
Manipulating Enzymes Properties with DNA Nanostructures


Abstract
:1. Introduction
2. Chemical Strategies for the Synthesis and Purification of DNA-Protein Conjugates
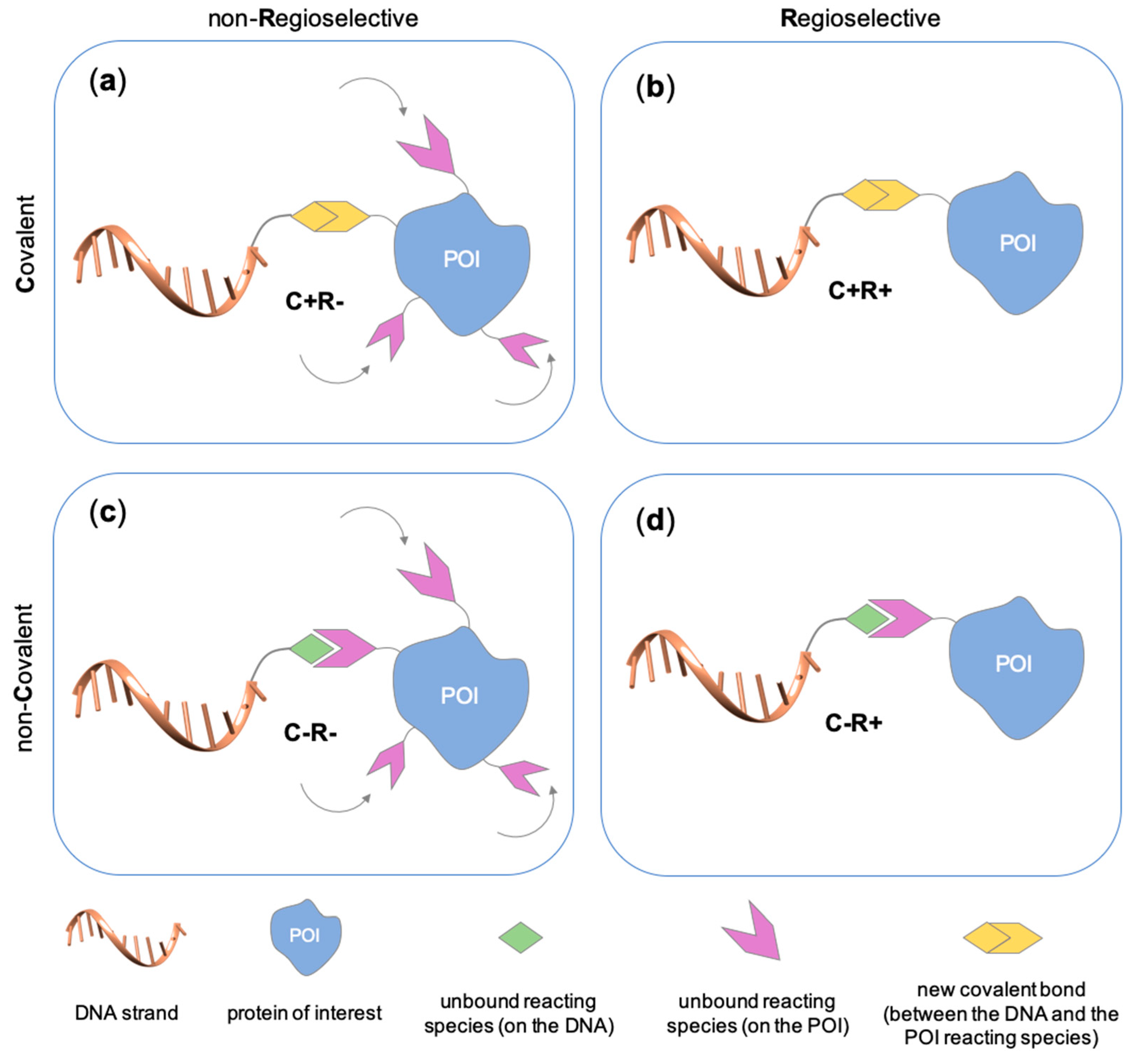
2.1. Covalent Methods
2.1.1. Covalent and Non-Regioselective Methods (C+R-)
2.1.2. Covalent and Regioselective Methods (C+R+)
