Achiral Zeolites as Reaction Media for Chiral Photochemistry

Abstract
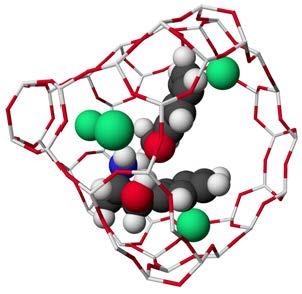
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Beginnings of Asymmetric Photochemistry
3. Zeolites as Media for Asymmetric Photoreactions: Chiral and Achiral Zeolites
4. Achiral Zeolites Rendered Chiral
5. Chiral Inductor as an Active Co-guest: Photoreduction Prompted by Electron Transfer
6. Chiral Inductor as a Passive Co-guest: Chiral Induction on Photoproducts within Zeolites
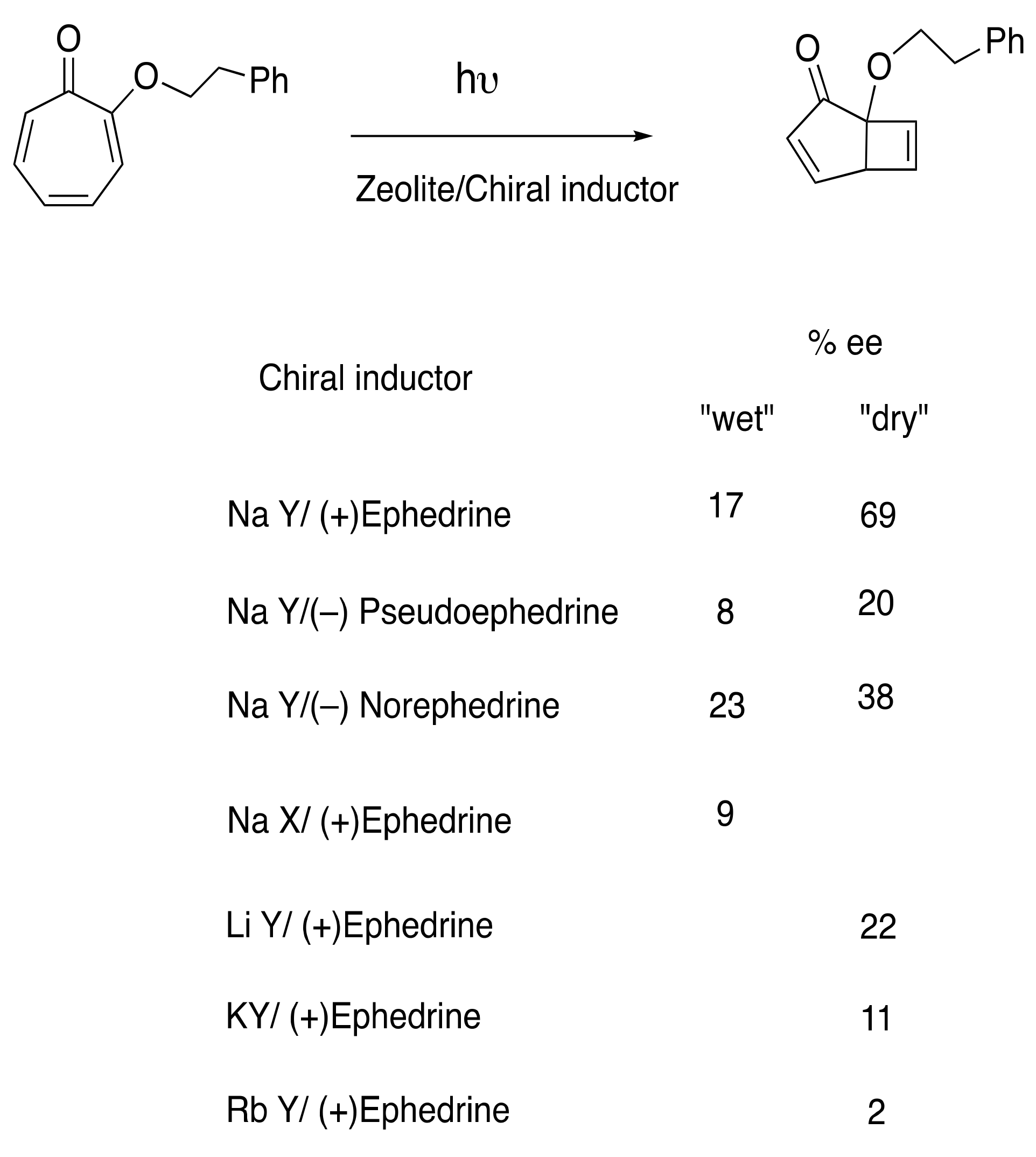
6.1. Photocyclization of Tropolones
6.2. Photocyclization of Pyridones
6.3. Photoisomerization of 1, 2-Diphenylcyclopropanes
6.4. Oxa-di-π-Methane Rearrangement of Cyclohexadienones
6.5. Norrish-Yang Photocyclizations
7. Summary
Funding
Acknowledgments
Conflicts of Interest
References
- Turro, N.J.; Ramamurthy, V.; Scaiano, J.C. Modern Molecular Photochemistry of Organic Molecules; University Science Books: Sausalito, CA, USA, 2010. [Google Scholar]
- De Mayo, P.; Reid, S.T. Photochemical rearrangements and related transformations. Q. Rev. Chem. Soc. 1961, 15, 393–417. [Google Scholar] [CrossRef]
- Von Ginsburg, D. Solid State Photochemistry; Verlag Chemie: Weinheim, Germany, 1976. [Google Scholar]
- Schonberg, A. Preparative Organic Photochemistry; Springer-Verlag New York Inc.: New York, NY, USA, 1968. [Google Scholar]
- Yoon, T.P. Photochemical Stereocontrol Using Tandem Photoredox-Chiral Lewis Acid Catalysis. Acc. Chem. Res. 2016, 49, 2307–2315. [Google Scholar] [CrossRef] [PubMed]
- McCusker, C.E.; Castellano, F.N. Materials Integrating Photochemical Upconversion. Top. Curr. Chem. 2016, 374, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Ramamurthy, V.; Natarajan, A.; Kaanumalle, L.S.; Karthikeyan, S.; Sivaguru, J.; Shailaja, J.; Joy, A. Chiral photochemistry within zeolites. In Chiral Photochemistry; Inoue, Y., Ramamurthy, V., Eds.; Marcel Dekker, Inc.: New York, NY, USA, 2004; Volume 11, pp. 563–631. [Google Scholar]
- Ramamurthy, V. Controlling photochemical reactions via confinement: Zeolites. J. Photochem. Photobiol. C 2000, 1, 145–166. [Google Scholar] [CrossRef]
- Rau, H. Asymmetric Photochemistry in Solution. Chem. Rev. 1983, 83, 535–547. [Google Scholar] [CrossRef]
- Inoue, Y. Asymmetric Photochemical Reactions in Solution. Chem. Rev. 1992, 92, 741–770. [Google Scholar] [CrossRef]
- Brenninger, C.; Jolliffe, J.D.; Bach, T. Chromophore Activation of α,β-Unsaturated Carbonyl Compounds and Its Application to Enantioselective Photochemical Reactions. Angew. Chem. Int. Ed. 2018, 57, 14338–14349. [Google Scholar] [CrossRef]
