Schizandrin Protects against OGD/R-Induced Neuronal Injury by Suppressing Autophagy: Involvement of the AMPK/mTOR Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. SA Protects Cells against OGD/R Injury in PC12 Cells
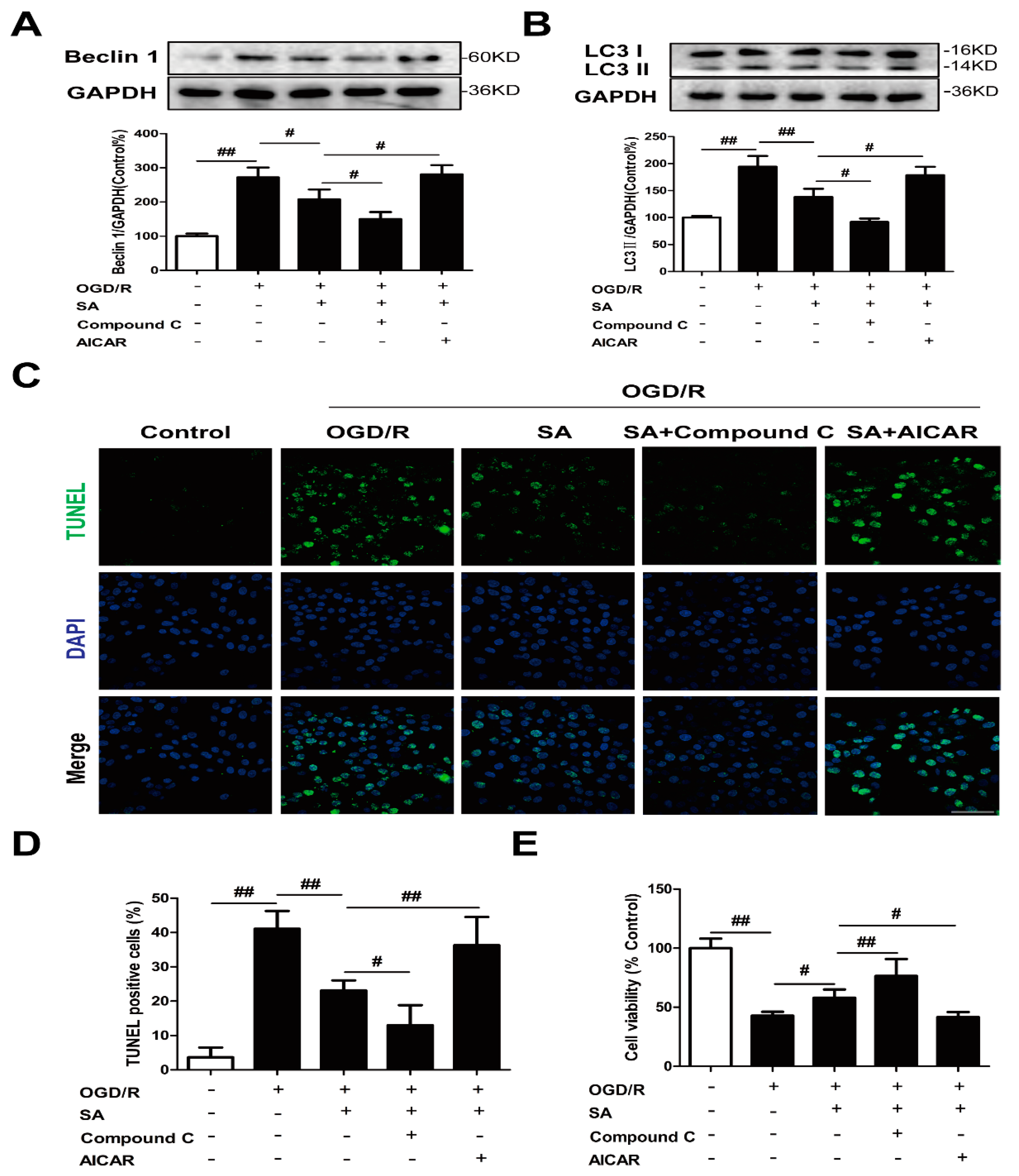
2.2. SA Inhibits Autophagy Following OGD/R in PC12 Cells
2.3. Autophagy Contributes to the Neuroprotective Effects of SA in PC12 Cells
2.4. Involvement of the AMPK-mTOR/Autophagy Pathway in SA-Induced Protection in PC12 Cells
2.5. SA Inhibits AMPK/mTOR Pathway and Autophagy Following Cerebral Ischemia/Reperfusion Injury in Mice
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Animals
4.3. Cell Culture
4.4. OGD/R and Drug Treatments
4.5. TUNEL Staining
4.6. Quantification of GFP-LC3 Puncta
4.7. Cell Viability
4.8. In Vivo Cerebral Ischemia Model
4.9. In Vivo Immunofluorescence
4.10. Western Blot Analysis
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sun, B.; Ou, H.; Ren, F.; Huan, Y.; Zhong, T.; Gao, M.; Cai, H. Propofol inhibited autophagy through Ca2+/CaMKKβ/AMPK/mTOR pathway in OGD/R-induced neuron injury. Mol. Med. 2018, 24, 58. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, S.F.; Sureda, A.; Sanches-Silva, A.; Pandima Devi, K.; Ahmed, T.; Shahid, M.; Sobarzo-Sánchez, E.; Dacrema, M.; Daglia, M.; Braidy, N.; et al. Novel therapeutic strategies for stroke: The role of autophagy. Crit. Rev. Clin. Lab. Sci. 2019, 56, 182–199. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Yang, L.; Wang, S.; Cai, M.; Sun, S.; Dong, H.; Xiong, L. TREK-2 mediates the neuroprotective effect of isoflurane preconditioning against acute cerebral ischemia in the rat. Rejuvenation Res. 2019, 22, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Pandian, J.D.; Gall, S.L.; Kate, M.P.; Silva, G.S.; Akinyemi, R.O.; Ovbiagele, B.I.; Lavados, P.M.; Gandhi, D.B.C.; Thrift, A.G. Prevention of stroke: A global perspective. Lancet 2018, 392, 1269–1278. [Google Scholar] [CrossRef]
- Dai, S.H.; Chen, T.; Li, X.; Yue, K.Y.; Luo, P.; Yang, L.K.; Zhu, J.; Wang, Y.H.; Fei, Z.; Jiang, X.F. Sirt3 confers protection against neuronal ischemia by inducing autophagy: Involvement of the AMPK-mTOR pathway. Free Radic. Biol. Med. 2017, 108, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.M.; Zhang, T.; Wang, M.M.; Wang, X.X.; Qin, Y.Y.; Wu, J.; Han, R.; Sheng, R.; Wang, Y.; Chen, Z.; et al. TIGAR alleviates ischemia/reperfusion-induced autophagy and ischemic brain injury. Free Radic. Biol. Med. 2019, 137, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Yu, M.; He, X.; Wen, L.; Bu, Z.; Feng, J. KCNQ1OT1 promotes autophagy by regulating miR-200a/FOXO3/ATG7 pathway in cerebral ischemic stroke. Aging Cell 2019, 18, e12940. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.D.; Sheng, R.; Zhang, L.S.; Han, R.; Zhang, X.; Zhang, X.D.; Han, F.; Fukunaga, K.; Qin, Z.H. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy 2008, 4, 762–769. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell. Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.Y.; Gertner, M.; Pontarelli, F.; Court-Vazquez, B.; Bennett, M.V.; Ofengeim, D.; Zukin, R.S. Global ischemia induces lysosomal-mediated degradation of mTOR and activation of autophagy in hippocampal neurons destined to die. Cell Death Differ. 2017, 24, 317–329. [Google Scholar] [CrossRef]
