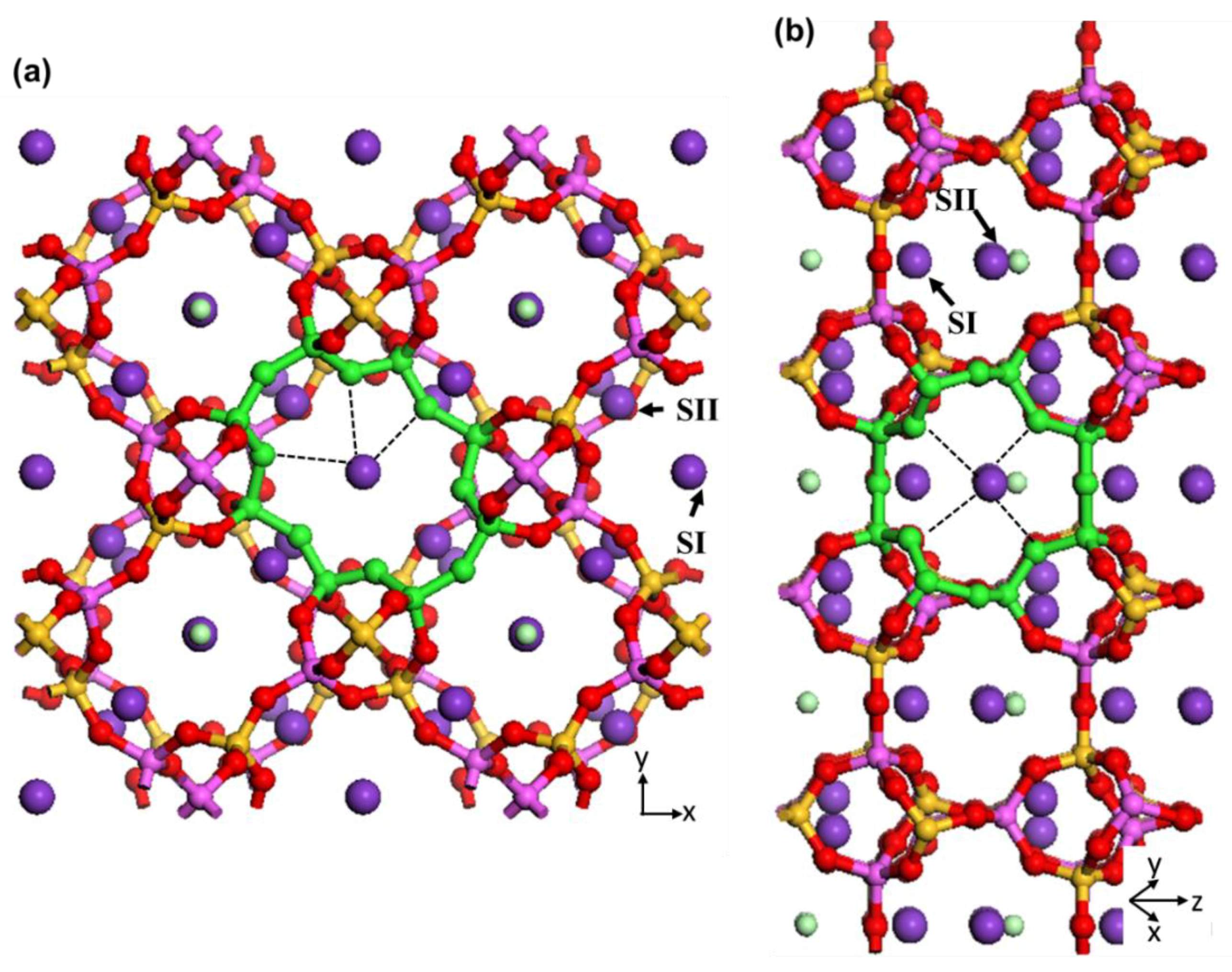
The intersection of these two channels creates cages that surround the potassium and chloride extra-framework ions and water molecules. Two different T sites for Si and Al framework atoms (T1 and T2), and five for the O atoms constructing the framework, are identified by X-ray diffraction [
2]. Two different sites are identified for the potassium extra-framework cations (SI and SII) [
2]. SI is located in the middle of the eight-membered rings along the [001] direction (K1) and SII is located in the middle of the other eight-membered rings along the [110] direction.
Figure 7 illustrates the position of these sites in the zeolite N unit cell. The extra-framework K1 has interaction with three framework oxygens (noted by black dashed lines in
Figure 7a), one chloride and two oxygens of water molecules. The extra-framework K2 has interaction with four framework oxygens (noted by black dashed lines in
Figure 7b), one chloride and two oxygens of water molecules. These loose van der Waals interactions can easily breakdown during exchange processes resulting in potassium cations leaving their structural sites for locations of more favourable energy for zeolite N.
