Design, Synthesis and Molecular Docking Analysis of Flavonoid Derivatives as Potential Telomerase Inhibitors


 and
and
Abstract
1. Introduction
2. Results and Discussion
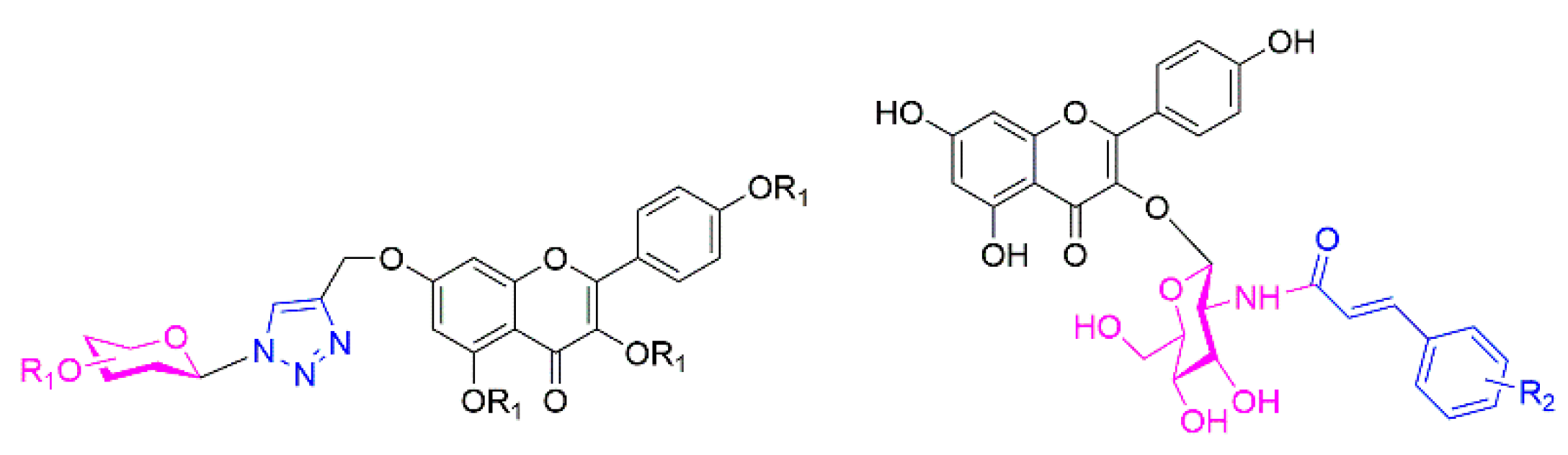
2.1. Chemistry
2.2. Biological Activity
2.2.1. Telomerase Inhibitory Assay
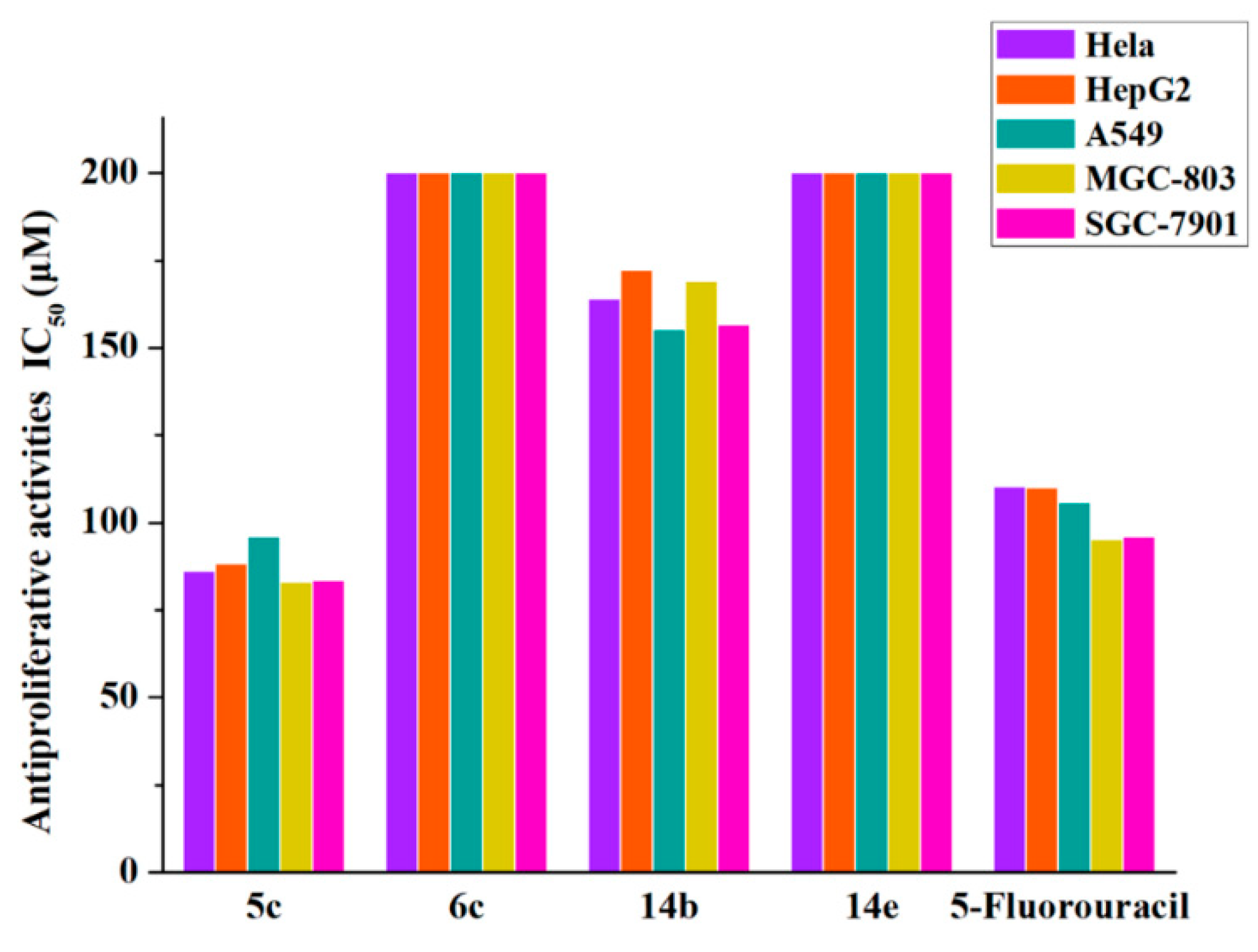
2.2.2. Antiproliferation Assay
2.3. Molecular Simulation Analyses
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure for the Preparation of Compounds 5a–5c
3.1.2. General Procedure for the Preparation of Compounds 6a–6c
3.1.3. General Procedure for the Preparation of Compounds 14a–14f
3.2. Biological Activity
3.2.1. Telomerase Activity Assays
3.2.2. Cell Proliferation Assays
3.3. Molecular Simulation Assays
3.3.1. Molecular Docking
3.3.2. MD Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Elshemy, H.A.H.; Zaki, M.A. Design and synthesis of new coumarin hybrids and insight into their mode of antiproliferative action. Bioorg. Med. Chem. 2017, 25, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer. J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity withimmortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cheng, F.X.; Yuan, X.L.; Tang, W.J.; Shi, J.B.; Liao, C.Z.; Liu, X.H. Dihydropyrazole derivatives as telomerase inhibitors: Structure-based design, synthesis, SAR and anticancer evaluation in vitro and in vivo. Eur. J. Med. Chem. 2016, 112, 231–251. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, K.; Xu, B. Telomerase inhibitors from natural products and their anticancer potential. Int. J. Mol. Sci. 2017, 19, 13. [Google Scholar] [CrossRef] [PubMed]
- De, C.A.; Lacroix, L.; Douarre, C.; Temime-Smaali, N.; Trentesaux, C.; Riou, J.F.; Mergny, J.L. Targeting telomeres and telomerase. Biochimie 2008, 90, 131–155. [Google Scholar]
- Sun, D.; Thompson, B.; Cathers, B.E.; Salazar, M.; Kerwin, S.M.; Trent, J.O.; Jenkins, T.C.; Neidle, S.; Hurley, L.H. Inhibition of human telomerase by a G-quadruplex-interactive compound. J. Med. Chem. 1997, 40, 2113–2116. [Google Scholar] [CrossRef]
- Paul, A.; Maji, B.; Misra, S.K.; Jain, A.K.; Muniyappa, K.; Bhattacharya, S. Stabilization and structural alteration of the G-quadruplex DNA made from the human telomeric repeat mediated by Troger’s base based novel benzimidazole derivatives. J. Med. Chem. 2012, 55, 7460–7471. [Google Scholar] [CrossRef]
