Abstract
In the era of personalized precision medicine, positron emission tomography (PET) and related hybrid methods like PET/CT and PET/MRI gain recognition as indispensable tools of clinical diagnostics. A broader implementation of these imaging modalities in clinical routine is closely dependent on the increased availability of established and emerging PET-tracers, which in turn could be accessible by the development of simple, reliable, and efficient radiolabeling procedures. A further requirement is a cGMP production of imaging probes in automated synthesis modules. Herein, a novel protocol for the efficient preparation of 18F-labeled aromatics via Cu-mediated radiofluorination of (aryl)(mesityl)iodonium salts without the need of evaporation steps is described. Labeled aromatics were prepared in high radiochemical yields simply by heating of iodonium [18F]fluorides with the Cu-mediator in methanolic DMF. The iodonium [18F]fluorides were prepared by direct elution of 18F− from an anion exchange resin with solutions of the corresponding precursors in MeOH/DMF. The practicality of the novel method was confirmed by the racemization-free production of radiolabeled fluorophenylalanines, including hitherto unknown 3-[18F]FPhe, in 22–69% isolated radiochemical yields as well as its direct implementation into a remote-controlled synthesis unit.
1. Introduction
Widespread implementation of molecular imaging techniques, especially positron emission tomography (PET) and related hybrid methods like PET/CT and PET/MRI in clinical practice have significantly contributed to a considerable increase of diagnostic accuracy in recent years. PET offers the unique opportunity to visualize physiological and pathological processes on the molecular level. This imaging modality utilizes probes labeled with positron emitting radionuclides (PET-tracers) interacting specifically with molecular targets or biochemical processes of interest. Biodistribution of such probes can be determined by the detection of antiparallel γ-photons originating from electron–positron annihilation. PET probes are used for accurate diagnosis and staging of diseases as well as monitoring of therapy success (e.g., for tumors, neurological or cardiac disorders). In addition to clinical applications, PET is a powerful tool in drug development which provides fast and precise assessment of pharmacological properties of drug candidates in vivo. The main challenge of PET-chemistry is the short half-life of the majority of commonly used PET radionuclides like 11C (20.4 min), 13N (10 min) and 15O (2 min) which severely limits the scope of reactions which can be used for radiolabeling.
18F is the most widely used PET radionuclide owing to the accessibility of no-carrier-added (n.c.a.) 18F– in multi-Curie amounts at low and medium energy cyclotrons from [18O]H2O via the high-yielding 18O(p,n)18F nuclear reaction. 18F has a longer half-life (109.8 min) which enables extended PET acquisition protocols and shipping of radiofluorinated tracers to remote PET centers as well as favorable decay characteristics like a high positron branching ratio of 97% and a low positron energy (Eβmax 0.63 MeV). The latter enables imaging with high spatial resolution.
The vast majority of 18F-labeled PET radiotracers are produced by SN2 and SNAr radiofluorination reactions. In order to be applied for these reactions 18F– produced from [18O]H2O has to be transformed into anhydrous [18F]F- with high nucleophilicity. Conventionally, in order to separate the bulk of [18O]water, 18F– is trapped on an anion exchange resin. It is recovered using an aqueous solution of suitable bases like K2CO3, Cs2CO3 or tetraalkylammonium hydrogen carbonates. [18O]H2O is then removed by repeatedly time-consuming azeotropic drying with MeCN. Finally, the residual anhydrous [18F]fluoride salt is taken up in a solution of the radiolabeling precursor in an aprotic polar solvent and heated for a short time affording the desired radiolabeled product. If potassium salts are used, K+ chelators like K2.2.2 or 18-crown-6 which increase the solubility of 18F– in organic solvents and its nucleophilicity by virtue of the charge separation, are typically added. Whereas a broad spectrum of labeled aliphatic probes can be prepared, only highly electron deficient (hetero)aromatics containing electron-withdrawing groups in o- or p-position to the leaving group can be labeled using such approach. Recently developed protocols, e.g., for Pd- [1,2,3], Ni- [4,5], and especially Cu-mediated radiofluorination enabled to overcome this limitation and efficiently prepare electron-neutral and even -rich 18F-labeled (hetero)aromatics. Initially described for (aryl)(mesityl)iodonium salts (Scheme 1) [6], Cu-mediated radiofluorination was also applied for labeling of easily accessible aryl pinacol boronates (ArBPin) [7,8], aryl boronic acids [9], aryl trialkyl stannanes [10] as well as (aryl)(mesityl)iodonium salts generated in situ from electron-rich (hetero)aromatics [11] and directed CH-18F-fluorination [12]. The original procedures, while working well at a small scale, were in some cases inoperative for batch PET-tracer production. Several radiolabeling protocols, which allowed overcoming this limitation, were published [13,14,15,16,17,18,19]. Moreover, the “minimalist protocol” originally developed for labeling of suitable onium salt precursors using only [18F]fluoride [20] was successfully implemented into Cu-mediated radiofluorination of (aryl)(mesityl)iodonium salts. Additionally, this protocol was also transferred to an automated synthesis [17,21]. This enabled the high-yielding preparation of several clinically relevant PET-tracers like, 4-[18F]fluorophenylalanine, 6-[18F]fluorodopamine and [18F]DAA1106 on a preparative scale using only Cu-catalyst, iodonium salt and [18F]fluoride. The method obviates the need for azeotropic drying, base and any other additives. Accordingly, [18F]fluoride is directly eluted from a anion exchange resin with a solution of the corresponding iodonium salt precursor in MeOH. Low-boiling MeOH is evaporated; the residue is taken up in a solution of Cu(MeCN)4OTf in DMF and heated furnishing the desired radiolabeled product. However, solvent evaporation before 18F-fluorination still has to be carried out. Noteworthy, in the case of remote-controlled radiosyntheses, complete removal of MeOH is a prerequisite to obtain high radiolabeling yields. In contrast, the “alcohol-enhanced” Cu-mediated radiofluorination not only enables to obtain considerably higher radiochemical yields (RCYs) but also substantially simplifies the production of 18F-fluorinated (hetero)aromatics by the elimination of any evaporation steps. Furthermore, a broad scope of stannyl and boronyl substrates could be efficiently radiolabeled under general reaction conditions [22,23]. Hence, 18F– is directly eluted with an alcoholic (usually nBuOH) solution of a suitable salt like Et4NHCO3, Bu4POMs, K2CO3/K2.2.2, etc., into a solution of the appropriate precursor and Cu(py)4(OTf)2 in DMA or DMF. The resulting solution is briefly heated affording the 18F-labeled probe. The efficacy of Cu-mediated 18F-fluorination in alcoholic media markedly contradicted previous observations concerning the deleterious effect of protic solvents, including alcohols, on SnAr fluorination. Noteworthy, quite recently the efficient metal-free 18F-fluorination of electron-deficient (hetero)aromatics in pure EtOH was published [24]. Herein, we disclose a novel evaporation step free protocol for Cu-mediated radiofluorination of (aryl)(mesityl)iodonium salts which combines advantages of the “alcohol-enhanced” with the “minimalist” approach. We applied the procedure for radiolabeling of model substrates and also for the preparation of [18F]fluorophenylalanines ([18F]FPhe) including hitherto unknown 3-[18F]FPhe.
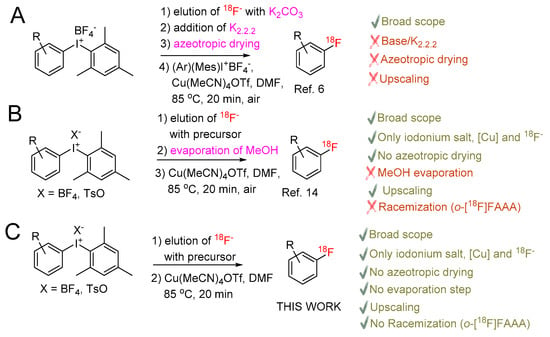
Scheme 1.