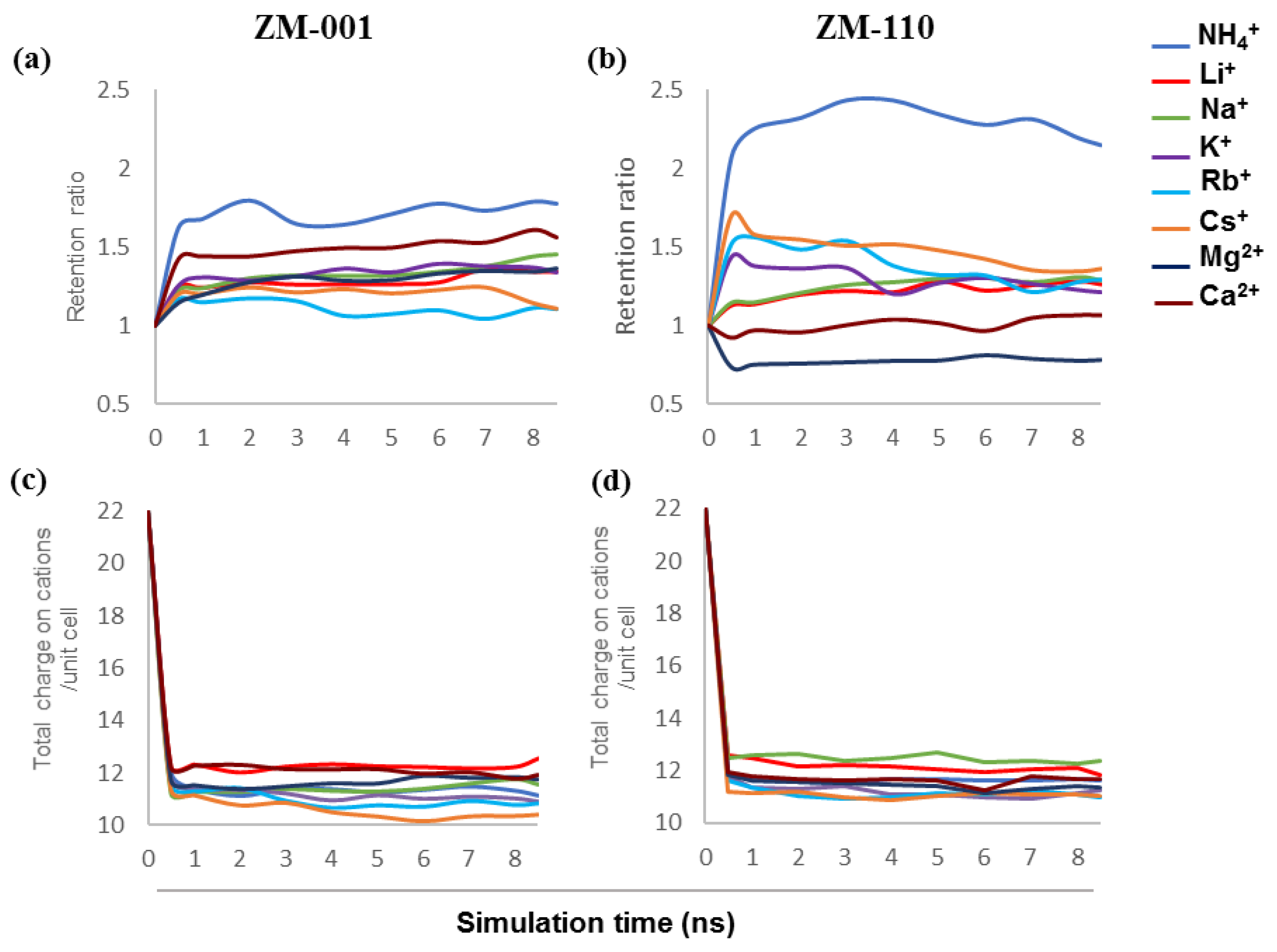
As mentioned in the Introduction, experimental investigations present valuable data on the ion-exchange capability and comportment of zeolite N. However, the exchange mechanism of cations within the zeolite N structure is unclear. In this study, we investigated the ion exchange characteristics of mono- and divalent cations in the zeolite N structure by molecular dynamics calculations. Here, we discuss the retention, structural arrangement and mobility of monovalent NH4+, Li+, Na+, K+, Rb+ and Cs+ as well as divalent Mg2+ and Ca2+ cations inside zeolite N membranes. This allows exploration of the exchange and diffusion mechanism of cations inside channels along the [001] and [110] directions of zeolite N.
3.1. Ion Retention
The chemical formula of the zeolite N unit cell indicates 12 exchangeable cations to compensate for the negatively charged −10e of the aluminosilicate framework and two Cl anions [
2]. The results show that the total charge compensation on cations per unit cell in most exchange systems is below 12, except for Li
+ in ZM-001 and Na
+ and Li
+ in ZM-110. The potassium-rich ZM-001 and ZM-110 membranes without guest cations, release 8% and 11%, respectively, of their K to solution and thus, contain less than 12 cations per unit cell. Over the simulation time, each membrane loses up to one K
+ per unit cell. This calculated outcome is consistent with experimental observations that show a neutral water solution will record a pH ~ 9 (or greater) with addition of zeolite N at room temperature. Measurements of this solution with zeolite N also show the presence of K
+ ions.
Our previous simulation outcomes [
10] show similar conditions regarding the total number of cations retained in the membrane. These results suggest that the zeolite N structure prefers to hold no more K or other cations than an approximately equilibrium value. The one exception to this preference is for the small cation, Li
+, for which zeolite N is able to adsorb more ions than the charge compensating capacity.
Experimental studies provide valuable data on the tendency of zeolite N to uptake ammonium ions from aqueous solutions [
5,
6,
9]. Moreover, Zwingmann et al. [
7] showed that ammonium exchanged zeolite N is an ideal slow release fertiliser for sandy soils due to the high retention capacity of zeolite N for NH
4+ ions. Our computational results from previous [
10] and this study for NH
4+ retention are consistent with these experimental results. This study shows that NH
4+ has the highest retention between all exchanging systems in zeolite N membranes along both crystallographic directions. The NH
4+ retention in ZM-110 is higher than ZM-001 (
Figure S1a,b). However, the total number of cations that remain in the structure are similar for both membranes. The small sized channel openings in ZM-110 does not allow NH
4+ ions to leave the membrane and thus, ensures capture in 3D cages formed at the intersection of the (001) and (110) planes. Release of K
+ ions from the membrane provides additional space for retained NH
4+ ions. Our calculations suggest that only NH
4+ ions close to the membrane surfaces can leave the ZM-110 structure. In contrast, NH
4+ ions can more readily leave the ZM-001 membrane due to the larger size of the channel openings.
