d, J = 8.7 Hz, 1H); 4.78 (ddd, J = 8.8, 5.7, 1.9 Hz, 1H); 4.51 (t, J = 9.2 Hz, 1H); 4.43 (dd, J = 15.0, 1.9 Hz, 1H); 4.00–4.03 (m, 1H); 3.95 (dd, J = 9.3, 2.1 Hz, 1H); 3.81 (dd, J = 15.0, 3.6 Hz, 1H); 0.56 (s, 9H). 13C-NMR δ = 152.8 (C); 138.8 (C); 134.2 (C); 133.59 (C); 133.55 (C); 132.1 (C); 131.8 (C); 130.7 (C); 130.2 (CH); 129.9 (CH); 128.6 (CH); 128.3 (CH); 128.0 (CH); 127.9 (C); 127.5 (CH); 126.52 (CH); 126.45 (CH); 126.2 (CH);125.8 (CH); 123.7 (CH); 122.5 (CH); 82.8 (C); 72.7 (CH); 68.6 (CH); 56.4 (CH2); 49.2 (CH2); 27.2 (CH3). HRMS: calcd for C30H28N2NaO5 [M + Na]+: 519.1896; found: 519.1897.
Similar treatment of epoxide 12 (TFA, DCM, r.t., 22 h) afforded a mixture of diaminoepoxide 14 and hydroxyaziridine 15: (1aR*,17aR*)-1a,2,3,16,17,17a-Hexahydrodinaphtho[2,1-b:1′,2′-d]oxireno[2,3-h][1,6]diazecine (14): 9 mg (31% yield, 0.08 mmol scale); colorless oil. 1H-NMR δ = 7.96 (d, J = 8.8 Hz, 2H); 7.91 (br.d, J = 8.1 Hz, 2H); 7.41–7.45 (m, 2H); 7.42 (d, J = 8.6 Hz, 2H); 7.35 (ddd, J = 8.3, 6.7, 1.4 Hz, 2H); 7.28 (dm, J = 8.4 Hz, 2H); 3.56 (dd, J = 13.7, 3.0 Hz, 2H); 2.88 (br.s, 2H); 2.63–2.72 (br.m, 2H); 2.03–2.08 (m, 2H). 13C-NMR δ = 144.6 (C); 133.7 (C); 130.3 (C); 129.9 (CH); 128.3 (CH); 127.5 (CH); 125.0 (CH); 124.8 (CH); 124.6 (C); 123.7 (CH); 55.1 (CH); 52.2 (CH2). HRMS: calcd for C24H21N2O [M + H]+: 353.1648; found: 353.1651.
(13R*,13aS*)-12,13,13a,14-Tetrahydro-11H-azirino[1,2-a]dinaphtho[2,1-f:1′,2′-h][1,5]diazonin-13-ol (15): 9 mg (30% yield, 90% purity, 0.08 mmol scale). 1H-NMR δ = 7.94 (d, J = 8.8 Hz, 1H); 7.93 (d, J = 8.7 Hz, 1H); 7.90 (br.d, J = 8.3 Hz, 1H); 7.84 (br.d, J = 8.0 Hz, 1H); 7.47 (d, J = 8.9 Hz, 1H); 7.39 (ddd, J = 8.0, 5.3, 2.7 Hz, 1H); 7.25-7.27 (m, 3H); 7.25 (d, J = 8.8 Hz, 1H); 7.16 (ddd, J = 8.1, 6.7, 1.2 Hz, 1H); 6.88 (d, J = 8.4 Hz, 1H); 3.33–3.40 (m, 2H); 2.86 (td, J = 9.3, 4.5 Hz, 1H); 2.86 (br.s, ~1H); 2.34 (d, J = 4.6 Hz, 1H); 2.23 (d, J = 3.0 Hz, 1H); 2.12 (ddd, J = 8.8, 4.5, 3.0 Hz, 1H). 13C-NMR δ = 145.6 (C); 145.4 (C); 134.3 (C); 132.7 (C); 130.4 (C); 130.3 (C); 129.9 (CH); 129.3 (CH); 128.1 (CH); 128.0 (CH); 127.1 (CH); 126.7 (CH); 126.01 (C); 125.1 (CH); 124.8 (CH); 124.6 (CH); 124.0 (CH); 122.4 (CH); 120.5 (CH); 73.8 (CH); 55.2 (br.CH2); 44.4 (CH); 29.2 (CH2). HRMS: calcd for C24H21N2O [M + H]+: 353.1648; found: 353.1651.