Cu-mediated [18F]fluorination of (mesityl)(aryl)iodonium salts: A—original protocol of Ichiishi et al. [6]; B—“minimalist” approach [17,21]; C—This work. o-[18F]FAAA–ortho-[18F]fluorinated aromatic amino acids.
2. Results and Discussion
(Mesityl)[(4-methoxy)phenyl]iodonium tosylate and tetrafluoroborate (1·OTs and 1·BF4) were selected as model substrates (Scheme 2). First, the elution of [18F]fluoride from an anion exchange resin with 1·OTs or 1·BF4 in anhydrous or 80% aqueous DMF as well as in a mixture of DMF with different alcohols (20% alcohol content) was examined (Figure 1). Experiments were carried out either manually or in a self-made semi-automated synthesis module. The highest recovery of 18F–, was obtained with aqueous and methanolic DMF, amounting to 83% and 73%, respectively (with 1·BF4). In contrast to the recovery of [18F]fluoride with solutions of ammonium and phosphonium salts in ROH/DMF,[22] 18F– recovery with solutions of iodonium salts in mixtures of DMF and higher alcohols was only moderate and amounted to 50% and 31% for EtOH and nPrOH, respectively (with 1·BF4). Elution efficacy with 1·BF4 in 20% nBuOH in DMF was comparably low as with a solution of this precursor in pure DMF (about 20%). Notably, 18F– recovery with 1·OTs in MeOH or EtOH was up to 20% higher than recovery with 1·BF4 in the same solvents. The eluates were directly added to solutions of Cu(MeCN)4OTf in DMF (10% final concentration of ROH) and heated at 85 °C for 20 min.
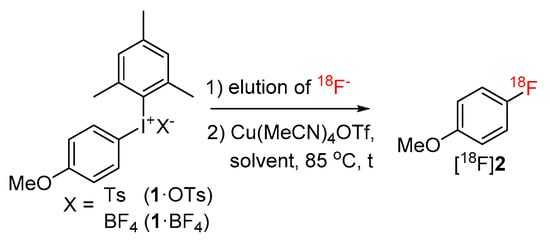
Scheme 2.
Model labeling reaction used for the optimization of the reaction parameters.
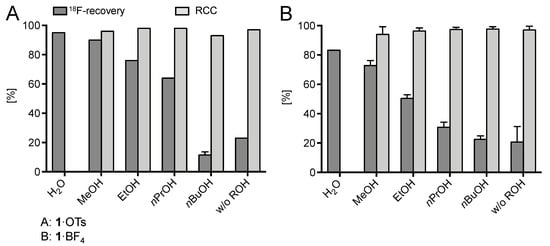
Figure 1.
18F-Recovery and 18F-incorporation (RCC) using different alcohols as co-solvents. Conditions: [18F]Fluoride (50–200 MBq) was loaded on a QMA-CO3 cartridge (not conditioned) from the male side and the cartridge was rinsed with MeOH (5 mL) in the same direction. Afterwards, [18F]fluoride was eluted with a solution of 1·OTs or 1·BF4 (21 µmol) in ROH/DMF (120/500 μL; 20% ROH) from the female side directly into a solution of Cu(MeCN)4OTf (7.9 mg, 21 µmol) in DMF (600 µL) and the resulting solution was heated at 85 °C for 20 min. The reaction mixture was diluted with H2O (2 mL) and RCCs were determined by TLC (for 1·OTs; experiments were carried out in Jülich) or HPLC (for 1·BF4; experiments were carried out in Saint-Petersburg). Experiments with 1·OTs were carried out once (except for MeOH, n > 20 and nBuOH, n = 2) and with 1·BF4 in triplicate. Error bars represent standard deviations (SDs). Experiments with 1·OTs were carried out in the self-made semi-automated synthesis module (Saint-Petersburg) and with 1·BF4 manually (in Jülich).
Whereas almost quantitative 18F-incorporation yields (>94%) were observed for both radiolabeling substrates using DMF and ROH/DMF mixtures as reaction medium, no radiofluorination took place in aqueous DMF, presumably, owing to the decomposition of Cu(MeCN)4OTf in the presence of water and/or unfavorably strong solvation of 18F–. In trying to further improve 18F– recovery, the MeOH content of the elution solution was increased to 30% (12% final concentration of MeOH in the reaction mixture; Figure 2). This modification led to substantially improved elution efficacy (90%), while radiochemical incorporation (RCC) remained high (86%). A further increase of the MeOH content was detrimental and caused a rapid decrease of the 18F-incorporation rate owing to the formation of sTable 18F–(MeOH)n clusters (n = 2–8, mainly 3, 4). Cluster formation substantially decreases the nucleophilicity of fluoride [25].
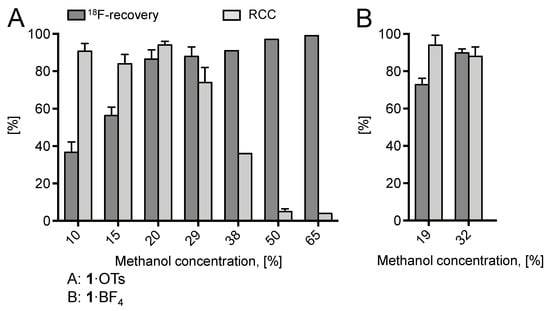
Figure 2.
18F-Recovery and 18F-incorporation (RCC) using different concentrations of MeOH. Conditions: A [18F]Fluoride (50–200 MBq) was eluted from the respective anion exchange cartridge (see legend of Figure 1) with a solution of 1·OTs (9.2 mg, 21 µmol) in MeOH/DMF (620 µL; different ratios of MeOH/DMF) directly to a solution of Cu(MeCN)4OTf (7.9 mg, 21 µmol) in DMF (600 µL) and the resulting solution was heated at 85 °C for 20 min. The reaction mixture was diluted with H2O (2 mL) and RCCs were determined by TLC. All reactions were carried out with the self-made semi-automated synthesis module. B: [18F]Fluoride (50–200 MBq) was eluted from the respective anion exchange cartridge (see legend of Figure 1) with a solution of 1·BF4 (9.2 mg, 21 µmol) in MeOH/DMF (120/500 µL or 120/250 µL) directly to a solution of Cu(MeCN)4OTf (7.9 mg, 21 µmol) in DMF (600 µL) and the resulting solution was heated at 85 °C for 20 min. The reaction mixture was diluted with H2O (2 mL) and RCCs were determined by HPLC. These radiolabeling experiments were carried out manually. Experiments were carried out in triplicate. Error bars represent standard deviations (SDs).
Next, the influence of aprotic solvents was studied. Replacement of DMF by CH3CN, DMSO, DMA, DMI, DMPU or sulfolane had no significant influence on 18F- recovery (73–90%). However, surprisingly, no formation of 4-[18F]fluoroanisole ([18F]2) was observed in any of the examined solvents including DMA, a higher analog of DMF which was the best suited solvent for Cu-mediated radiofluorination of boronate and stannyl substrates (data not shown). This somewhat unexpected observation could be possibly explained by the fact that DMF, in contrast to all other studied solvents, is a monodentate O-donor ligand which can stabilize the intermediately formed (Ar)(MesI)Cu(III)18F complex and facilitate its reductive elimination [26].
Interestingly, in contrast to earlier observations,[17] aerobic conditions were not a prerequisite for successful radiolabeling. The same RCCs were obtained under air and N2 indicating a direct oxidation of the Cu(I) complex by the iodonium salt precursor (data not shown).
Additionally, radiolabeling kinetics was briefly studied. Incorporation yields of >90 % were already observed after 5 min incubation at 85 °C (Figure 3).