- Tang, W.J.; Wang, J.; Tong, X.; Shi, J.B.; Liu, X.H.; Li, J. Design and synthesis of celastrol derivatives as anticancer agents. Eur. J. Med. Chem. 2015, 95, 166–173. [Google Scholar] [CrossRef]
- Lee, C.C.; Huang, K.F.; Lin, P.Y.; Huang, F.C.; Chen, C.L.; Chen, T.C.; Guh, J.H.; Lin, J.J.; Huang, H.S. Synthesis, antiproliferative activities and telomerase inhibition evaluation of novel asymmetrical 1,2-disubstituted amidoanthraquinone derivatives. Eur. J. Med. Chem. 2012, 47, 323–336. [Google Scholar] [CrossRef]
- Zheng, D.S.; Chen, L.S. Triterpenoids from Ganoderma lucidum inhibit the activation of EBV antigens as telomerase inhibitors. Exp. Ther. Med. 2017, 14, 3273–3278. [Google Scholar] [CrossRef] [PubMed]
- Imad Naasani, F.O.; Tomoko, O.; Wan, Y.F.; Johnston, J.; Chan, K.; Tsuruo, T. Blocking telomerase by dietary polyphenols is a major mechanism for limiting the growth of human cancer cells in vitro and in vivo. Cancer Res. 2003, 63, 824–830. [Google Scholar]
- Menichincheri, M.; Ballinari, D.; Bargiotti, A.; Bonomini, L.; Ceccarelli, W.; D’Alessio, R.; Fretta, A.; Moll, J.; Polucci, P.; Soncini, C.; et al. Catecholic flavonoids acting as telomerase inhibitors. J. Med. Chem. 2004, 47, 6466–6475. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.K.; Kao, T.Y.; Wu, M.F.; Ko, J.L.; Tzeng, Y.M. Identification of small molecule inhibitors of telomerase activity through transcriptional regulation of hTERT and calcium induction pathway in human lung adenocarcinoma A549 cells. Bioorg. Med. Chem. 2010, 18, 6987–6994. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Song, B.A.; Zhao, H.J.; Qi, X.B.; Huang, Y.J.; Liu, X.H. Novel myricetin derivatives: Design, synthesis and anticancer activity. Eur. J. Med. Chem. 2015, 97, 155–163. [Google Scholar] [CrossRef]
- Hou, Z.; Lin, B.; Bao, Y.; Yan, H.N.; Zhang, M.; Chang, X.W.; Zhang, X.X.; Wang, Z.J.; Wei, G.F.; Cheng, M.S.; et al. Dual-tail approach to discovery of novel carbonic anhydrase IX inhibitors by simultaneously matching the hydrophobic and hydrophilic halves of the active site. Eur. J. Med. Chem. 2017, 132, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Thirumurugan, P.; Matosiuk, D.; Jozwiak, K. Click chemistry for drug development and diverse chemical-biology applications. Chem. Rev. 2013, 113, 4905–4979. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Liu, W.; Lu, Y.; Wang, Y.; Chen, X.; Bai, S.; Zhao, Y.; He, T.; Lao, F.; Shang, Y.; et al. Naturally occurring cinnamic acid sugar ester derivatives. Molecules 2016, 21, 1402–1449. [Google Scholar] [CrossRef] [PubMed]