Li
+ and Na
+ are the next monovalent ions that show high and similar retention behaviour in membranes. The K
+/Li
+ and K
+/Na
+ systems in this study hold more cations compared to other systems due to the small size of Li
+ and Na
+ ions. In the ZM-001 membrane Li
+ and Na
+ show similar retention rates (
Figure S1a,b). The retention of Na
+ in the ZM-001 membrane is the same as simulations shown previously under similar conditions [
10]. However, the K
+/Li
+ system retains more K
+ in the membrane compared to the K
+/Na
+ system during the simulation. In the ZM-110 membrane, Na
+ retention is slightly higher than Li
+. However, both K
+/Li
+ and K
+/Na
+ simulations show that a similar amount of K
+ is retained in the ZM-110 membrane.
The K
+/Rb
+ and K
+/Cs
+ systems show the lowest retention and total number of cations per unit cell among monovalent cations in both membranes due to the large size of these ions. The retention of Rb
+ and Cs
+ is higher in ZM-110 than ZM-001 (
Figure S1a,b). Similar to the case for NH
4+, the large size of these ions does not allow passage through the small opening of the ZM-110 membrane channels to the solute. The retention behaviour of Rb
+ and Cs
+ compared to other monovalent cations in this study for ZM-001 is similar to our previous outcomes [
10]. However, in this study, we found lower retention ratios over time for Rb
+ and Cs
+.
Experimental data show that the potassic form of zeolite N can take up to three times more Ca
2+ than Mg
2+ from a mixed solution of NH
4+, Mg
2+ and Ca
2+ (with low ammonium concentration, 30 mg/L) and this uptake of divalent ions is 10× lower than the NH
4+ uptake [
9]. These data indicate a high preference of zeolite N for monovalent cations over divalent ions. Computational results from this study for retention of Mg
2+ and Ca
2+ ions in zeolite N are in good agreement with experimental outcomes [
9]. For example, the number of guest cations for the K
+/Mg
2+ and K
+/Ca
2+ systems show the lowest value for total retained cations in the membranes (
Figure S1a,b). Accordingly, in these systems the number of K
+ ions retained in the membrane are higher than Mg
2+ or Ca
2+.
Based on the experimentally determined chemical formula for zeolite N, each unit cell contains two Cl
− anions [
2]. However, our simulations show that both membranes release up to one Cl
− per unit cell into solution during the exchange process (