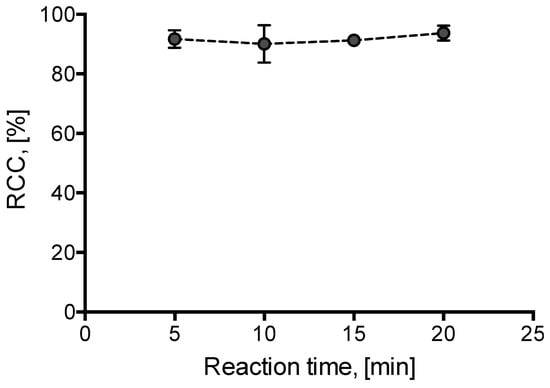
Figure 3.
Dependency of RCC on reaction time. [18F]Fluoride (50–200 MBq) was eluted from the respective anion exchange cartridge (see legend of Figure 1) with a solution of 1·OTs (11 mg, 21 µmol) in MeOH/DMF (120/500 µL) directly into a solution of Cu(MeCN)4OTf (7.9 mg, 21 µmol) in DMF (600 µL) and the resulting solution was heated at 85 °C for a given time. The reaction mixture was diluted with H2O (2 mL) and RCCs were determined by TLC. All reactions were carried out in the self-made semi-automated synthesis module. All experiments were carried out in duplicate. Error bars represent standard deviations (SDs).
Furthermore, the dependency of 18F– elution efficiency and RCC of [18F]2 at 85 °C after 20 min reaction time on the amount of 1·OTs precursor was studied (Figure 4). In these experiments equimolar amounts of Cu(MeCN)4OTf were used. While ≥90% 18F- recovery was observed for the different precursor amounts (3.5–21 µmol), maximum RCCs >90% were obtained with 21 and 14 µmol of 1·OTs. Minimization of the amount of iodonium salt from 7 to 3.5 µmol still produced [18F]2 in RCCs of 81% and 44 %, respectively.
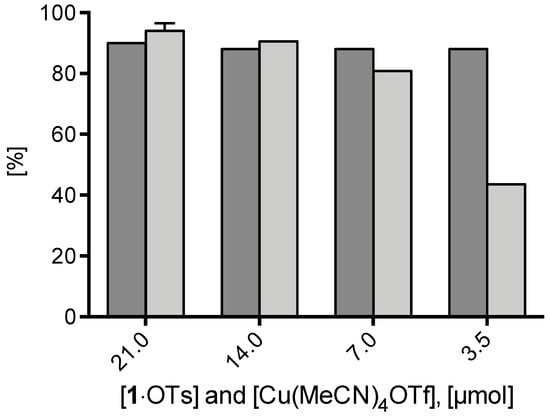
Figure 4.
18F-Recovery and 18F-incorporation (RCC) using different amounts of 1·OTs and Cu(MeCN)4OTf. Conditions: [18F]Fluoride (50–200 MBq) was eluted from the respective anion exchange cartridge (see legend of Figure 1) with a solution of a given amount 1·OTos in MeOH/DMF (120/500 µL) directly into a solution of equimolar amount of Cu(MeCN)4OTf in DMF (600 µL) and the resulting solution was heated at 85 °C for 20 min. The reaction mixture was diluted with H2O (2 mL) and RCCs were determined by TLC. All reactions were carried out in the self-made semi-automated synthesis module.
The scope of the novel radiofluorination protocol was further evaluated using phenyl- and (3-carbonyl)phenyl-substituted (mesityl)iodonium tetrafluoroborates as a model for an electron-neutral and an electron-deficient substrate, respectively. Gratifyingly, both precursors were successfully radiolabeled affording [18F]fluorobenzene ([18F]3) and 3-[18F]fluorobenzaldehyde ([18F]4) in RCCs of >90% and >72%, respectively (Table 1). Accordingly, using the novel protocols, model compounds were prepared in RCCs comparable to or even better than those obtained applying the original “minimalist” protocol for Cu-mediated radiofluorination [17].

Table 1.
Substrate scope of alcohol-supported Cu-mediated radiofluorination of (aryl)(mesityl)iodonium salts.
Being particularly interested in fast and efficient procedures for the preparation of 18F-labeled aromatic amino acids, we applied the novel protocol for the synthesis of 2–4-[18F]FPhes ([18F]8–10) (Table 1, Scheme 3), which should be potentially suitable for imaging of tumors and neurological disorders.
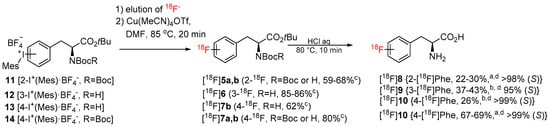
Scheme 3.
Preparation of 2-4-[18F]FPhes (2-4-[18F]-8–10). Conditions for manual synthesis (a): radiolabeling step: refer to caption of Table 1; deprotection step: the reaction mixture was cooled to 65 °C, diluted with H2O (4.5 mL), and loaded onto a C18 SPE cartridge. The cartridge was washed with H2O (10 mL) and radiolabeled intermediates were eluted with MeOH (2 mL). MeOH was evaporated to 1 mL at 85 °C within 1 min and the residual solution was diluted with 12 n HCl (0.5 mL) and heated at 80 °C for 10 min. The reaction mixture was cooled and 18F-labeled FPhes were isolated by HPLC. Conditions for the semi-automated synthesis (b): refer to GP3 in Materials and Methods c–RCC; d–RCY (EOB). Each radiosynthesis, except the preparation of 4-[18F]FPhe from 13, was carried out at least in duplicate.
The precursor of 3-[18F]FPhe 12 was prepared as follows (Scheme 4). (S)-Ni-BPB-Gly [27] was alkylated with 3-iodobenzyl bromide, affording the Ni(II) complex 14 as a single (S,S)-isomer. 14 was decomposed by transchelation with the ditetraethylammonium salt of DTPA [28] furnishing 3-IPhe, which was N-Boc and O-tBu protected in two sequential steps to give Boc-3-IPhe-OtBu 15 in 56% over four steps with a single chromatographic purification. The latter was transformed into Boc-3-(SnMe3)Phe-OtBu which in turn was allowed to react with MesI(OH)OTs affording the iodonium salt 12 in 20% yield over six steps.
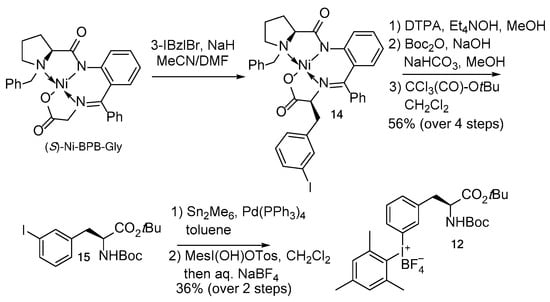
Scheme 4.
Synthesis of radiolabeling precursor 12.
N-mono-Boc protected 12 and 13 as well as N,N-di-Boc protected precursors of 2-[18F]FPhe and 4-[18F]FPhe, 11 and 14 [28], were successfully radiolabeled affording protected radiolabeled FPhes, in RCCs of 85-86%, 62%, 59–68%, and 80%, respectively (Table 1; Scheme 3). Finally, deprotection with 12 n HCl and HPLC purification furnished 2–4-[18F]FPhe in isolated RCYs of 22–30%, 37–43%, and 67–69%, respectively, within 110 min. The molar activity determined for 4-[18F]FPhe (2.22 GBq), which was produced from the iodonium salt 13, amounted to 207 GBq/µmol. Importantly, the novel procedure enabled the production of 3- and 4-[18F]FPhes in a remote-controlled synthesis unit. Remarkably, whereas, the application of the conventional “minimalist” protocol led to significant loss of enantiopurity [formation of up to 20% of the (R)-isomer] in the case of 2-[18F]FPhe [28], the novel procedure delivered this radiotracer with >95% enantiomeric excess (ee). The observed suppression of racemization in the case 2-[18F]FPhe could be explained as follows. The thermally unstable iodonium bicarbonate and carbonate iodonium salts could form as a result of a partial anion exchange with the anion exchange resin in HCO3– and/or CO32– form during the elution of [18F]fluoride. In the subsequent MeOH evaporation step CO2 and H2O will be lost affording, in the case of the ortho-iodonium salt 9, the corresponding betaine with concurrent loss of stereointegrity (refer to [28] for a more detailed discussion). Consequently, avoiding MeOH evaporation suppresses racemization. Accordingly, the novel radiolabeling method could be beneficial also for the production of other ortho-[18F]fluoro-substituted aromatic amino acids such as 6-[18F]FDOPA, 2-[18F]FTyr and 6-[18F]FMT with high enantiomeric purity.