- Brimioulle, R.; Lenhart, D.; Maturi, M.M.; Bach, T. Enantioselective Catalysis of Photochemical Reactions. Angew. Chem. Int. Ed. 2015, 54, 3872–3890. [Google Scholar] [CrossRef]
- Grosch, B.; Bach, T. Chiral Photochemistry. In Chiral Photochemistry; Inoue, Y., Ramamurthy, V., Eds.; Marcel Dekker: New York, NY, USA, 2004; pp. 315–340. [Google Scholar]
- Müller, C.; Bach, T. Chirality Control in Photochemical Reactions: Enantioselective Formation of Complex Photoproducts in Solution. Aus. J. Chem. 2008, 61, 557–564. [Google Scholar] [CrossRef]
- Poplata, S.; Troster, A.; Zou, Y.Q.; Bach, T. Recent Advances in the Synthesis of Cyclobutanes by Olefin [2 + 2] Phtocycloaddition Reactions. Chem. Rev. 2016, 116, 9748–9815. [Google Scholar] [CrossRef]
- Zou, Y.-Q.; Hoffmann, F.M.; Bach, T. Iminium and enamine catalysis in enantioselectivephotochemical reactions. Chem. Soc. Rev. 2018, 47, 278–290. [Google Scholar] [CrossRef]
- Kumarasamy, E.; Ayitou, A.J.-L.; Vallavoju, N.; Raghunathan, R.; Iyer, A.; Clay, A.; Kandappa, S.K.; Sivaguru, J. Tale of Twisted Molecules. Atropselective Photoreactions: Taming Light Induced Asymmetric Transformations through Non-biaryl Atropisomers. Acc. Chem. Res. 2016, 49, 2713–2724. [Google Scholar] [CrossRef] [PubMed]
- Pete, J.-P. Asymmetric Photoreactions of Conjugated Enones and Esters. In Advances in Photochemistry; Neckers, D.C., Volman, D.H., Von Bunan, G., Eds.; John Wiley & Sons, Inc.: Hobaken, NJ, USA, 1996. [Google Scholar]
- Pete, J.-P.; Hoffmann, N. Diastereodifferentiating Photoreactions. In Chiral Photochemistry; Inoue, Y., Ramamurthy, V., Eds.; Marcel Dekker: New York, NY, USA, 2004; pp. 179–233. [Google Scholar]
- Griesbeck, A.G.; Meierhenrich, U.J. Asymmetric Photochemistry and Photochirogenesis. Angew. Chem. Int. Ed. 2002, 41, 3147–3154. [Google Scholar] [CrossRef]
- Buschman, H.; Scharf, H.-D.; Hoffmann, N.; Esser, P. The Isoinversion Principle- A General Model of Chemical Selectivity. Angew. Chem. Int. Ed. Engl. 1991, 30, 477–515. [Google Scholar] [CrossRef]
- Yang, C.; Inoue, Y. Supramolecular Photochirogenesis. Chem. Soc. Rev. 2014, 43, 4123–4143. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Inoue, Y. Supramolecular Photochirogenesis. In Supramolecular Photochemistry; Ramamurthy, V., Inoue, Y., Eds.; John Wiley& Sons, Inc.: Hobaken, NJ, USA, 2011; pp. 115–154. [Google Scholar]
- Inoue, Y.; Ramamurthy, V. Chiral Photochemistry; Marcel Dekker: New York, NY, USA, 2004. [Google Scholar]