Figure S1e,f). A larger cation size results in greater reduction in the number of chlorides inside each membrane. Consistent with this, systems with a lower amount of cations per unit cell require a lower (or equivalent) amount of anions. These results are in also agreement with experimental data [
9].
Experimental and computational studies show that hydrated zeolite N contains 8 water molecules per cage [
2,
10]. The results from this study reveal that zeolite N membranes along different directions show different hydration behaviour during the exchange process. For example, the number of water molecules per cage changes during the ion-exchange process depending on the guest ions. A ZM-001 membrane releases zeolitic water into solution at early stages of the exchange process in all K
+/M
n+ systems and subsequently continuously adsorbs water molecules over time. This feature is noteworthy for the K
+/Cs
+ system of the ZM-001 membrane. Concentration profiles confirm that this increase is due to adsorbed water molecules in the opening of the pores to the membrane. However, the total number of water molecules inside the membrane cages is constant (
Figure S9). All exchanging systems in ZM-110 adsorb more than 8 water molecules per cage during the simulation except for the K
+/Rb
+ and K
+/Cs
+ systems. Zeolite N channels are not large enough to accommodate K
+, Rb
+ and Cs
+ cations together with water molecules. As with ZM-001, water molecules concentrate at the pore openings of the zeolite N membrane.
3.2. Ion Localization
Ion concentration profiles, electron density field maps and RDF results provide different perspectives on the localization of guest and host ions within zeolite N membranes.
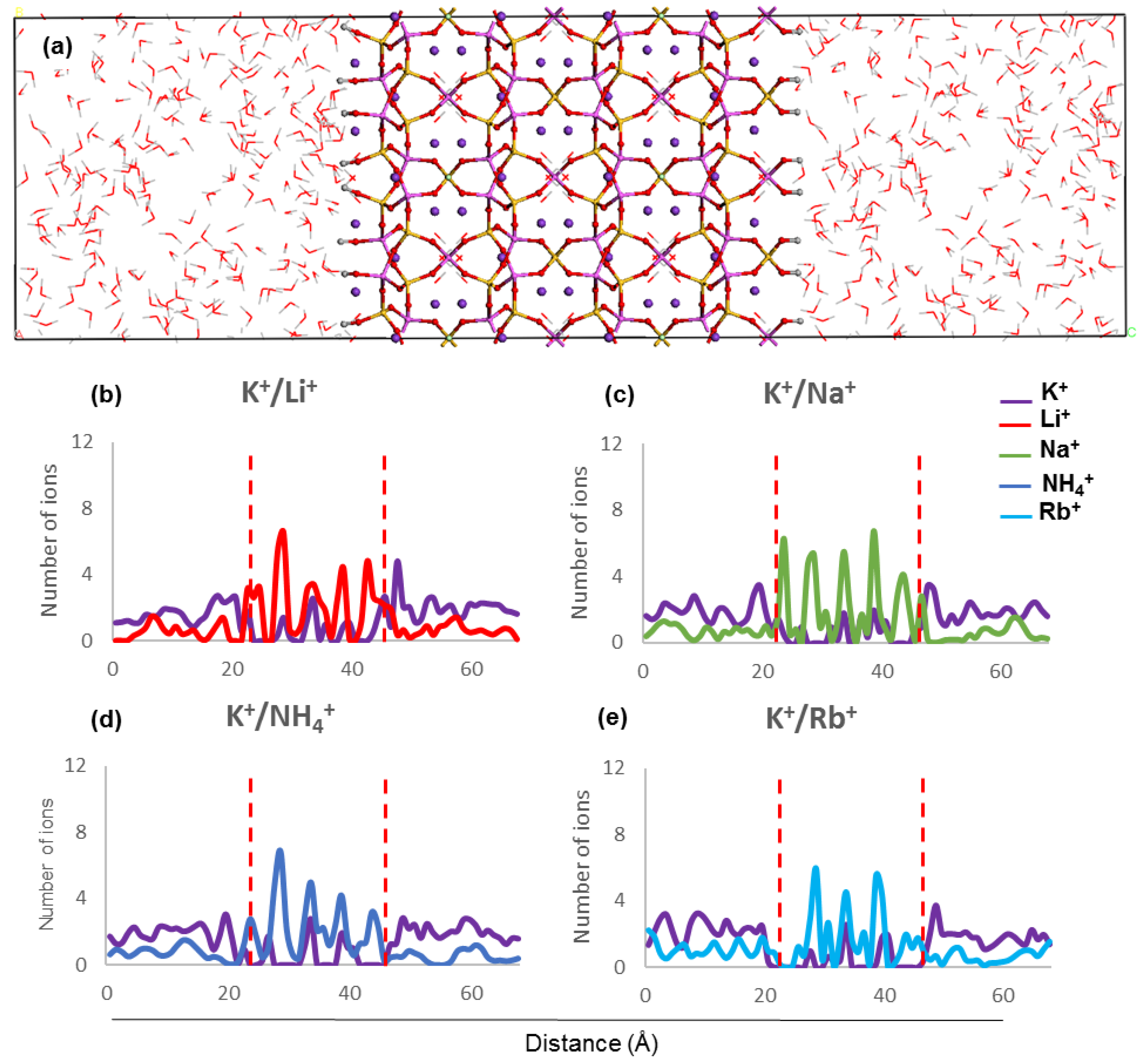
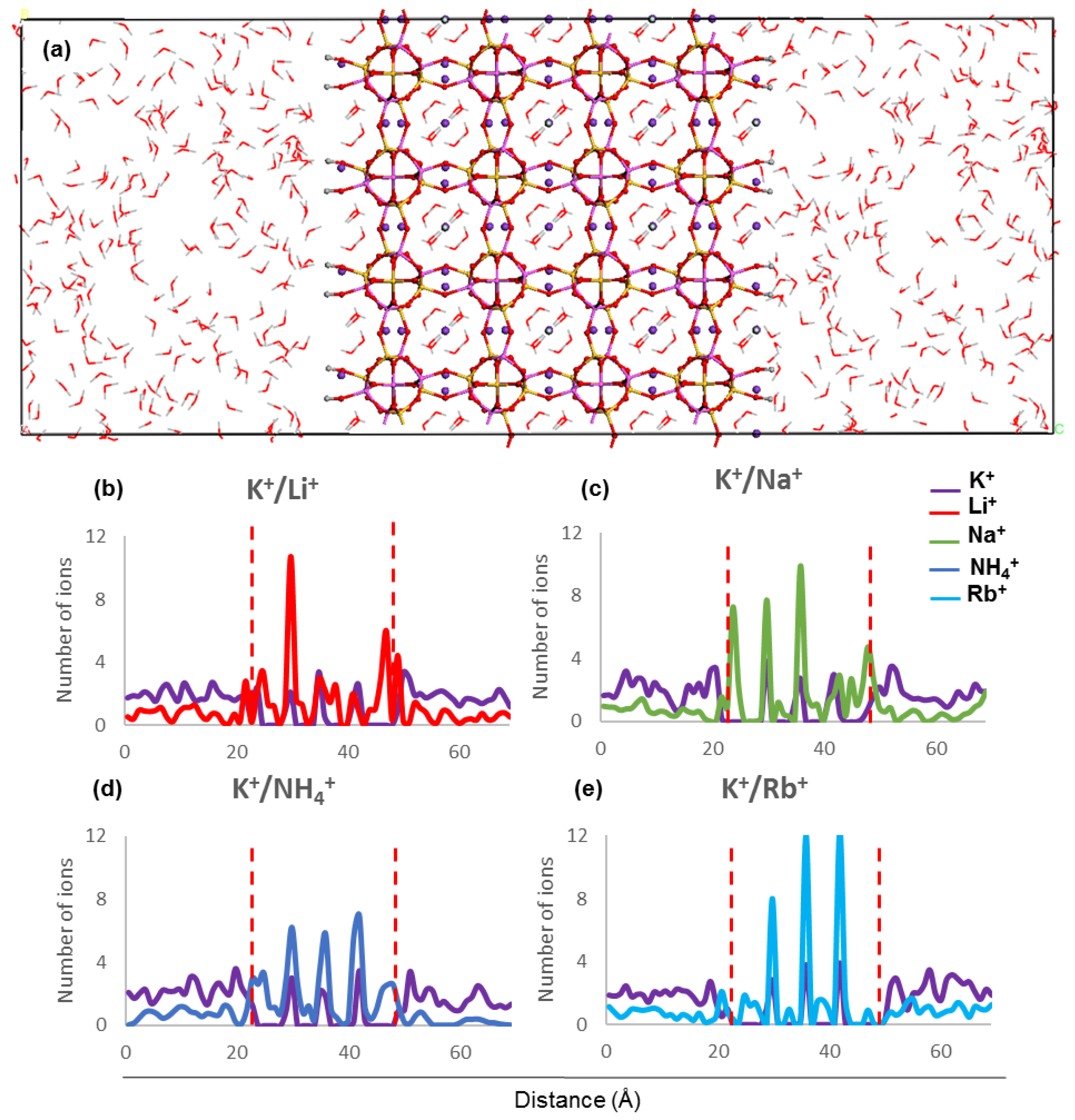
The concentration profiles show that ions undergo similar localisation behaviour within the ZM-001 and ZM-110 membranes (
Figure 2 and
Figure 3,
Figures S2 and S3). The ionic size predominantly affects the distribution of ions inside membranes whereby larger ions, such as NH
4+, K
+, Rb
+ and Cs
+, are distributed more uniformly compared with smaller ions such as, Li
+, Na
+, Mg
2+ and Ca
2+. On the other hand, these simulations show that NH
4+, Li
+, Na
+, Mg
2+ and Ca
2+ ions concentrate in channel openings at the surface of the membrane due to their strong interaction with framework oxygens.