3. Materials and Methods
3.1. General
Chemicals and solvents were purchased from Sigma-Aldrich GmbH (Steinheim, Germany), Fluka AG (Buchs, Switzerland), TCI EUROPE N.V. (Zwijndrecht, Belgium), ChemPUR GmbH (Karlsruhe, Germany), Merck KGaA (Darmstadt, Germany) and ABCR GmbH (Karlsruhe, Germany) and used as delivered. Anhydrous solvents were purchased from Sigma-Aldrich GmbH (Steinheim, Germany) and stored under argon.
3.2. Nuclear Magnetic Resonance
1H-NMR spectra: Bruker Avance III (400 MHz) and Varian INOVA 400 (400 MHz). 1H chemical shifts are reported in ppm relative to residual peaks of deuterated solvents. The observed signal multiplicities are characterized as follows: s = singlet, d = doublet, t = triplet, m = multiplet, and br = broad. Coupling constants (J) were reported in Hertz (Hz). 13C-NMR spectra [additional APT (Attached Proton Test) or DEPT (Distortionless Enhancement by Polarization Transfer)]: Bruker Avance III (101 MHz) and Varian INOVA 400 (101 MHz). 13C chemical shifts are reported in ppm relative to residual peaks of deuterated solvents. 19F-NMR spectra: and Bruker DPX Avance 200 (188 MHz). Copies of the 1H and 13C NMR spectra are available in the Supplementary Materials.
3.3. Mass Spectroscopy
High-resolution mass spectra (HRMS) were measured on LTQ FT Ultra (Thermo Fisher Scientific Inc., Bremen, Germany). Copies of MS spectra are available in the Supplementary Materials.
3.4. Chemistry
All reactions were carried out with magnetic stirring, if not stated otherwise, and, if air or moisture sensitive, substrates and/or reagents were handled in flame-dried glassware under argon or nitrogen. Organic extracts were dried with anhydrous MgSO4.
Column chromatography: Merck silica gel, grade 60, 230–400 mesh. Solvent proportions are indicated in a volume/volume ratio.
Thin layer chromatography (TLC) was performed using precoated sheets, 0.25 mm Sil G/UV254 from Merck KGaA (Darmstadt, Germany). The chromatograms were viewed under UV light (λ = 254 nm).
(4-Methoxyphenyl)(mesityl)iodonium tosylate (1·OTs) [29], (4-methoxyphenyl)(mesityl)iodonium tetrafluoroborate (1·BF4) [17,29], (phenyl)(mesityl)iodonium tetrafluoroborate [30], (3-carbonylphenyl)(mesityl)iodonium tetrafluoroborate [30], 2-{2-[(2S)-2-{bis[(tert-butoxy)carbonyl]amino}-3-(tert-butoxy)-3-oxopropyl]phenyl}(2,4,6-trimethylphenyl)iodanium tetrafluoroborate (11) [28], 4-{2-[(2S)-2-{[(tert-butoxy)carbonyl]amino}-3-(tert-butoxy)-3-oxopropyl]phenyl}(2,4,6-trimethylphenyl)iodanium tetrafluoroborate (13) [17], 4-{2-[(2S)-2-{bis[(tert-butoxy)carbonyl]amino}-3-(tert-butoxy)-3-oxopropyl]phenyl}(2,4,6-trimethylphenyl)iodanium tetrafluoroborate [28], (S)-Ni-BPB-Gly [27] hydroxy{[(4-methylphenyl)sulfonyl]oxy}(2,4,6-trimethylphenyl)-λ3–iodine [31] were prepared according to the literature or modified literature procedures.
3.4.1. (S,S)-Ni-BPB-3-IPhe (14)
A suspension of (S)-Ni-BPB-Gly (5 g, 10.04 mmol) in a mixture of DMF (5 mL) and MeCN (10 mL) was degassed with three freeze-pump-thaw cycles at −70 °C. Afterwards, NaH (520 mg, 60 % in oil, 13.0 mmol) and 3-iodobenzyl bromide (3 g, 10.10 mmol) were sequentially added. The cooling bath was replaced by another one (–10 °C; ice–salt bath), and the reaction mixture was vigorously stirred for 40 min. Aqueous AcOH (5 mL + 8 mL H2O) was carefully added, the mixture was poured into ice water (1 L) and the resulting mixture was stirred 16 h. The formed fine crystalline precipitate was filtered off, washed with H2O, dried and carefully washed with Et2O to give 14 as a red solid [6.4 g, 89%, 5% of (S,R)-isomer according to the 1H-spectrum]. 1H NMR (400 MHz, CDCl3) δ 1.77–1.89 (m, 1 H) 1.97–2.08 (m, 1 H) 2.32–2.46 (m, 2 H) 2.46–2.63 (m, 1 H) 2.82 (dd, J = 13.8, 6.0 Hz, 1 H) 3.03 (dd, J = 13.8, 4.4 Hz, 1 H) 3.14–3.24 (m, 1 H) 3.35 (dd, J = 9.8, 7.2 Hz, 1 H) 3.45–3.52 (m, 1 H) 4.23 (dd, J = 6.0, 4.4 Hz, 1 H) 4.30 (d, J = 12.6 Hz, 1 H) 6.68 (d, J = 3.9 Hz, 2 H) 6.86 (d, J = 7.6 Hz, 1 H) 7.10–7.13 (m, 2 H) 7.13–7.20 (m, 2 H) 7.28–7.35 (m, 3 H) 7.42–7.48 (m, 2 H) 7.54–7.58 (m, 2 H) 7.72 (ddd, J = 5.3, 3.6, 1.8 Hz, 1 H) 8.02 (d, J = 7.1 Hz, 2 H) 8.26 (d, J = 8.6 Hz, 1 H). 13C NMR (101 MHz, CDCl3) δ 180.3, 178.2, 171.3, 142.9, 139.1, 138.3, 136.5, 134.0, 133.5, 133.2 (×2), 132.51, 131.47, 130.3, 129.9, 129.7, 129.2, 129.0, 128.9, 128.80, 128.77, 127.7, 127.1, 125.92, 123.4, 120.6, 95.1, 71.1, 70.3, 63.3, 57.3, 39.4, 30.8, 23.4. HRMS (ESI): m/z [M + H]+ calcd for C34H31O3N3INi+: 714.07584; found: 714.07572, [M + Na]+ calcd for C34H31O3N3INiNa+: 736.05778; found: 736.05790. Correct isotopic pattern.