- Sowndhararajan, K.; Deepa, P.; Kim, M.; Park, S.J.; Kim, S. An overview of neuroprotective and cognitive enhancement properties of lignans from Schisandra chinensis. Biomed. Pharmacother. 2018, 97, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Li, X.; Sun, Y.; Zhang, X. Schizandrin A inhibits proliferation, migration and invasion of thyroid cancer cell line TPC-1 by down regulation of microRNA-429. Cancer Biomark. 2019, 24, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xie, R.; Jiang, C.; Liu, M. Schizandrin a alleviates LPS-induced injury in human keratinocyte cell HaCaT through a microRNA-127-dependent regulation. Cell Physiol. Biochem. 2018, 49, 2229–2239. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.J.; Kim, S.R.; Jung, U.J. Schizandrin A supplementation improves nonalcoholic fatty liver disease in mice fed a high-fat and high-cholesterol diet. Nutr. Res. 2019, 64, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.P.; Li, G.C.; Shi, Y.W.; Zhang, X.C.; Li, J.L.; Wang, Z.W.; Ding, F.; Liang, X.M. Neuroprotective effect of schizandrin A on oxygen and glucose deprivation/reperfusion- induced cell injury in primary culture of rat cortical neurons. J. Physiol. Biochem. 2014, 70, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Wang, M.; Ju, J.; Wang, Y.; Liu, Z.; Zhao, X.; Yan, Y.; Yan, S.; Luo, X.; Fang, Y. Schizandrin A protects against cerebral ischemia-reperfusion injury by suppressing inflammation and oxidative stress and regulating the AMPK/Nrf2 pathway regulation. Am. J. Transl. Res. 2019, 11, 199–209. [Google Scholar]
- Guo, Z.; Cao, G.; Yang, H.; Zhou, H.; Li, L.; Cao, Z.; Yu, B.; Kou, J. A combination of four active compounds alleviates cerebral ischemia–reperfusion injury in correlation with inhibition of autophagy and modulation of AMPK/mTOR and JNK pathways. J. Neurosci. Res. 2014, 92, 1295–1306. [Google Scholar] [CrossRef]
- Spicer, Z.; Millhorn, D.E. Oxygen sensing in neuroendocrine cells and other cell types: Pheochromocytoma (PC12) cells as an experimental model. Endocr. Pathol. 2003, 14, 277–291. [Google Scholar] [CrossRef]
- Yang, Y.; Xu, K.; Koike, T.; Zheng, X. Transport of autophagosomes in neurites of PC12 cells during serum deprivation. Autophagy 2008, 4, 243–245. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Wang, J.; Feng, J. The neuroprotective effects of curcumin are associated with the regulation of the reciprocal function between autophagy and HIF-1α in cerebral ischemia-reperfusion injury. Drug Des. Devel. Ther. 2019, 13, 1135–1144. [Google Scholar] [CrossRef]
- Ji, H.; Xu, L.; Wang, Z.; Fan, X.; Wu, L. Effects of thymosin β4 on oxygen-glucose deprivation and reoxygenation-induced injury. Int. J. Mol. Med. 2018, 41, 1749–1755. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Zeng, K.; Liao, L.; Yu, Q.; Tu, P.; Wang, X. Schizandrin A inhibits microglia-mediated neuron inflammation through inhibiting TRAF6-NF-κB and Jak2-Stat3 signaling pathways. PLoS ONE 2016, 11, e0149991. [Google Scholar]