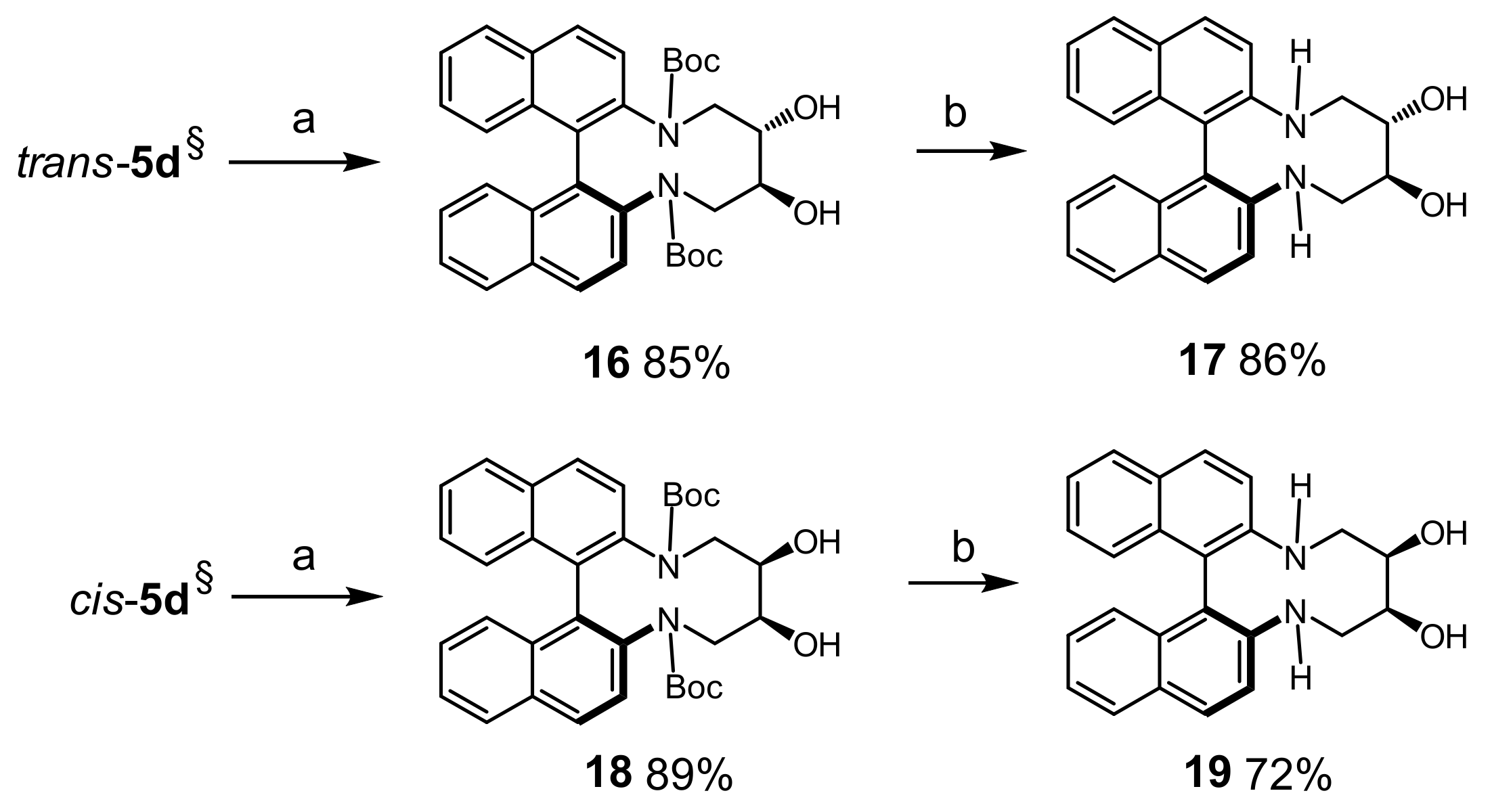
Di-tert-butyl(9S*,10S*)-9,10-dihydroxy-8,9,10,11-tetrahydrodinaphtho[2,1-b:1′,2′-d][1,6]diazecine- 7,12-dicarboxylate (16): To a solution of trans-5d (54 mg, 0.1 mmol) in THF/water (10:1, 2 mL) was added 2 equ. of NMO (50% in water, 25 mg) and K2OsO4∙H2O (0.01 mmol, 3.7 mg). After stirring for 24 h at r.t., solid Na2S2O3 (17 mg) was added and stirring was continued for 1 h. The mixture was diluted with DCM (19 mL), dried (MgSO4), filtered, and concentrated. MPLC (EtOAc(50→100%)/heptane) afforded 48 mg (85%) of 16 as colorless solid; m.p.: 201–205 °C. 1H-NMR δ = 7.94 (br.d, J = 8.6 Hz, 2H); 7.87 (d, J = 7.9 Hz, 2 H); 7.45 (ddd, J = 8.3, 6.2, 2.0 Hz, 2H); 7.33–7.46 (br.m, 2H); 7.19–7.26 (br. m, 4H); 3.40–3.86 (br.m, 4H); 3.31 (br.s, 2H); 2.27–2.95 (br.s, 2H); 1.18 (br.s, 18H). 13C-NMR δ = 138.9 (br.C); 133.1 (C); 132.3 (C); 129.3 (br.CH); 127.8 (br.CH); 127.5 (br.CH); 126.2 (2CH); 126.1 (CH); 80.7 (CH2); 73.9 (br.CH); 28.0 (CH3). HRMS: calcd for C34H38N2NaO6 [M + Na]+: 593.2628; found: 593.2624.
(9S*,10S*)-7,8,9,10,11,12-Hexahydrodinaphtho[2,1-b:1′,2′-d][1,6]diazecine-9,10-diol (17): To a solution of diol 16 (57 mg, 0.1 mmol) in DCM (1 mL) was added TFA (1 mL) and the reaction was stirred for 2 h at r.t. The mixture was concentrated under reduced pressure and the residue was dissolved in EtOAc (10 mL). Solid Na2CO3 was added and the mixture was stirred for 30 min. After filtration and evaporation of solvent the crude material was purified by MPLC (EtOAc(50→100%)/heptane) to afforded 32 mg (86%) of 17 as colorless crystalline solid; m.p: >165 °C (dec.). 