- Sova, M. Antioxidant and antimicrobial activities of cinnamic acid derivatives. Mini. Rev. Med. Chem. 2012, 12, 749–767. [Google Scholar] [CrossRef]
- De, P.; Bedos-Belval, F.; Vanucci-Bacque, C.; Baltas, M. Cinnamic acid derivatives in tuberculosis, malaria and cardiovascular diseases—A Review. Cur. Org. Chem. 2012, 16, 747–768. [Google Scholar]
- Sharma, P. Cinnamic acid derivatives: A new chapter of various pharmacological activities. J. Chem. Pharm. Res. 2011, 3, 403–423. [Google Scholar]
- Li, H.N.; Wang, H.; Wang, Z.P.; Yan, H.N.; Zhang, M.; Liu, Y.; Cheng, M.S. Synthesis, antitumor activity evaluation and mechanistic study of novel hederacolchiside A1 derivatives bearing an aryl triazole moiety. Bioorg. Med. Chem. 2018, 26, 4025–4033. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.F.; Wang, S.; Luan, W.J.; Cui, S.S.; Li, F.R.; Liu, Y.X.; Liu, Y.; Cheng, M.S. A library of 1,2,3-triazole-substituted oleanolic acid derivatives as anticancer agents: Design, synthesis, and biological evaluation. Org. Biomol. Chem. 2015, 13, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Cui, S.; Luan, W.; Wang, S.; Hou, Z.; Liu, Y.; Liu, Y.; Cheng, M. Natural product-based design, synthesis and biological evaluation of Albiziabioside A derivatives that selectively induce HCT116 cell death. Eur. J. Med. Chem. 2016, 113, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Sun, J.; Hou, Z.; Luan, W.; Wang, S.; Cui, S.; Cheng, M.; Liu, Y. Novel antitumor compound optimized from natural saponin Albiziabioside A induced caspase-dependent apoptosis and ferroptosis as a p53 activator through the mitochondrial pathway. Eur. J. Med. Chem. 2018, 157, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.H.D.; Tam, J.; Wu, R.A.; Greber, B.J.; Toso, D.; Nogales, E.; Collins, K. Cryo-EM structure of substrate-bound human telomerase holoenzyme. Nature 2018, 557, 190–195. [Google Scholar] [CrossRef]
- Bryan, C.; Rice, C.; Hoffman, H.; Harkisheimer, M.; Sweeney, M.; Skordalakes, E. Structural Basis of Telomerase Inhibition by the Highly Specific BIBR1532. Structure 2015, 23, 1934–1942. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Y.; Kong, D.; Jiang, H.; Wang, J.; Cheng, M. In silico exploration of aryl sulfonamide analogs as voltage-gated sodium channel 1.7 inhibitors by using 3D-QSAR, molecular docking study, and molecular dynamics simulations. Comput. Biol. Chem. 2018, 77, 214–225. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, B.; Peng, Y.; Xiong, X.; Jing, W.; Wang, J.; Gao, H. In Silico Exploration of the Molecular Mechanism of Cassane Diterpenoids on Anti-inflammatory and Immunomodulatory Activity. J. Chem. Inf. Model. 2019, 59, 2309–2323. [Google Scholar] [CrossRef]