3.4.2. Boc-3IPhe-OtBu (15)
A solution of DTPA(Et4N)2–3 [108 mL; prepared by dissolution of DTPA (28.1 g, 71.43 mmol) and Et4NHCO3 (38.2 g, 199.7 mmol) in 100 mL H2O] was added to a solution of 13 (6.3 g, 8.84 mmol) in MeOH (200 mL) and the resulting dark red mixture was stirred at 70 °C for 24 h. At this point, the TLC control (CHCl3:acetone = 3:1, visualization: UV and ninhydrin) showed the almost complete decomposition of the Ni complex 13 (color change: dark red to blue). MeOH was distilled off under reduced pressure, the pH of the resulting suspension was adjusted to 9 with 1 M NaOH, the precipitate was filtered, washed with an ice-cold H2O and dried affording (S)-2-[N-(N’-benzyl-prolyl)amino]benzophenone (BPB). The filtrate was extracted with Et2O (×3; if the precipitation of the Et2O insoluble 3-IPhe was observed, more 1 m NaOH was added until the amino acid was completely dissolved). The organic extracts were washed with brine (×2), dried and concentrated under reduced pressure to give the second crop of BPB. 10% NaHCO3 (30 mL) was added to the aqueous fraction followed by Boc2O (5.7 g, 26.15 mmol). Afterwards MeOH was added until a homogenous solution was obtained and the resulting mixture was stirred for 16 h. MeOH was removed under reduced pressure, the residual aqueous solution was extracted with Et2O (×3), the organic fractions were discarded and the aqueous fraction was carefully acidified to pH 2 with solid NaHSO4 (vigorous evolution of CO2). The formed emulsion was extracted with Et2O (×2), the organic fraction washed with 1 m NaHSO4 (×3), H2O (×3), brine (×2), dried and concentrated under reduced pressure affording crude Boc-3IPhe-OH (2.5 g, 72%) as a colorless oil gradually solidified into a colorless solid which was used in the next step without further purification. Rf: 0.3 [EtOAc:hexane = 1:1.5 (3% AcOH)]. 1H NMR (400 MHz, DMSO-d6, mixture of two rotamers; only spectrum of the major rotamer is provided) δ ppm 1.32 (s, 9 H) 2.77 (dd, J = 13.6, 10.8 Hz, 1 H) 2.99 (dd, J = 13.8, 4.3 Hz, 1 H) 4.08 (dd, J = 19.0, 4.4 Hz, 1 H) 4.08 (d, J = 2.4 Hz, 1 H) 7.02–7.18 (m, 2 H) 7.27 (d, J = 7.8 Hz, 1 H) 7.57 (d, J = 7.8 Hz, 1 H) 7.62 (s, 1 H) 12.24–13.03 (br, 1 H).
tert-Butyl 2,2,2-trichloroacetimidate (2.77 mL, 3.38 g, 15.49 mmol) was added to a suspension of Boc-3IPhe-OH (2.4 g, 6.13 mmol) in anhydrous CH2Cl2 (10 mL) and the reaction mixture was stirred at 40–45 °C for 72 h. The mixture was cooled to ambient temperature, diluted with pentane (40 mL) cooled to −20 °C and filtered. The filtrate was washed with 5% NaHCO3 (×3), 1 m NaHSO4 (×3), H2O (×3), brine (×2), dried and concentrated under reduced pressure. The residual oil was dried at 60 °C and 1 mbar to remove a bulk of the unreacted tert-butyl 2,2,2-trichloroacetimidate and the residue was purified by column chromatography (EtOAc: hexane = 1:5) and the product-containing fractions were concentrated under reduced pressure to give colorless oil which was dissolved in pentane. The resulting solution was cooled to 5 °C, filtered and concentrated affording 15 as colorless oil which gradually solidified into a colorless solid. Finally, recrystallization from hexane (the mother liquor was concentrated under reduced pressure and the residue recrystallized from hexane; ×2) furnished the pure 15 [32] (2.37 g, 56% over 4 steps). Rf: 0.5 (EtOAc:hexane = 1:6). 1H NMR (400 MHz, CDCl3; mixture of two rotamers; only spectrum of the major rotamer is provided) δ 1.42 (s, 9 H) 1.44 (s, 8 H) 3.01 (d, J = 5.50 Hz, 2 H) 4.33–4.54 (m, 1 H) 5.04 (d, J = 6.75 Hz, 1 H) 6.97–7.09 (m, 1 H) 7.16 (d, J = 7.63 Hz, 1 H) 7.53 (s, 1 H) 7.57 (d, J = 7.88 Hz, 1 H). 13C NMR (101 MHz, CDCl3) δ 170.5, 155.0, 138.9, 138.5, 135.8, 130.0, 128.8, 94.2, 82.4, 79.8, 54.7, 38.0, 28.3, 29.0. HRMS (ESI): m/z [M + H]+ calcd for C18H27INO4+: 448.09793; found: 448.09800, [M + Na]+ calcd for C18H26INO4Na+: 470.07987; found: 470.07993.
3.4.3. 3-{2-[(2S)-2-{[(tert-Butoxy)carbonyl]amino}-3-(tert-butoxy)-3-oxopropyl]phenyl}(2,4,6-trime-thylphenyl)iodanium tetrafluoroborate (12)
Sn2Me6 (1.27 mL, 2.01 g, 6.15 mmol) was added to a solution of Boc-3-IPhe-OtBu (14) (1.1 g, 2.46 mmol) and Pd(PPh3)4 (0.284 g, 0.25 mmol) in anhydrous 1,4-dioxane (5 mL) in a glove box. The reaction mixture was stirred at 80 °C for 16 h and thereafter at 110 °C for 3 h, cooled to ambient temperature, filtered through Celite® and concentrated under reduced pressure. The residue was purified by column chromatography (EtOAc:hexane = 1:6 on silica gel with 0.1% CaO) to give Boc-3-(SnMe3)Phe-OtBu as a colorless oil (1.1 g, 92%) which was used in the next step without further purification and characterisation. Rf = 0.6, EtOAc:hexane = 1:6.
Hydroxy{[(4-methylphenyl)sulfonyl]oxy}-(2,4,6-trimethylphenyl)-λ3–iodine (0.98 g, 2.25 mmol) was added to a solution of Boc-3-(SnMe3)Phe-OtBu (1.09 g, 2.25 mmol) in CH2Cl2 (10 mL) and the reaction mixture was stirred for 90 min. After that the mixture was concentrated under reduced pressure. The residue was purified by column chromatography (CH2Cl2:MeOH = 10:1) to give 3-{2-[(2S)-2-{[(tert-butoxy)carbonyl]amino}-3-(tert-butoxy)-3-oxopropyl]phenyl}(2,4,6-trimethylphenyl)iodanium tosylate as a viscous yellow oil which was immediately used in the next step. Rf = 0.4, CH2Cl2:MeOH = 10:1.
To a solution of tosylate salt in CH2Cl2 (20 mL) the saturated solution of NaBF4 (10 mL) was added and the mixture was vigorously stirred for 10 min. The aqueous solution and precipitate were separated off, the saturated NaBF4 (10 mL) was added and the mixture was vigorously stirred for 10 min (×5). The organic fraction was dried and concentrated under reduced pressure. The residue was recrystallized from CH2Cl2/Et2O to give after drying under reduced pressure 12 (0.67 g, 36% over two steps) as a faint yellow solid. 1H NMR (400 MHz, CD3CN) δ 1.32 (s, 9 H) 1.36 (s, 9 H) 2.34 (s, 3 H) 2.62 (s, 6 H) 2.89 (dd, J = 14.0, 9.0 Hz, 1 H) 3.10 (dd, J = 13.9, 5.3 Hz, 1 H) 4.22 (d, J = 5.7 Hz, 1 H) 7.76 (s, 1H) 5.53 (d, J = 6.7 Hz, 1 H) 7.22 (s, 2 H) 7.42 (t, J = 7.8 Hz, 1 H) 7.51 (d, J = 7.5 Hz, 1 H), 7.73 (d, J = 8.3 Hz, 1 H) 7.76 (s, 1 H). 13C NMR (101 MHz, CD3CN) δ 171.4, 156.3, 145.9, 143.9, 143.6, 135.9, 134.8, 133.6, 133.2, 131.4, 121.4, 112.6, 82.5, 80.0, 56.1, 38.0, 28.4, 28.1, 27.3, 21.0. 19F NMR (376 MHz, CD3CN) δ −151.32 (10BF4−), −151.37 (11BF4−). HRMS (ESI): m/z [M]+ calcd for C27H37INO4+: 566.1754; found: 566.1756.