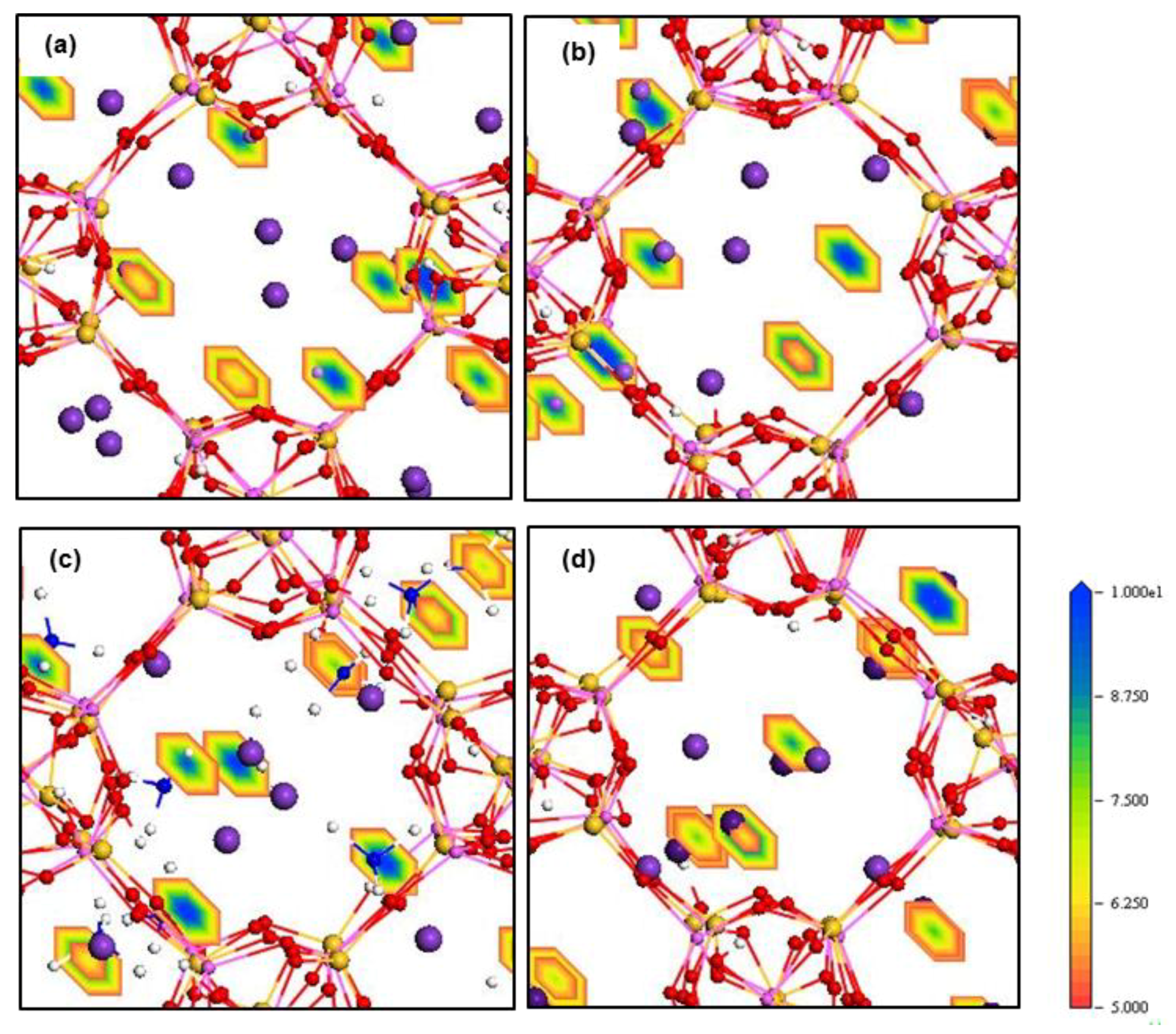
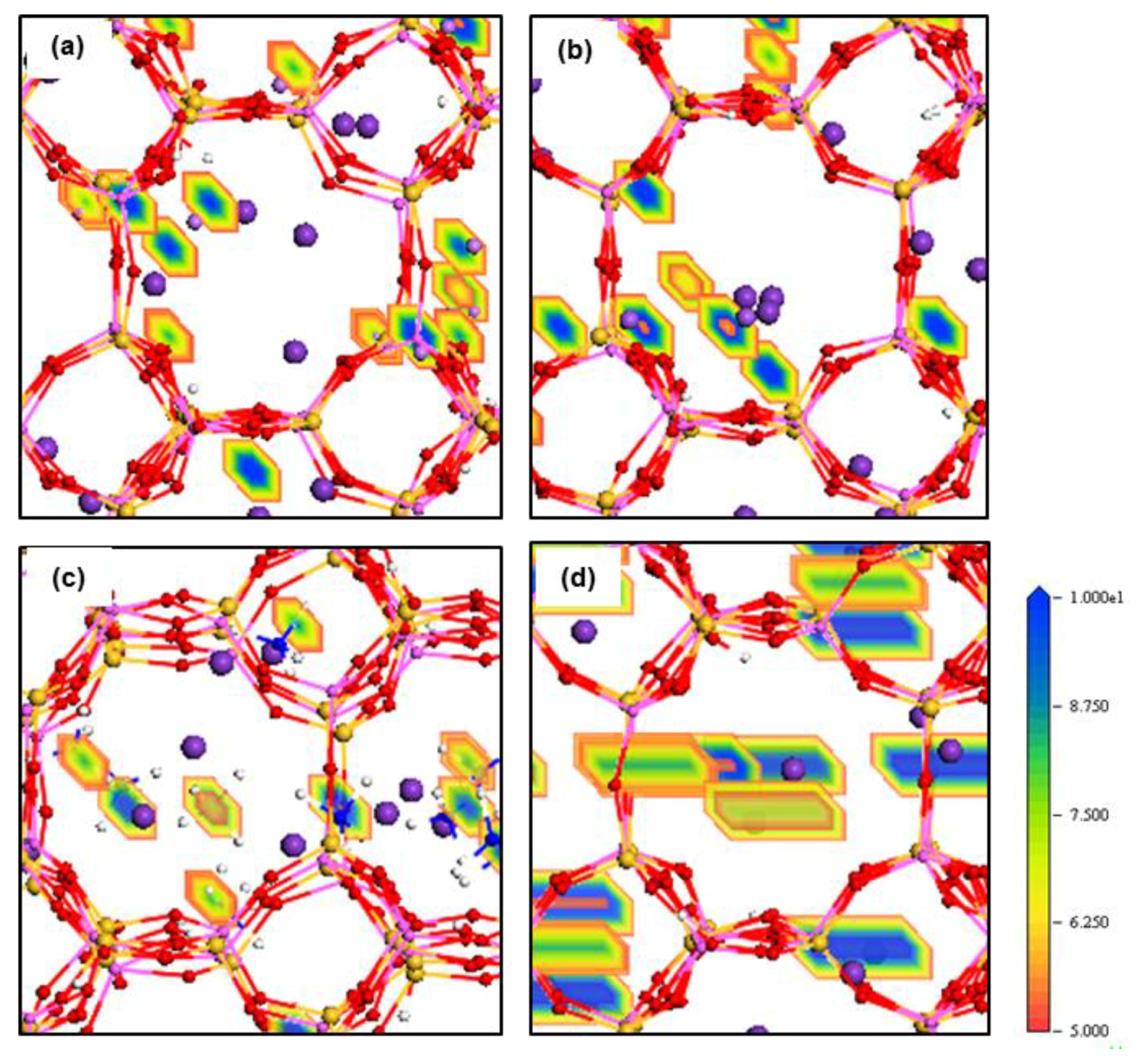
The electron density field maps indicate that for ion localisations inside both types of membrane cages, K
+ and NH
4+ ions localise at SI, SII and slightly shifted positions close to these two sites (
Figure 4 and
Figure 5,
Figures S4 and S5). However, ions larger than K
+, Rb
+ and Cs
+, exactly occupy both SI and SII sites. Ions smaller than K
+, including Li
+, Na
+, Mg
2+ and Ca
2+, reside in disordered crystallographic positions closer to the framework rather than at crystallographic sites. These ions localise differently in ZM-001 and ZM-110. The Li
+, Na
+, Mg
2+ and Ca
2+ ions occupy disordered crystallographic positions in cages of ZM-001 that are closer to SII sites. However, their locations in ZM-110 cages are closer to SI sites. The Na
+ and Ca
2+ ions are located at further distances to the framework compared to Li
+ and Mg
2+ ions, due to their comparatively larger ionic size.
The RDF values in
Table 2 indicate that all guest cations have stronger interaction with framework oxygen than oxygen of the water molecules or with Cl
− anions inside the membranes. The RDF results show that K
+ ions are located at the same distances to the framework in all exchanging systems, though the membranes contain different guest ions with different ionic sizes. The average calculated K distances to O, Si and Al of the framework, Cl anions and oxygen of water molecules inside the membranes are in good agreement with XRD data [