- E, Q.; Tang, M.; Zhang, X.; Shi, Y.; Wang, D.; Gu, Y.; Li, S.; Liang, X.; Wang, Z.; Wang, C. Protection of seven dibenzocyclooctadiene lignans from Schisandra chinensis against serum and glucose deprivation injury in SH-SY5Y cells. Cell Biol. Int. 2015, 39, 1418–1424. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Lee, M.K.; Koo, K.A.; Kim, S.H.; Sung, S.H.; Lee, N.G.; Markelonis, G.J.; Oh, T.H.; Yang, J.H.; Kim, Y.C. Dibenzocyclooctadiene lignans from Schisandra chinensis protect primary cultures of rat cortical cells from glutamate-induced toxicity. J. Neurosci. Res. 2004, 76, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Vindis, C. Autophagy: An emerging therapeutic target in vascular diseases. Br. J. Pharmacol. 2015, 172, 2167–2178. [Google Scholar] [CrossRef] [PubMed]
- Leidal, A.M.; Levine, B.; Debnath, J. Autophagy and the cell biology of age-related disease. Nat. Cell Biol. 2018, 20, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; DiFiglia, M.; Heintz, N.; Nixon, R.A.; Qin, Z.H.; Ravikumar, B.; Stefanis, L.; Tolkovsky, A. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy 2005, 1, 11–22. [Google Scholar] [CrossRef]
- Wang, P.; Shao, B.Z.; Deng, Z.; Chen, S.; Yue, Z.; Miao, C.Y. Autophagy in ischemic stroke. Prog. Neurobiol. 2018, 163, 98–117. [Google Scholar] [CrossRef]
- McCrary, M.R.; Jiang, M.Q.; Giddens, M.M.; Zhang, J.Y.; Owino, S.; Wei, Z.Z.; Zhong, W.; Gu, X.; Xin, H.; Hall, R.A.; et al. Protective effects of GPR37 via regulation of inflammation and multiple cell death pathways after ischemic stroke in mice. FASEB J. 2019, 3, fj201900070R. [Google Scholar] [CrossRef]
- Wang, J.F.; Mei, Z.G.; Fu, Y.; Yang, S.B.; Zhang, S.Z.; Huang, W.F.; Xiong, L.; Zhou, H.J.; Tao, W.; Feng, Z.T. Puerarin protects rat brain against ischemia/reperfusion injury by suppressing autophagy via the AMPK-mTOR-ULK1 signaling pathway. Neural. Regen. Res. 2018, 13, 989–998. [Google Scholar]
- Jiang, J.; Dai, J.; Cui, H. Vitexin reverses the autophagy dysfunction to attenuate MCAO-induced cerebral ischemic stroke via mTOR/Ulk1 pathway. Biomed. Pharmacother. 2018, 99, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.H.; Noh, A.R.; Kim, K.A.; Akram, M.; Shin, Y.J.; Kim, E.S.; Yu, S.W.; Majid, A.; Bae, O.N. Modulation of mitochondrial function and autophagy mediates carnosine neuroprotection against ischemic brain damage. Stroke 2014, 45, 2438–2443. [Google Scholar] [CrossRef] [PubMed]
- Pengyue, Z.; Tao, G.; Hongyun, H.; Liqiang, Y.; Yihao, D. Breviscapine confers a neuroprotective efficacy against transient focal cerebral ischemia by attenuating neuronal and astrocytic autophagy in the penumbra. Biomed. Pharmacother. 2017, 90, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Li, L.; Zhou, K.; Wang, Y.; Guan, T.; Chai, C.; Kou, J.; Yu, B.; Yan, Y. Shengmai injection attenuates the cerebral ischemia/reperfusion induced autophagy via modulation of the AMPK, mTOR and JNK pathways. Pharm. Biol. 2016, 54, 2288–2297. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Wang, W.J.; Song, Y.Z.; Liang, Z.Q. The protective mechanism of schisandrin A in d-galactosamine-induced acute liver injury through activation of autophagy. Pharm. Biol. 2014, 52, 1302–1307. [Google Scholar] [CrossRef] [PubMed]