3.5. Radiochemistry
3.5.1. General
All radiosyntheses were carried out using anhydrous DMF (Aldrich) and MeOH (Aldrich or Acros). Cu(MeCN)4OTf (Aldrich) was stored under argon. QMA cartridges (Sep-Pak Accell Plus QMA Carbonate Plus Light Cartridge) were obtained from Waters (Waters GmbH, Eschborn, Germany) and used without any preconditioning. RP-cartridges (Strata™-X 33 µm polymeric reversed phase, 200 mg/3 mL, tube) were from Phenomenex (Phenomenex Ltd., Aschaffenburg, Germany).
[18F]Fluoride was produced by the 18O(p,n)18F reaction by bombardment of enriched [18O]water with 16.5 MeV protons at the BC1710 cyclotron (The Japan Steel Works Ltd., Shinagawa, Japan) of the INM-5 (Forschungszentrum Jülich) or PETtrace 4 cyclotron (GE Healthcare, Uppsala, Sweden) at the IHB (Saint-Petersburg). If not otherwise noted, all radiolabeling experiments were carried out under ambient or synthetic air.
Standard deviations were calculated using standard formulae.
3.5.2. TLC Analysis
TLC analysis was carried out on silica gel plates 60 F254 (Merck), TLC Al foils (Sigma Aldrich) or Polygram SIL G/UV254 (Macherey Nagel); radioactivity distribution was measured using a MiniGita radioTLC scanner (Raytest). An aliquot of the quenched reaction mixture (2–3 µL) was applied to a TLC plate; the plate was eluted with 4:1 hexane/EtOAc (A) or 9:1 hexane/EtOAc (B). After developing the plate was immediately covered by tape to prevent the losses of the volatile radioactive products. The Rf values of the different compounds are compiled in Table 2. The RCC (radiochemical conversion) was determined by dividing of the product peak area by the total peak area. TL-chromatograms can be found in the Supplementary Materials.
Table 2.
Rf values of reference compounds.
3.5.3. HPLC Analysis
Before the determination of radiochemical conversions (RCCs), reaction mixtures were diluted with H2O (1–4 mL) (or 50% MeOH) to dissolve any 18F-fluoride adsorbed onto the reaction vessel walls. The loss of radioactivity on the vessel walls did not exceed 13% ± 2% from the starting activity (n > 100). All radiochemical yields (RCYs) were decay corrected and radiochemical purities (RCPs) were determined after purification.
High-performance liquid chromatography (HPLC) was carried out using a system equipped with: a Knauer pump, a Knauer K-2500 UV/VIS detector (Knauer), a manual Rheodyne injector (20 μL loop) and a NaI(Tl) well-type scintillation detector (EG&G Ortec; model 276 Photomultiplier Base) with an ACE Mate Amplifier and BIAS supply (all from Ortec Ametek). Table 3 contains the different applied HPLC methods (column and eluent).
Table 3.
Applied HPLC-methods.
Data acquisition and interpretation were performed using Gina software (Raytest). The dead times of the HPLC columns were determined using thiourea. UV and radioactivity detectors were connected in series, giving a time delay of 0.05–0.3 min depending on the flow rate. 18F-Labeled compounds were identified by co-injection of the unlabeled reference compounds. The completeness of the radioactivity elution was checked by analyzing of the same sample amount choosing a column bypass (Figure 5).
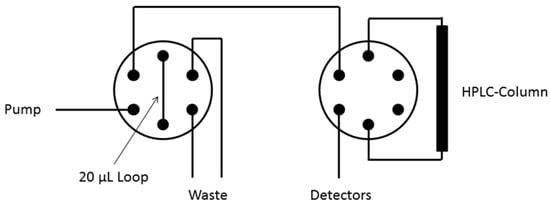
Figure 5.
Arrangement of HPLC valves (analytical HPLC).
Alternatively, analytical HPLC was performed on a Dionex ICS- 5000 system equipped with a gradient pump, a variable wavelength UV detector Dionex UV 254 and NaI(Tl) well-type scintillation detector (Carroll and Ramsey Associates, model 105-S). The k’ values of the different radiolabeled compounds with the used HPLC methods are compiled in Table 4. HPL chromatograms can be found in the Supplementary Materials.
Table 4.
Retention factors (k’)a of the reference compounds.
3.5.4. Determination of Enantiomeric Purity
Enantiomeric excess was determined by HPLC (method D (Table 3)); cf. Supplementary Materials.
3.5.5. Manual Radiosynthesis. Screening of the Reaction Conditions and Reaction Scope—General Procedure 1 (GP 1)
A solution of [18F]F– (0.05–0.5 GBq) in [18O]H2O was loaded from the male side on a QMA cartridge. The cartridge was flushed from the male side with MeOH (1 mL). 18F– was eluted from the female side of the cartridge with a solution of the respective radiolabeling precursor (21 µmol; if not otherwise stated) in a solution of 19% MeOH in DMF (620 µL, MeOH/DMF = 120/500 µL, if not otherwise stated) directly into a solution of Cu(MeCN)4OTf (7.9 mg, 21 µmol; if not otherwise stated) in DMF (600 µL; if not otherwise stated) and the resulting solution was heated at 85 °C for 20 min (if not otherwise stated). The reaction mixture was diluted with H2O or 50% MeOH (1–4 mL) and RCCs were determined by TLC and/or HPLC (method A and method B of Table 3).
3.5.6. Manual Radiosynthesis. Preparation of 2-[18F]FPhe ([18F]8) and 4-[18F]FPhe ([18F]10)–General Procedure 2 (GP 2)
A solution of [18F]F– (0.5–5 GBq) in [18O]H2O was loaded from the male side on a QMA cartridge. The cartridge was flushed from the male side with MeOH (1 mL). 18F– was eluted from the female side of the cartridge with a solution of the respective radiolabeling precursor (21 µmol) in 19% MeOH in DMF (620 µL, MeOH/DMF = 120/500 µL) directly into a solution of Cu(MeCN)4OTf (7.9 mg, 21 µmol) in DMF (600 µL) and the resulting solution was heated at 85 °C for 20 min. The reaction mixture was diluted with 50% MeOH (4 mL) and stirred for 30 sec. The resulting solution was loaded onto a RP cartridge, the reaction vial was rinsed with H2O (2 mL) which was passed through the cartridge. The cartridge was washed with H2O (4 mL) and radiolabeled intermediates were eluted MeCN (3 mL). All volatiles were removed under reduced pressure of Ar. 12 n HCl (0.8 mL) was added and the reaction mixture was heated for 10 min at 80 °C. Afterwards the mixture was concentrated under reduced pressure to approximately 0.3–0.4 mL and diluted with H2O (0.3 mL). The desired radiolabeled amino acids were isolated by HPLC using method B and C (Table 3).
3.5.7. Semi-Automated Synthesis of 18F-Labeled Products–General Procedure 3 (GP 3) (Figure 6)
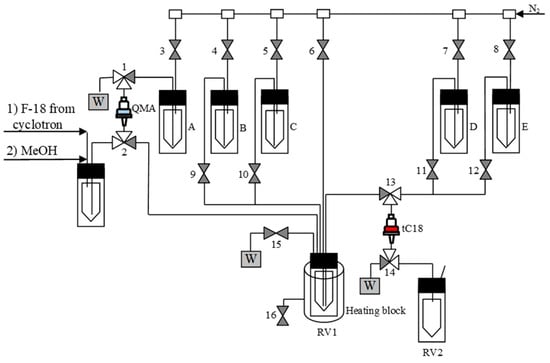
Figure 6.