2].
The nearest distance of Mn+ cations to the framework atoms are identified by the position of the first peak of the function g(r). The distances of Mn+ cations to the zeolite N framework atoms of ZM-001 and ZM-110 membranes are similar except for Rb+ and Cs+ cations. In ZM-110, most Rb+ and Cs+ cations tend to locate in the middle of cages at a further distance to the framework. However, in ZM-001 these ions are equally localised at both sites.
The nearest distances for NH
4+ to the framework oxygen and to Si or Al atoms of ZM-001 are larger and smaller, respectively, than previously obtained values [
10]. However, the RDF distances for Na
+, K
+, Rb
+ and Cs
+ are smaller than our previous results [
10]. These differences in nearest distance of atom pairs between these models of zeolite N exchange, are related to different partial charges on framework atoms used in simulations [
12].
Furthermore, the RDF graphs in this study show notable first peaks for Li
+ as well as for Na
+, Mg
2+ and Ca
2+ around the framework Si and Al atoms (
Figures S6 and S7). These nearest distances for Li
+, Na
+, Mg
2+ and Ca
2+ to framework Si and Al atoms are due to the small sizes of these cations as well as to the presence of two different Si and Al atomic positions (T1 and T2 sites) with different partial charges in the zeolite framework [
12]. RDF plots and density field maps indicate that these ions are closer to the Si/Al in T2 sites rather than the Si/Al in T1 sites.