- Hammond, G.S.; Cole, R.S. Asymmetric Induction during Energy Transfer. J. Am. Chem. Soc. 1965, 87, 3256–3257. [Google Scholar] [CrossRef]
- Elgavi, A.; Green, S.B.; Schmidt, G.M.J. Reactions in Chiral Crystals. Optically Active Heterophotodimer Formation from Chiral Single Crystals. J. Am. Chem. Soc. 1973, 95, 2058–2059. [Google Scholar] [CrossRef]
- Green, B.S.; Lahav, M.; Rabinovich, D. Asymmetric Synthesis via Reactions in Chiral Crystals. Acc. Chem. Res. 1979, 12, 191–197. [Google Scholar] [CrossRef]
- Addadi, L.; Cohen, M.D.; Lahav, M. Synthesis of Chiral Polymers by Reactions in Chiral Crystal structures. Mol. Cryst. Liq. Cryst. 1976, 32, 137–141. [Google Scholar] [CrossRef]
- Addadi, L.; Lahav, M. Towards the planning and execution of an “Absolute” Asymmetric synthesis of Chiral dimers and polymers with quantitative Enantiomeric Yield. Pure App.l. Chem. 1979, 51, 1269–1284. [Google Scholar] [CrossRef]
- Sakamoto, M. Absolute Asymmetric synthesis from Achiral Molecules in the Chiral Crystalline Environment. Chem. Eur. J. 1997, 3, 684–689. [Google Scholar] [CrossRef]
- Sakamoto, M. Absolute asymmetric photochemistry using spontaneous chiral crystallization. In Chiral Photochemistry; Inoue, Y., Ramamurthy, V., Eds.; Marcel Dekker, Inc.: New York, NY, USA, 2004; pp. 415–461. [Google Scholar]
- Caswell, L.; Garcia-Garibay, M.A.; Scheffer, J.R.; Trotter, J. Optical Activity can be created from “Nothing”. J. Chem. Edu. 1993, 70, 785–787. [Google Scholar] [CrossRef]
- Farina, M.; Audisio, G.; Natta, G. A New Kind of Asymmetric Synthesis. The Radiation Polymerization of trans-1,3-Pentadiene Included in Optically Active Perhydrotriphenylene. J. Am. Chem. Soc. 1967, 89, 5071. [Google Scholar] [CrossRef]
- Toda, F. Enantiocontrol of photoreactions in the solid state. Mol. Cryst. Liq. Cryst. 1988, 161, 355–362. [Google Scholar] [CrossRef]
- Toda, F. Reaction Control of Guest Compounds in Host-Guest Incluson Complexes. Top. Curr. Chem. 1988, 149, 211–238. [Google Scholar]
- Toda, F. Solid State Organic Chemistry: Efficient Reactions, Remarkable Yields, and Stereoselectivity. Acc. Chem. Res. 1995, 28, 480–486. [Google Scholar] [CrossRef]
- Scheffer, J.R. In the footsteps of Pasteur: Asymmetric induction in the photochemistry of crystalline ammonium carboxylate salts. Can. J. Chem. 2001, 79, 349–357. [Google Scholar] [CrossRef]
- Scheffer, J.R. Ionic Chiral Auxiliary Approach in Photochemistry. In Chiral Photochemistry; Inoue, Y., Ramamurthy, V., Eds.; Marcell Dekker: New York, NY, USA, 2004; pp. 463–483. [Google Scholar]