- Lai, Q.; Luo, Z.; Wu, C.; Lai, S.; Wei, H.; Li, T.; Wang, Q.; Yu, Y. Attenuation of cyclosporine A induced nephrotoxicity by schisandrin B through suppression of oxidative stress, apoptosis and autophagy. Int. Immunopharmacol. 2017, 52, 15–23. [Google Scholar] [CrossRef]
- Giridharan, V.V.; Thandavarayan, R.A.; Arumugam, S.; Mizuno, M.; Nawa, H.; Suzuki, K.; Ko, K.M.; Krishnamurthy, P.; Watanabe, K.; Konishi, T. Schisandrin B Ameliorates ICV-Infused Amyloid β Induced Oxidative Stress and Neuronal Dysfunction through Inhibiting RAGE/NF-κB/MAPK and Up-Regulating HSP/Beclin Expression. PLoS ONE 2015, 10, e0142483. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, Z.W.; Jin, H.; Hu, C.; He, Z.X.; Yu, Z.L.; Ko, K.M.; Yang, T.; Zhang, X.; Pan, S.Y.; et al. Schisandrin B inhibits cell growth and induces cellular apoptosis and autophagy in mouse hepatocytes and macrophages: Implications for its hepatotoxicity. Drug Des. Devel. Ther. 2015, 9, 2001–2027. [Google Scholar]
- Kim, J.S.; Yi, H.K. Schisandrin C enhances mitochondrial biogenesis and autophagy in C2C12 skeletal muscle cells: Potential involvement of anti-oxidative mechanisms. Naunyn. Schmiedebergs Arch Pharmacol. 2018, 391, 197–206. [Google Scholar] [CrossRef]
- Takanche, J.S.; Kim, J.S.; Kim, J.E.; Han, S.H.; Yi, H.K. Schisandrin C enhances odontoblastic differentiation through autophagy and mitochondrial biogenesis in human dental pulp cells. Arch. Oral. Biol. 2018, 88, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Huang, L.; Cao, C.; Yin, Q.; Liu, J. Inhibition of AMP-activated protein kinase alleviates focal cerebral ischemia injury in mice: Interference with mTOR and autophagy. Brain Res. 2016, 1650, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Xue, T.F.; Ding, X.; Ji, J.; Yan, H.; Huang, J.Y.; Guo, X.D.; Yang, J.; Sun, X.L. PD149163 induces hypothermia to protect against brain injury in acute cerebral ischemic rats. J. Pharmacol. Sci. 2017, 135, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hou, J.; Zhao, H.; Liu, J. Synergistic Use of Geniposide and Ginsenoside Rg1 Balance Microglial TNF-α and TGF-β1 following Oxygen-Glucose Deprivation In Vitro: A Genome-Wide Survey. Evid. Based Complement Alternat. Med. 2015, 2015, 756346. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, G.; Wang, T.; Zhang, Y.; Li, F.; Yu, B.; Kou, J. Schizandrin Protects against OGD/R-Induced Neuronal Injury by Suppressing Autophagy: Involvement of the AMPK/mTOR Pathway. Molecules 2019, 24, 3624. https://doi.org/10.3390/molecules24193624
Wang G, Wang T, Zhang Y, Li F, Yu B, Kou J. Schizandrin Protects against OGD/R-Induced Neuronal Injury by Suppressing Autophagy: Involvement of the AMPK/mTOR Pathway. Molecules. 2019; 24(19):3624. https://doi.org/10.3390/molecules24193624
Chicago/Turabian StyleWang, Guangyun, Tiezheng Wang, Yuanyuan Zhang, Fang Li, Boyang Yu, and Junping Kou. 2019. "Schizandrin Protects against OGD/R-Induced Neuronal Injury by Suppressing Autophagy: Involvement of the AMPK/mTOR Pathway" Molecules 24, no. 19: 3624. https://doi.org/10.3390/molecules24193624