The strength of the interaction between atom pairs is estimated from peak intensities of g(r). The peak heights for O-M
n+ for NH
4+, Li
+, Na
+, Mg
2+ and Ca
2+ are higher than Si/Al-M
n. Moreover, the O-M
n+ value for these ions is the nearest distance to the framework atoms that show strong interaction with framework oxygen atoms compared to Si and Al. The interaction increases from NH
4+ < Na
+ < Ca
2+ < Li
+ < Mg
2+. In contrast, the higher peaks for Si/Al-M
n+ where M = K
+, Rb
+ or Cs
+ are not the nearest distances and consequently have weaker interaction with framework oxygen atoms. The interaction strength decreases from K
+ > Rb
+ > Cs
+, in complete agreement with previous outcomes [
10]. RDF results using ion density profiles and ion density fields confirm that ions are localised inside the framework relative to their ionic size. An exception to this outcome is NH
4+ for which hydrogen bonding provides stronger interactions with framework oxygen atoms compared with other cations evaluated in these simulations [
10].
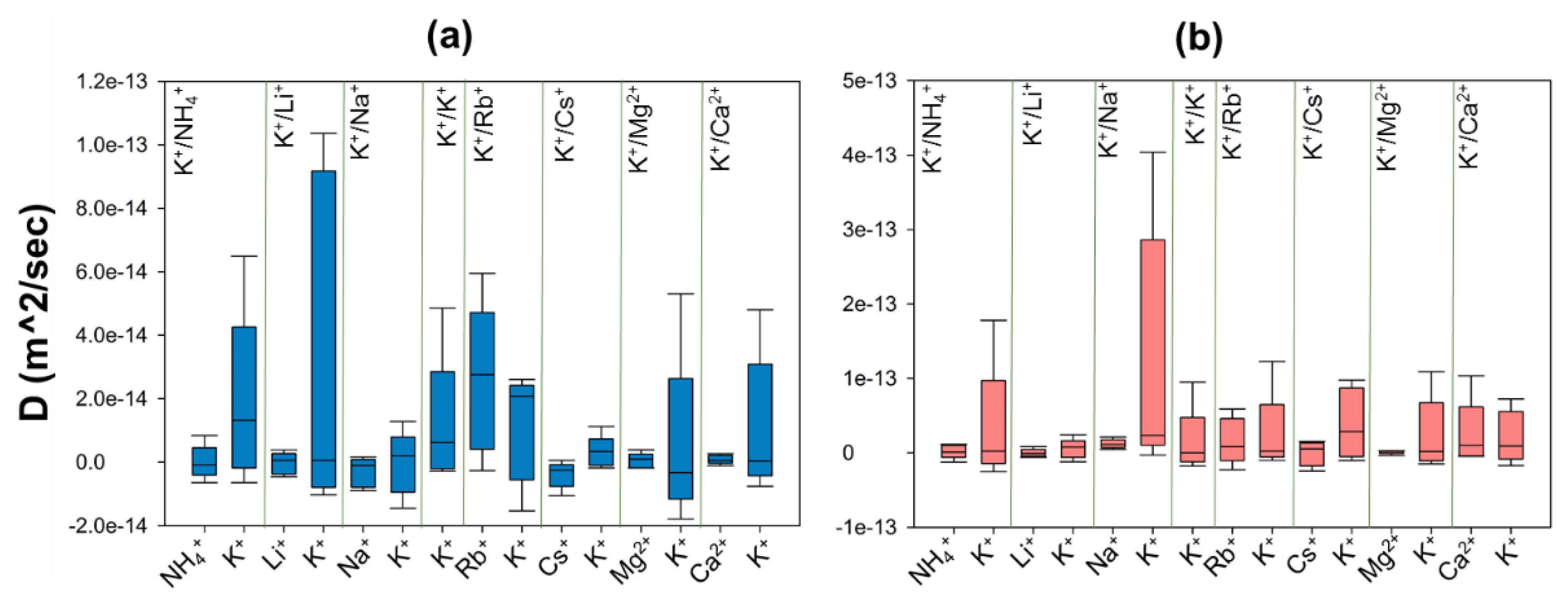
3.3. Ion Diffusion
We investigate the relative mobility of ions inside and outside zeolite N membranes by calculating the self-diffusion coefficient (D) of ions from their mean square displacement (MSD) over the simulation time. The diffusion behaviour of ions inside the confined geometry of zeolites is clearly very different from their bulk behaviour in solution. Simulations show that values of D for ions inside zeolite N membranes are smaller than values obtained in the electrolyte solution by several orders of magnitude (
Figure 6 and
Figure S8).
The D values for cations inside both membranes are close to zero. Moreover, the measured D values for NH4+, Li+, Na+ and Cs+ cations are negative inside ZM-001 as well as for Li+ and Cs+ inside ZM-110. Close inspection of MSD curves reveal that ions do not show diffuse behaviour while some curves show different behaviour regimes over time with positive and negative slopes. This behaviour means that movement of ions inside the membranes is significantly affected by a number of mechanisms. The small or negative diffusion of ions in this study indicates that these ions can not pass the free energy barriers inside zeolite N channels and jump from one low energy site to another. Therefore, these ions localise in a specific position within the structure and show an oscillatory behaviour.
These energy barriers are present in all directions and include dispersion-repulsion and electrostatic energies between ions and the framework as well as the activation energy that a particular ion requires to move between different crystallographic positions [
13]. The density field illustrations shown in
Figure 4 and
Figure 5,
Figures S4 and S5 are exemplars of this mechanistic interpretation for zeolite N. No systematic dependence on ionic size is observed for D values of ions inside zeolite N membranes.