- Scheffer, J.R.; Xia, W. Asymmetric Induction in Organic Photochemistry via the Solid-State Ionic Chiral Auxiliary Approch. Top. Curr. Chem. 2005, 254, 233–262. [Google Scholar]
- Gamlin, J.N.; Jones, R.; Leibovitch, M.; Patrick, B.; Scheffer, J.R.; Trotter, J. The Ionic Auxiliary Concept in Solid State Organic Photochemistry. Acc. Chem. Res. 1996, 29, 203–209. [Google Scholar] [CrossRef]
- Leibovitch, M.; Olovsson, G.; Scheffer, J.R.; Trotter, J. Absolute configuration correlation studies in solid state organic photochemistry. Pure Appl. Chem. 1997, 69, 815–823. [Google Scholar] [CrossRef]
- Thakur, T.S.; Dubey, R.; Desiraju, G.R. Crystal Structure and Prediction. Annu. Rev. Phys. Chem. 2015, 66, 21–42. [Google Scholar] [CrossRef] [PubMed]
- Tschernich, R.W. Zeolites of the World; Geoscience Press, Inc.: Phoenix, AZ, USA, 1992. [Google Scholar]
- Breck, D.W. Zeolite Molecular Sieves: Structure, Chemistry and Use; Wiley: New York, NY, USA, 1974. [Google Scholar]
- Bekkum, H.V.; Flanigen, E.M.; Jansen, J.C. Introduction to Zeolite Science and Practice; Elsevier: New York, NY, USA, 1991. [Google Scholar]
- Ramamurthy, V.; Weiss, R.G.; Hammond, G.S. A Model for the Influence of Organized Media on Photochemical Reactions. Adv. Photochem. 1993, 18, 67–234. [Google Scholar]
- Weiss, R.G.; Ramamurthy, V.; Hammond, G.S. Photochemistry ib Organized and Confining Media: A Model. Acc. Chem. Res. 1993, 26, 530–536. [Google Scholar] [CrossRef]
- Treacy, M.M.J.; Newsam, J.M. Two new three-dimensional twelve-ring zeolite framework of which zeolite beta is a disordered intergrowth. Nature 1988, 332, 249–251. [Google Scholar] [CrossRef]
- Newsam, J.M.; Treacy, M.M.J.; Koetsier, W.T.; De Gruyter, C.B. Structural characterisation of zeolite beta. Proc. R. Soc. Lond. A 1988, 420, 375–405. [Google Scholar] [CrossRef]
- Davis, M.E.; Lobo, R.F. Zeolite and molecular sieve synthesis. Chem. Mater. 1992, 4, 756–768. [Google Scholar] [CrossRef]
- Brand, S.K.; Schmidt, J.E.; Deem, M.W.; Daeyaert, F.; Ma, Y.; Terasaki, O.; Orazov, M.; Davis, M.E. Enantiomerically enriched, polycrystalline molecular sieves. Proc. Natl. Acad. Sci. USA 2017, 114, 5101–5106. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.E. A thirty year journey to the creation of the first enantiomerically enriched molecular sieve. ACS Catal. 2018, 8, 10082–10088. [Google Scholar] [CrossRef]
- Sundarababu, G.; Leibovitch, M.; Corbin, D.R.; Scheffer, J.R.; Ramamurthy, V. Zeolite as a host for chiral induction. Chem. Commun. 1996, 18, 2159–2160. [Google Scholar] [CrossRef]