Process flow diagram (PFD) for the semi-automated radiosynthesis of [18F]2–10. A: a solution of precursor in MeOH/DMF (0.12/0.5 mL); B: H2O (4 mL); C: 12 n HCl (0.5 mL); D: H2O (5 mL); E: MeOH (2.0 mL); RV1: Cu(MeCN)4OTf (21 µmol) in DMF (0.6 mL).
1. Loading of [18F]fluoride (0.1–10 GBq) onto a QMA ion exchange cartridge;
2. Washing of the cartridge with MeOH (2.0 mL);
3. Elution of [18F]fluoride from the ion exchange cartridge with a solution of the radiolabeling precursor (21 µmol) in 20% MeOH in DMF (0.6 mL) into RV1 with a solution of Cu(MeCN)4OTf (21 µmol) in DMF (0.6 mL);
4. Open valve 1, 2, 3 to completely transfer methanolic solution from QMA to RV1;
5. Heating of the reaction mixture in RV1 at 85 °C for 20 min;
6. Cooling of RV1 down to 50 °C;
7. Addition of water (4 mL) → in the case of [18F]-amino acids: precipitation of precursor;
For optimization experiments:
8. Analysis of the reaction mixture by TLC and/or HPLC.
For the production of radiolabeled amino acids [18F]8–10:
8. Loading of the mixture onto a tC18 long RP SPE cartridge;
9. Rinsing of the cartridge with H2O (5 mL);
10. Elution of the radiolabeled intermediate into RV2 using MeOH (2.0 mL);
11. Place vial RV2 in the heating block;
12. Evaporation of MeOH at 100 °C within 5 min using a flow of N2;
13. Addition of 12 n HCl (0.5 mL) and heating at 130 °C for 10 min;
14. Cooling of RV2 to 55 °C and addition of a 10 mM AcONa/50 mM AcOH/0,1 g/L ascorbic acid solution (3.5 mL);
15. Loading of the mixture in the loop of semi preparative HPLC system; HPLC: pump: SYCAM S1122 solvent delivery system, UV detector and radioactivity detector from the GE Tracerlab FX Cpro module; column: Ascentis RP-Amide 250 × 10 mm (Supelco).
16. Isolation of the product using 10 mM AcONa/50 mM AcOH/0,1 g/L ascorbic acid, flow rate 4 mL/min;
17. Manual collection of the product-containing fraction into a collection vial;
18. Transfer the product solution from a collection vial into a sterile, filter-vented final product vial via 0.22 µm sterile membrane filter using a flow of N2.
4. Conclusions
We demonstrated for the first time that alcohols are suitable co-solvents for Cu-mediated 18F-fluorination of iodonium salts. The developed evaporation-free radiofluorination protocol enables a simple, fast and efficient preparation of labeled aromatics and is amenable to automation. The practicality of the protocol was confirmed by the high-yielding production of clinical doses of 2–4-[18F]fluorophenylalanines as well as by its direct implementation in a remote-controlled synthesis unit. Additionally, the novel method avoids significant racemization observed if the conventional “minimalist” protocol is applied for the preparation of ortho-[18F]fluoro-substituted aromatic amino acids.
Supplementary Materials
The following are available online: 1H-, 13C-, 19F-NMR and MS spectra, and radio-TLC and HPLC-chromatograms.
Author Contributions
V.V.O., D.J.M., N.K., O.F.K., O.S.F., E.A.U. and B.D.Z. performed experiments, B.N., R.N.K. and B.D.Z. conceived and designed experiments, V.V.O., O.S.F., B.N., R.N.K. and B.D.Z. wrote the paper.
Funding
This work was supported by Russian Foundation of Basic Researches (RFBR), grant 16-54-12062/16, and Deutsche Forschungsgemeinschaft (DFG), grant ZL 65/1-1.
Acknowledgments
The authors thank H. Endepols for valuable help in the preparation of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lee, E.; Kamlet, A.S.; Powers, D.C.; Neumann, C.N.; Boursalian, G.B.; Furuya, T.; Choi, D.C.; Hooker, J.M.; Ritter, T. A Fluoride-Derived Electrophilic Late-Stage Fluorination Reagent for PET Imaging. Science 2011, 334, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Kamlet, A.S.; Neumann, C.N.; Lee, E.; Carlin, S.M.; Moseley, C.K.; Stephenson, N.; Hooker, J.M.; Ritter, T. Application of Palladium-Mediated 18F-Fluorination to PET Radiotracer Development: Overcoming Hurdles to Translation. PLoS ONE 2013, 8, e59187. [Google Scholar] [CrossRef] [PubMed]
- Hoover, A.J.; Lazari, M.; Ren, H.; Narayanam, M.K.; Murphy, J.M.; van Dam, R.M.; Hooker, J.M.; Ritter, T. A Transmetalation Reaction Enables the Synthesis of [18F]5-Fluorouracil from [18F]Fluoride for Human PET Imaging. Organometallics 2016, 35, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Hooker, J.M.; Ritter, T. Nickel-Mediated Oxidative Fluorination for PET with Aqueous [18F] Fluoride. J. Am. Chem. Soc. 2012, 134, 17456–17458. [Google Scholar] [CrossRef] [PubMed]
- Zlatopolskiy, B.D.; Zischler, J.; Urusova, E.A.; Endepols, H.; Kordys, E.; Frauendorf, H.; Mottaghy, F.M.; Neumaier, B. A Practical One-Pot Synthesis of Positron Emission Tomography (PET) Tracers via Nickel-Mediated Radiofluorination. ChemistryOpen 2015, 4, 457–462. [Google Scholar] [CrossRef]
- Ichiishi, N.; Brooks, A.F.; Topczewski, J.J.; Rodnick, M.E.; Sanford, M.S.; Scott, P.J.H. Copper-Catalyzed [18F]Fluorination of (Mesityl)(aryl)iodonium Salts. Org. Lett. 2014, 16, 3224–3227. [Google Scholar] [CrossRef]
- Tredwell, M.; Preshlock, S.M.; Taylor, N.J.; Gruber, S.; Huiban, M.; Passchier, J.; Mercier, J.; Génicot, C.; Gouverneur, V. A General Copper-Mediated Nucleophilic 18F-Fluorination of Arenes. Angew. Chem. Int. Ed. 2014, 53, 7751–7755. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.J.; Emer, E.; Preshlock, S.; Schedler, M.; Tredwell, M.; Verhoog, S.; Mercier, J.; Genicot, C.; Gouverneur, V. Derisking the Cu-Mediated 18F-Fluorination of Heterocyclic Positron Emission Tomography Radioligands. J. Am. Chem. Soc. 2017, 139, 8267–8276. [Google Scholar] [CrossRef]
- Mossine, A.V.; Brooks, A.F.; Makaravage, K.J.; Miller, J.M.; Ichiishi, N.; Sanford, M.S.; Scott, P.J.H. Synthesis of [18F]Arenes via the Copper-Mediated [18F]Fluorination of Boronic Acids. Org. Lett. 2015, 17, 5780–5783. [Google Scholar] [CrossRef]
- Makaravage, K.J.; Brooks, A.F.; Mossine, A.V.; Sanford, M.S.; Scott, P.J.H. Copper-Mediated Radiofluorination of Arylstannanes with [18F]KF. Org. Lett. 2016, 18, 5440–5443. [Google Scholar] [CrossRef]