1H-NMR (DMSO-d6) δ = 7.82 (d, J = 9.1 Hz, 2H); 7.77 (dd, J = 8.0, 1.1 Hz, 2H); 7.49 (d, J = 9.2 Hz, 2H); 7.14 (ddd, J = 7.9, 6.6, 1.2 Hz, 2H); 7.09 (ddd, J = 8.4, 6.7, 1.4 Hz, 2H); 6.79 (br.d, J = 8.4 Hz, 2H); 5.01–5.04 (m, 2H); 4.54 (d, J = 11.8 Hz, 2H); 4.11 (s, 2H); 3.80 (br.dd, J = 14.8, 12.6 Hz, 2H); 3.31 (d, J = 14.9 Hz, 2H). 13C-NMR (DMSO-d6) δ = 145.9 (C); 133.7 (C); 128.4 (CH); 128.0 (CH); 127.3 (C); 125.8 (CH); 124.0 (CH); 121.3 (CH); 117.6 (CH); 111.9 (C); 72.9 (CH); 48.0 (CH2). HRMS: calcd for C24H23N2O2 [M + H]+: 371.1760; found: 371.1745.
Di-tert-butyl (9R*,10S*)-9,10-dihydroxy-8,9,10,11-tetrahydrodinaphtho[2,1-b:1′,2′-d][1,6]diazecine- 7,12-dicarboxylate (18): A procedure similarly as described for 16 was applied to give 18; 51 mg (89% yield, colorless solid, 0.1 mmol scale); m.p.: 133–135 °C. 1H-NMR δ = 7.96 (d, J = 8.9 Hz, 1H); 7.94 (d, J = 8.9 Hz, 1H); 7.86 (d, J = 6.2 Hz, 1H); 7.84 (d, J = 6.2 Hz, 1H); 7.56 (br.d, J = 8.2 Hz, 1H); 7.39–7.45 (m, 2H); 7.29 (br.d, J = 8.7 Hz, 1H); 7.15–7.22 (m, 2H); 7.09–7.14 (br.m, 1H); 3.9–4.6 (br.s, ~2H); 4.04 (dd, J = 13.6, 4.6 Hz, 1H); 3.72–3.85 (m, 2H); 3.20–3.33 (m, 1H); 2.6–3.8 (br.s, ~2H); 1.01 (s, 9H); 0.93 (s, 9H). 13C-NMR δ = 140.0 (C); 137.3 (br.C); 133.7 (C); 133.5 (C); 132.5 (C); 132.0 (C); 131.8 (C); 130.1 (CH); 129.3 (CH); 128.7 (br.CH); 128.6 (br.CH); 127.5 (CH); 126.03 (CH); 125.95 (CH); 125.9 (CH); 125.7 (CH); 125.2 (br.CH); 80.8 (C); 80.7 (C); 54.5 (CH2); 48.3 (br.CH2); 27.9 (CH3); 27.7 (CH3). HRMS: calcd for C34H38N2NaO6 [M + Na]+: 593.2628; found: 593.2618.