- Leibovitch, M.; Olovsson, G.; Sundarababu, G.; Ramamurthy, V.; Scheffer, J.R.; Trotter, J. Asymmetric Induction in Photochemical Reactions Conducted in Zeolites and in the Crystalline State. J. Am. Chem. Soc. 1996, 118, 1219–1220. [Google Scholar] [CrossRef]
- Sivaguru, J.; Natarajan, A.; Kaanumalle, L.S.; Shailaja, J.; Uppili, S.; Joy, A.; Ramamurthy, V. Asymmetric Photoreactions Within Zeolites: Role of Confinement and Alkali Metal Ions. Acc. Chem. Res. 2003, 36, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Shailaja, J.; Ponchot, K.J.; Ramamurthy, V. Enantioselective Photoreduction of Arylalkyl Ketones via Restricting the Reaction to Chirally Modified Zeolite Cages. Org. Lett. 2000, 2, 937–940. [Google Scholar] [CrossRef] [PubMed]
- Shailaja, J.; Kaanumalle, L.S.; Sivasubramanian, K.; Natarajan, A.; Ponchot, K.; Pradhan, A.R.; Ramamurthy, V. Asymmetric induction during electron transfer mediated photoreduction of carbonyl compounds: Role of zeolites. Org. Biomol. Chem. 2006, 4, 1561–1571. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, A.R.; Uppili, S.; Shailaja, J.; Sivaguru, J.; Ramamurthy, V. Zeolite-coated quartz fibers as media for photochemical and photophysical studies. Chem. Commun. 2002, 6, 596–597. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.G.; Parola, A.; Parsons, G.H. Photoreduction by amines. Chem. Rev. 1973, 73, 141–161. [Google Scholar] [CrossRef]
- Kaanumalle, L.S.; Sivaguru, J.; Arunkumar, N.; Karthikeyan, S.; Ramamurthy, V. Cation-p interactions as a tool to enhance the power of a chiral auxiliary during asymmetric photoreactions within zeolites. Chem. Commun. 2003, 7, 116–117. [Google Scholar] [CrossRef] [PubMed]
- Lakshminarasimhan, P.; Sunoj, R.B.; Chandrasekhar, J.; Ramamurthy, V. Cation-π Interaction Controlled Selective Geometric Photoisomerization of Diphenylcyclopropane. J. Am. Chem. Soc. 2000, 122, 4815–4816. [Google Scholar] [CrossRef]
- Joy, A.; Scheffer, J.R.; Corbin, D.R.; Ramamurthy, V. Enantioselective photoelectrocyclization within zeolites: Tropolone methyl ether in chirally modified NaY. Chem. Commun. 1998, 13, 1379–1380. [Google Scholar] [CrossRef]
- Joy, A.; Ramamurthy, V. Chiral photochemistry within zeolites. Chem. Eur. J. 2000, 6, 1287–1293. [Google Scholar] [CrossRef]
- Shailaja, J.; Sivaguru, J.; Uppili, S.; Joy, A.; Ramamurthy, V. Use of confined space (zeolite) in enantio and diasteroselective photoreactions. Microporous Mesoporous Mater. 2001, 48, 319–328. [Google Scholar] [CrossRef]
- Joy, A.; Kaanumalle, L.S.; Ramamurthy, V. Role of cations and confinement in asymmetric photochemistry: Enantio- and diastereo-selective photocyclization of tropolone derivatives within zeolites. Org. Biomol. Chem. 2005, 3, 3045–3053. [Google Scholar] [CrossRef] [PubMed]