- McCammant, M.S.; Thompson, S.; Brooks, A.F.; Krska, S.W.; Scott, P.J.H.; Sanford, M.S. Cu-Mediated C–H 18F-Fluorination of Electron-Rich (Hetero)arenes. Org. Lett. 2017, 19, 3939–3942. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Makaravage, K.J.; Brooks, A.F.; Scott, P.J.H.; Sanford, M.S. Copper-Mediated Aminoquinoline-Directed Radiofluorination of Aromatic C−H Bonds with K18F. Angew. Chem. Int. Ed. 2019, 58, 3119–3122. [Google Scholar] [CrossRef] [PubMed]
- Antuganov, D.; Zykov, M.; Timofeev, V.; Timofeeva, K.; Antuganova, Y.; Orlovskaya, V.; Fedorova, O.; Krasikova, R. Copper-Mediated Radiofluorination of Aryl Pinacolboronate Esters: A Straightforward Protocol by Using Pyridinium Sulfonates. Eur. J. Org. Chem. 2019, 2019, 918–922. [Google Scholar] [CrossRef]
- Antuganov, D.; Zykov, M.; Timofeeva, K.; Antuganova, Y.; Orlovskaya, V.; Krasikova, R. Effect of Pyridine Addition on the Efficiency of Copper-Mediated Radiofluorination of Aryl Pinacol Boronates. ChemistrySelect 2017, 2, 7909–7912. [Google Scholar] [CrossRef]
- Preshlock, S.; Calderwood, S.; Verhoog, S.; Tredwell, M.; Huiban, M.; Hienzsch, A.; Gruber, S.; Wilson, T.C.; Taylor, N.J.; Cailly, T.; et al. Enhanced copper-mediated 18F-fluorination of aryl boronic esters provides eight radiotracers for PET applications. Chem. Commun. 2016, 52, 8361–8364. [Google Scholar] [CrossRef]
- Zarrad, F.; Zlatopolskiy, B.D.; Krapf, P.; Zischler, J.; Neumaier, B. A Practical Method for the Preparation of 18F-Labeled Aromatic Amino Acids from Nucleophilic [18F]Fluoride and Stannyl Precursors for Electrophilic Radiohalogenation. Molecules 2017, 22, 2231. [Google Scholar] [CrossRef]
- Zlatopolskiy, B.D.; Zischler, J.; Krapf, P.; Zarrad, F.; Urusova, E.A.; Kordys, E.; Endepols, H.; Neumaier, B. Copper-Mediated Aromatic Radiofluorination Revisited: Efficient Production of PET Tracers on a Preparative Scale. Chem. Eur. J. 2015, 21, 5972–5979. [Google Scholar] [CrossRef]
- Mossine, A.V.; Brooks, A.F.; Bernard-Gauthier, V.; Bailey, J.J.; Ichiishi, N.; Schirrmacher, R.; Sanford, M.S.; Scott, P.J.H. Automated synthesis of PET radiotracers by copper-mediated 18F-fluorination of organoborons: Importance of the order of addition and competing protodeborylation. J. Labelled Compd. Radiopharm. 2018, 61, 228–236. [Google Scholar] [CrossRef]
- Mossine, A.V.; Brooks, A.F.; Ichiishi, N.; Makaravage, K.J.; Sanford, M.S.; Scott, P.J.H. Development of Customized [18F]Fluoride Elution Techniques for the Enhancement of Copper-Mediated Late-Stage Radiofluorination. Sci. Rep. 2017, 7, 233. [Google Scholar] [CrossRef]
- Richarz, R.; Krapf, P.; Zarrad, F.; Urusova, E.A.; Neumaier, B.; Zlatopolskiy, B.D. Neither azeotropic drying, nor base nor other additives: A minimalist approach to 18F-labeling. Org. Biomol. Chem. 2014, 12, 8094–8099. [Google Scholar] [CrossRef]
- Zischler, J.; Krapf, P.; Richarz, R.; Zlatopolskiy, B.D.; Neumaier, B. Automated synthesis of 4-[18F]fluoroanisole, [18F]DAA1106 and 4-[18F]FPhe using Cu-mediated radiofluorination under “minimalist” conditions. Appl. Radiat. Isot. 2016, 115, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Zischler, J.; Kolks, N.; Modemann, D.; Neumaier, B.; Zlatopolskiy, B.D. Alcohol-Enhanced Cu-Mediated Radiofluorination. Chem. Eur. J. 2017, 23, 3251–3256. [Google Scholar] [CrossRef] [PubMed]
- Zlatopolskiy, B.D.; Zischler, J.; Schäfer, D.; Urusova, E.A.; Guliyev, M.; Bannykh, O.; Endepols, H.; Neumaier, B. Discovery of 7-[18F]Fluorotryptophan as a Novel Positron Emission Tomography (PET) Probe for the Visualization of Tryptophan Metabolism in Vivo. J. Med. Chem. 2018, 61, 189–206. [Google Scholar] [CrossRef] [PubMed]
- Zlatopolskiy, B.D.; Zischler, J.; Krapf, P.; Richarz, R.; Lauchner, K.; Neumaier, B. Minimalist approach meets green chemistry: Synthesis of 18F- labeled (hetero)aromatics in pure ethanol. J. Labelled Compd. Radiopharm. 2019, 62, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Corbett, C.A.; Martínez, T.J.; Lisy, J.M. Solvation of the Fluoride Anion by Methanol. J. Phys. Chem. A 2002, 106, 10015–10021. [Google Scholar] [CrossRef]
- Ichiishi, N.; Canty, A.J.; Yates, B.F.; Sanford, M.S. Mechanistic Investigations of Cu-Catalyzed Fluorination of Diaryliodonium Salts: Elaborating the CuI/CuIII Manifold in Copper Catalysis. Organometallics 2014, 33, 5525–5534. [Google Scholar] [CrossRef] [PubMed]
- Belokon, Y.N.; Tararov, V.I.; Maleev, V.I.; Savel’eva, T.F.; Ryzhov, M.G. Improved procedures for the synthesis of (S)-2-[N-(N′-benzylprolyl)amino]benzophenone (BPB) and Ni(II) complexes of Schiff’s bases derived from BPB and amino acids. Tetrahedron: Asymmetry 1998, 9, 4249–4252. [Google Scholar] [CrossRef]
- Modemann, D.J.; Zlatopolskiy, B.D.; Urusova, E.A.; Zischler, J.; Craig, A.; Ermert, J.; Guliyev, M.; Endepols, H.; Neumaier, B. 2-[18F]Fluorophenylalanine: Synthesis by Nucleophilic 18F-Fluorination and Preliminary Biological Evaluation. Synthesis 2019, 51, 664–676. [Google Scholar] [CrossRef]
- Lindstedt, E.; Reitti, M.; Olofsson, B. One-Pot Synthesis of Unsymmetric Diaryliodonium Salts from Iodine and Arenes. J. Org. Chem. 2017, 82, 11909–11914. [Google Scholar] [CrossRef]
- Ichiishi, N.; Canty, A.J.; Yates, B.F.; Sanford, M.S. Cu-Catalyzed Fluorination of Diaryliodonium Salts with KF. Org. Lett. 2013, 15, 5134–5137. [Google Scholar] [CrossRef]
- Merritt, E.A.; Carneiro, V.M.T.; Silva, L.F.; Olofsson, B. Facile Synthesis of Koser’s Reagent and Derivatives from Iodine or Aryl Iodides. J. Org. Chem. 2010, 75, 7416–7419. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Murai, Y.; Yoshida, T.; Ishida, A.; Masuda, K.; Sakihama, Y.; Hashidoko, Y.; Hatanaka, Y.; Hashimoto, M. Alternative One-Pot Synthesis of (Trifluoromethyl)phenyldiazirines from Tosyloxime Derivatives: Application for New Synthesis of Optically Pure Diazirinylphenylalanines for Photoaffinity Labeling. Org. Lett. 2015, 17, 616–619. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of all compounds are available from the authors. |



















© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).