(9R*,10S*)-7,8,9,10,11,12-Hexahydrodinaphtho[2,1-b:1′,2′-d][1,6]diazecine-9,10-diol (19): A procedure as described similarly for 17 was applied to give 19; yield: 21 mg (72%, colorless solid, 0.08 mmol scale); m.p.: 240-245 °C (dec.). 1H-NMR (DMSO-d6) δ = 7.93 (d, J = 9.0 Hz, 1H); 7.83 (dm, J = 7.9 Hz, 1H); 7.81 (d, J = 9.1 Hz, 1H); 7.78 (dm, J = 7.9 Hz, 1H); 7.49 (d, J = 9.1 Hz, 1H); 7.48 (d, J = 9.1 Hz, 1H); 7.09–7.21 (m, 4H); 6.83 (dm, J = 8.4 Hz, 1H); 6.79 (dm, J = 8.3 Hz, 1H); 5.01 (d, J = 4.1 Hz, 1H); 4.79 (d, J = 4.4 Hz, 1H); 4.07 (dd, J = 12.0, 2.3 Hz, 1H); 3.91 (ddd, J = 14.6, 12.0, 2.6 Hz, 1H); 3.71–3.79 (m, 2H); 3.59–3.68 (m, 1H); 3.06–3.14 (m, 2H). 13C-NMR (DMSO-d6) δ = 146.2 (C); 144.9 (C); 134.0 (C); 133.6 (C); 129.4 (CH); 128.3 (CH); 128.2 (CH); 128.0 (CH); 127.8 (C); 127.6 (C); 126.1 (CH); 125.9 (CH); 123.9 (CH); 123.7 (CH); 121.7 (CH); 121.6 (CH); 118.5 (CH); 116.5 (CH); 113.7 (C); 112.6 (C); 79.7 (CH); 70.1 (CH); 50.1 (CH2); 47.7 (CH2). HRMS: calcd for C24H23N2O2 [M + H]+: 371.1760; found: 371.1754.
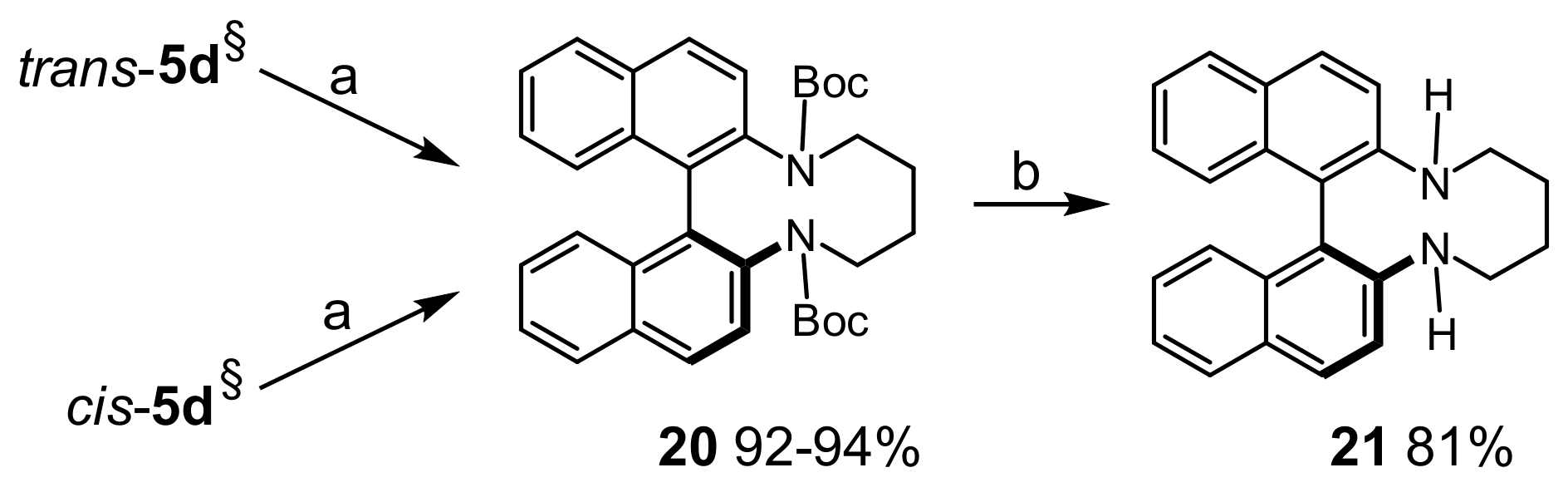
Di-tert-butyl8,9,10,11-tetrahydrodinaphtho[2,1-b:1′,2′-d][1,6]diazecine-7,12-dicarboxylate (20): To a solution of cis- or trans-5d (54 mg, 0.1 mmol) in THF/water (3 + 3 mL) was added Pd/C (10%, 5 mg) and the mixture was stirred under H2 (2 bar) at r.t. for 2 h. After filtration and concentration, the crude product was purified by MPLC (EtOAc(25→40%)/heptane) to afforded 49 mg (92% from cis-5d), and 51 mg (94% from trans-5d) of 20, respectively as colorless crystaline solid; m.p.: 172–173 °C. 1H-NMR δ = 7.93 (d, J = 8.7 Hz, 2H); 7.84 (br.d, J = 8.1 Hz, 2H); 7.33–7.44 (br.m, 4H); 7.14–7.21 (br.m, 4H); 3.84–3.93 (br.m, 2H); 3.50–3.66 (br.m, 2H); 1.78 (br.s, 2H); 1.52–1.63 (br.m, 2H); 0.99 (s, 18H). 