- Bach, T.; Bergmann, H.; Harms, K. Enantioselective Photochemical Reactions of 2-Pyridones in Solution. Org. Lett. 2001, 3, 601–603. [Google Scholar] [CrossRef] [PubMed]
- Sivasubramanian, K.; Kaanumalle, L.S.; Uppili, S.; Ramamurthy, V. Value of zeolites in asymmetric induction during photocyclization of pyridones, cyclohexadienones and naphthalenones. Org. Biomol. Chem. 2007, 5, 1569–1576. [Google Scholar] [CrossRef] [PubMed]
- Sivaguru, J.; Wada, T.; Origane, Y.; Inoue, Y.; Ramamurthy, V. Mechanism of photoisomerization of optically pure trans-2,3-dipehnylcyclopropane-1-carboxylic acid derivatives. Photochem. Photobiol. Sci. 2005, 4, 119–127. [Google Scholar] [CrossRef]
- Sivaguru, J.; Jockusch, S.; Turro, N.J.; Ramamurthy, V. Photoisomerization of 2,3-diphenylcyclopropane-1-carboxylic acid derivatives. Photochem. Photobiol. Sci. 2003, 2, 1101–1106. [Google Scholar] [CrossRef] [PubMed]
- Kaanumalle, L.S.; Sivaguru, J.; Sunoj, R.B.; Lakshminarasimhan, P.H.; Chandrasekhar, J.; Ramamurthy, V. Light-Induced Geometric Isomerization of 1,2-Diphenylcyclopropanes Included within Y Zeolites: Role of Cation-Guest Binding. J. Org. Chem. 2002, 67, 8711–8720. [Google Scholar] [CrossRef]
- Chong, K.C.W.; Sivaguru, J.; Shichi, T.; Yoshimi, Y.; Ramamurthy, V.; Scheffer, J.R. Use of Chirally Modified Zeolites and Crystals in Photochemical Asymmetric Synthesis. J. Am. Chem. Soc. 2002, 124, 2858–2859. [Google Scholar] [CrossRef]
- Jayaraman, S.; Uppili, S.; Natarajan, A.; Joy, A.; Chong, K.C.W.; Netherton, M.R.; Zenova, A.; Scheffer, J.R.; Ramamurthy, V. The influence of chiral auxiliaries is enhanced within zeolites. Tetrahedron Lett. 2000, 41, 8231–8235. [Google Scholar] [CrossRef]
- Uppili, S.; Ramamurthy, V. Enhanced Enantio- and Diastreoselctivities via Confinement: Photorearrangement of 2,4-cyclohexadienones included in Zeolites. Org. Lett. 2002, 4, 87–90. [Google Scholar] [CrossRef]
- Natarajan, A.; Ramamurthy, V. Asymmetric induction during photocyclization of chiral and achiral α-oxoamides within achiral zeolites. Org. Biomol. Chem. 2006, 4, 4533–4542. [Google Scholar] [CrossRef]
- Natarajan, A.; Joy, A.; Kaanumalle, L.S.; Scheffer, J.R.; Ramamurthy, V. Enhanced Enantio- and Diastereoselectivity via Confinement and Cation Binding: Yang Photocyclization of 2-Benzoyladamantane Derivatives within Zeolites. J. Org. Chem. 2002, 67, 8339–8350. [Google Scholar] [CrossRef] [PubMed]