- Wang, M.; Li, W.; Wang, Y.; Song, Y.; Wang, J.; Cheng, M. In silico insight into voltage-gated sodium channel 1.7 inhibition for anti-pain drug discovery. J. Mol. Graph. Model. 2018, 84, 18–28. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 5a–5c, 6a–6c and 14a–14f are available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 Values (μM) | ||||
|---|---|---|---|---|---|
| Hela | HepG2 | A549 | MGC-803 | SGC-7901 | |
| 5c | 86.183 ± 2.444 | 88.149 ± 0.761 | 95.842 ± 0.926 | 82.992 ± 3.149 | 83.421 ± 1.593 |
| 6c | >200 | >200 | >200 | >200 | >200 |
| 14b | 163.971 ± 2.253 | 172.18 ± 1.881 | 155.323 ± 6.627 | 169.063 ± 1.676 | 156.527 ± 0.914 |
| 14e | >200 | >200 | >200 | >200 | >200 |
| 5-FU | 110.164 ± 1.147 | 110.013 ± 0.603 | 105.712 ± 1.905 | 95.172 ± 2.618 | 96.011 ± 1.785 |
| Compound | IC50 values (μM) | |
|---|---|---|
| Hacat | BEAS-2B | |
| 5c | >200 | >200 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Z.-F.; Ho, S.-T.; Wen, R.; Fu, Y.; Zhang, L.; Wang, J.; Hu, C.; Shaw, P.-C.; Liu, Y.; Cheng, M.-S. Design, Synthesis and Molecular Docking Analysis of Flavonoid Derivatives as Potential Telomerase Inhibitors. Molecules 2019, 24, 3180. https://doi.org/10.3390/molecules24173180
Fan Z-F, Ho S-T, Wen R, Fu Y, Zhang L, Wang J, Hu C, Shaw P-C, Liu Y, Cheng M-S. Design, Synthesis and Molecular Docking Analysis of Flavonoid Derivatives as Potential Telomerase Inhibitors. Molecules. 2019; 24(17):3180. https://doi.org/10.3390/molecules24173180
Chicago/Turabian StyleFan, Zhan-Fang, Sai-Tim Ho, Rui Wen, Ya Fu, Lei Zhang, Jian Wang, Chun Hu, Pang-Chui Shaw, Yang Liu, and Mao-Sheng Cheng. 2019. "Design, Synthesis and Molecular Docking Analysis of Flavonoid Derivatives as Potential Telomerase Inhibitors" Molecules 24, no. 17: 3180. https://doi.org/10.3390/molecules24173180
APA StyleFan, Z.-F., Ho, S.-T., Wen, R., Fu, Y., Zhang, L., Wang, J., Hu, C., Shaw, P.-C., Liu, Y., & Cheng, M.-S. (2019). Design, Synthesis and Molecular Docking Analysis of Flavonoid Derivatives as Potential Telomerase Inhibitors. Molecules, 24(17), 3180. https://doi.org/10.3390/molecules24173180