There are few experimental studies calculating the self-diffusion of cations inside different zeolites (e.g., analcite [
14], chabazite [
15], mordenite [
16] and clinoptilolite [
17]) by measuring all activation, dispersion-repulsion and coloumbic energies. These studies calculated the self-diffusion coefficient values for monovalent cations NH
4+, Na
+, K
+, Rb
+, and Cs
+, in different zeolites, in the range of 10
−11−10
−26 m
2·sec
−1 and for divalent cations in the range of 10
−15−10
−17 m
2·sec
−1. The self-diffusion coefficient values for cations obtained in this study are of similar magnitude to experimentally measured values of self-diffusion for cations in other zeolites at 25 °C [
14,
15,
16,
17].
3.4. Ion Exchange Mechanism
In general, the ion exchange property of zeolites and of cation diffusion in zeolites depend on various parameters, including cation size, incipient charge on cations, the smallest free diameter of channel pore-openings, the number of oxygens involved in the pore-openings, the Si/Al ratio and finally, the water flux inside the zeolite structure and its interaction with cations. Therefore, the difference between ZM-001 and ZM-110 ion exchange properties is due to the difference between their pore-opening diameter and the shape of the pore-opening that interfaces with the ambient solution, since all other parameters are the same for both membrane types. However, the interconnection of [001] and [110] channels in both membranes complicates the intracrystalline exchange and diffusion of cations.
Ammonium ions have the highest retention in these simulations and the closest distances to framework atoms in both membranes, even though the ionic size for NH
4+ is larger than K
+. In our previous simulations, we indicated that hydrogen bonding influences the selectivity of NH
4+ in comparison to other monovalent cations [
10]. The hydrogen in NH
4+ interacts with between one and three framework oxygen atoms and between one and two water molecules or other NH
4+ ions. Of these, the interaction between the NH
4+ hydrogen and framework oxygen atoms is the most stable.
These hydrogen bonds result in completely different diffusive behaviour for NH4+ compared to other cations. The formation and elimination of hydrogen bonding facilitates the movement of NH4+ ions within zeolite channels. However, K+ cations leave the membranes more quickly than NH4+ ions due to loose van der Waals interactions with framework and water molecules. As we have seen, the self-diffusion of K+ cations is higher than NH4+ ions in K+/NH4+ systems.
In this study, we consider nitrogen as the centre of the NH4+ ion for calculation of the ammonium self-diffusion. The D value for the total NH4+ ion is larger than the D value of N by several orders of magnitude, as a result of changes in hydrogen bonding. For example, the nitrogen and total NH4+ self-diffusions in ZM-001 are −1.1 × 10−17 m2·sec−1 and 7.2 × 10−12 m2·sec−1, respectively. Although the NH4+ interaction is the same in both membranes, the ZM-110 membrane retains more NH4+. The small diameter of channels along the [110] direction increases the energy barrier for NH4+ ions leaving the membrane with water molecules. Therefore, NH4+ ions localise within ZM-110 membrane channels along the a axis with limited or no access to the solution outside the membrane.
In general, the exchange of monovalent cations and their location to the framework inside zeolite N membranes depends on their ionic size and, respectively, decrease and increase with increase in cation size. However, the exchanges of Li+ and Cs+ do not follow this general principle for the ZM-110 membrane. Li+ and Na+ cations have lower mobility compared to K extra-framework cations, due to their stronger electrostatic interactions with the zeolite N framework. The Li+ cations show higher levels of interaction than Na+ cations due to a higher charge density. Na+ cations show between two and three electrostatic interactions with framework oxygen atoms, one van der Waals interaction with water molecules and/or one interaction with chloride anions. However, Li+ cations show interactions with two water molecules while they have the same number of electrostatic interactions with framework oxygen and chloride atoms. As a result, Li+ can be more mobile within zeolite N channels compared to Na+.
Rb
+ and Cs
+ ions seem to follow similar diffusion mechanisms in zeolite N. These cations prefer to localise at the SI and SII sites, which are the lowest energy sites within the zeolite N structure. Rb
+ and Cs
+ have between three and four van der Waals interactions with framework oxygens and between two and three interactions with water molecules and/or one chloride ion. As a result of these many interactions, and their large ionic size, these ions oscillate at their site positions and show limited diffusivity. In comparison to K
+, Rb
+ shows a higher self-diffusion value in the ZM-001 membrane. The diffusion of Rb
+ and Cs
+ in the ZM-110 membrane is anisotropic. Rb and Cs in the ZM-110 membrane cannot transport through channels to the external solution due to their ionic size in comparison to the small pore opening sizes of this membrane direction. Thus, these larger ions prefer to move through channels along the
a axis inside the ZM-110 membrane. This attribute is evident by the rectangular shape of the density fields for these cations shown in
Figure 4d and
Figure S5b.
The behaviour of divalent Mg
2+ and Ca
2+ cations gives the impression that they follow a similar exchange and diffusion mechanism as monovalent Li
+ and Na
+ cations, respectively. As shown in
Figure 4 and
Figure 5,
Figures S4 and S5, the localisation behaviour and structural arrangements around framework oxygen atoms are similar, especially for Mg
2+ and Li
+. However, the exchange and diffusion processes are completely different. For example, the Mg
2+ and Ca
2+ mobilities inside zeolite N membranes are higher than monovalent Li
+ and Na
+ cations even though they show the same number of electrostatic interactions with framework oxygen atoms as Li
+ and Na
+. However, Mg
2+ and Ca
2+ interact with more water molecules compared to Li
+ and Na
+. As a result, these associated water molecules enhance the mobility of Mg
2+ and Ca
2+ inside zeolite channels. Nevertheless, in comparison to extra-framework K
+, the diffusion of Mg
2+ and Ca
2+, is less facile due to the higher coulombic interaction with framework oxygen atoms. Thus, for the ZM-001 membrane, these simulations suggest that divalent Mg
2+ and Ca
2+ show analogous retention behaviour to monovalent Li
+ and Na
+.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}