13C-NMR δ = 139.0 (br.C); 133.8 (C); 132.9 (br.C); 132.1 (C); 129.3 (CH); 128.9 (br.CH); 127.2 (br.CH); 125.6 (CH); 125.5 (CH); 125.1 (br.CH); 79.9 (CH2); 48.9 (CH2); 27.9 (CH3). HRMS: calcd for C34H39N2O4 [M + H]+: 539.2910; found: 539.2909.
7,8,9,10,11,12-Hexahydrodinaphtho[2,1-b:1′,2′-d][1,6]diazecine (21): To 20 (53 mg, 0.1 mmol) dissolved in DCM (1 mL) was added TFA (1 mL) and the solution was stirred at r.t. for 2 h. The solvents were removed under vacuum and the crude product was dissolved in DCM (10 mL). Solid Na2CO3 was added and the mixture was stirred for 30 min. After filtration and concentration the pure product was obtained by MPLC (EtOAc(10→80%)/heptane); yield: 27 mg (81%, colorless crystals); m.p.: 275–278 °C. 1H-NMR δ = 7.92 (d, J = 8.9 Hz, 2H); 7.82 (dm, J = 7.9 Hz, 2H); 7.40 (d, J = 8.9 Hz, 2H); 7.28 (ddd, J = 8.1, 6.8, 1.3 Hz, 2H); 7.19 (ddd, J = 8.4, 6.8, 1.4 Hz, 2H); 7.08 (dm, J = 8.4 Hz, 2H); 4.00 (br.d, J = 11.7 Hz, 2H); 3.74 (br.t, J = 12.7 Hz, 2H); 2.76 (br.t, J = 13.5 Hz, 2H); 1.69–1.78 (m, 2H); 1.32–1.40 (m, 2H). 13C-NMR δ = 144.3 (C); 134.5 (C); 129.8 (CH); 129.0 (C); 128.0 (CH); 126.8 (CH); 124.7 (CH); 123.1 (CH); 117.9 (CH); 117.7 (C); 46.5 (CH2); 25.9 (CH2). HRMS: calcd for C24H23N2 [M + H]+: 339.1861; found: 339.1855.

 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}