- Sivaguru, J.; Sunoj, R.B.; Wada, T.; Origane, Y.; Inoue, Y.; Ramamurthy, V. Enhanced Diastereoselectivity via Confinement: Diastereoselective Photoisomerization of 2,3-Diphenyl-1-benzoylcyclopropane Derivatives within Zeolites. J. Org. Chem. 2004, 69, 5528–5536. [Google Scholar] [CrossRef] [PubMed]
- Sivaguru, J.; Sunoj, R.B.; Wada, T.; Origane, Y.; Inoue, Y.; Ramamurthy, V. Enhanced Diastereoselectivity via Confinement: Diastereoselective Photoisomerization of 2,3-Diphenylcyclopropane-1-carboxylic Acid Derivatives within Zeolites. J. Org. Chem. 2004, 69, 6533–6547. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, A.; Wang, K.; Ramamurthy, V.; Scheffer, J.R.; Patrick, B. Control of Enantioselectivity in the Photochemical Conversion of a-Oxoamides into b-Lactam Derivatives. Org. Lett. 2002, 4, 1443–1446. [Google Scholar] [CrossRef] [PubMed]
- Sivaguru, J.; Scheffer, J.R.; Chandrasekhar, J.; Ramamurthy, V. Confined Space and Cations enhance the power of a chiral auxiliary: Photochemistry of 1,2-diphenylcyclopropane derivatives. Chem. Commun. 2002, 8, 830–831. [Google Scholar] [CrossRef]
- Sivaguru, J.; Shichi, T.; Ramamurthy, V. Reactive-State Spin-Dependent Diastereoselective Photoisomerization of trans,trans-2,3-Diphenylcyclopropane-1- carboxylic Acid Derivatives Included in Zeolites. Org. Lett. 2002, 4, 4221–4224. [Google Scholar] [CrossRef]
- Cheung, E.; Chong, K.C.W.; Jayaraman, S.; Ramamurthy, V.; Scheffer, J.R.; Trotter, J. Enantio- and Diastereo-differentiating cis, trans-Photoisomerization of 2b, 3b-Diphenylcyclopropane-1a-carboxylic Acid Derivatives in Organized Media. Org. Lett. 2000, 2, 2801–2804. [Google Scholar] [CrossRef]
- Joy, A.; Uppili, S.; Netherton, M.R.; Scheffer, J.R.; Ramamurthy, V. Photochemistry of a Tropolone ether and 2,2-dimethyl-1-(2H)-naphthalenones within a Zeolite: Enhanced diastereoselectivity via confinement. J. Am. Chem. Soc. 2000, 122, 728–729. [Google Scholar] [CrossRef]















© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramamurthy, V. Achiral Zeolites as Reaction Media for Chiral Photochemistry. Molecules 2019, 24, 3570. https://doi.org/10.3390/molecules24193570
Ramamurthy V. Achiral Zeolites as Reaction Media for Chiral Photochemistry. Molecules. 2019; 24(19):3570. https://doi.org/10.3390/molecules24193570
Chicago/Turabian StyleRamamurthy, Vaidhyanathan. 2019. "Achiral Zeolites as Reaction Media for Chiral Photochemistry" Molecules 24, no. 19: 3570. https://doi.org/10.3390/molecules24193570
APA StyleRamamurthy, V. (2019). Achiral Zeolites as Reaction Media for Chiral Photochemistry. Molecules, 24(19), 3570. https://doi.org/10.3390/molecules24193570