2.2. Non-Covalent Chemical Strategies
2.2.1. Non-Covalent and Non-Regioselective Methods (C-R-)
2.2.2. Non-Covalent and Regioselective Methods (C-R+)
2.3. From Single Stranded DNA-Protein Conjugates to Large DNA-Protein Nanostructures
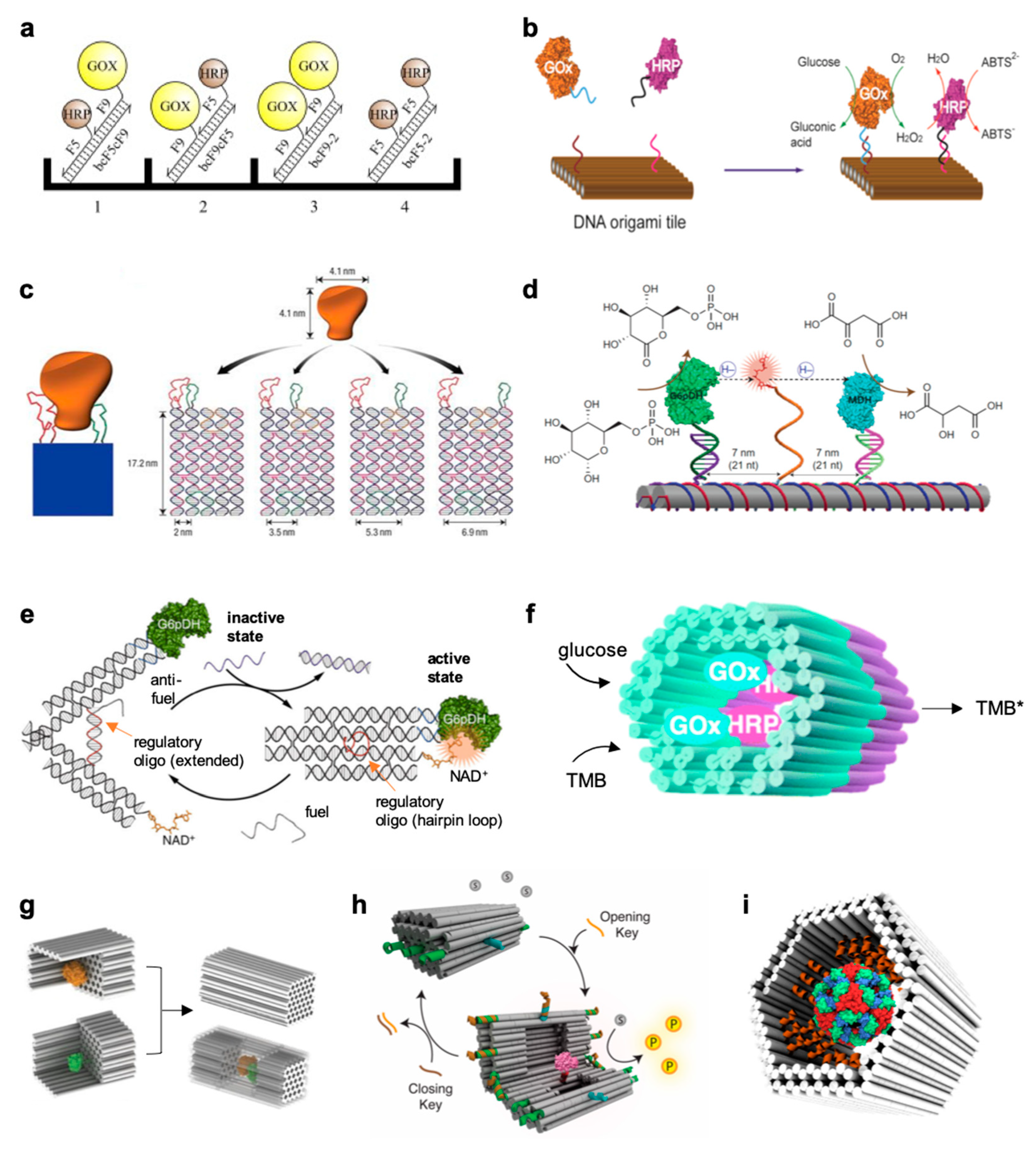
3. Geometry of the DNA-Protein Nanostructure
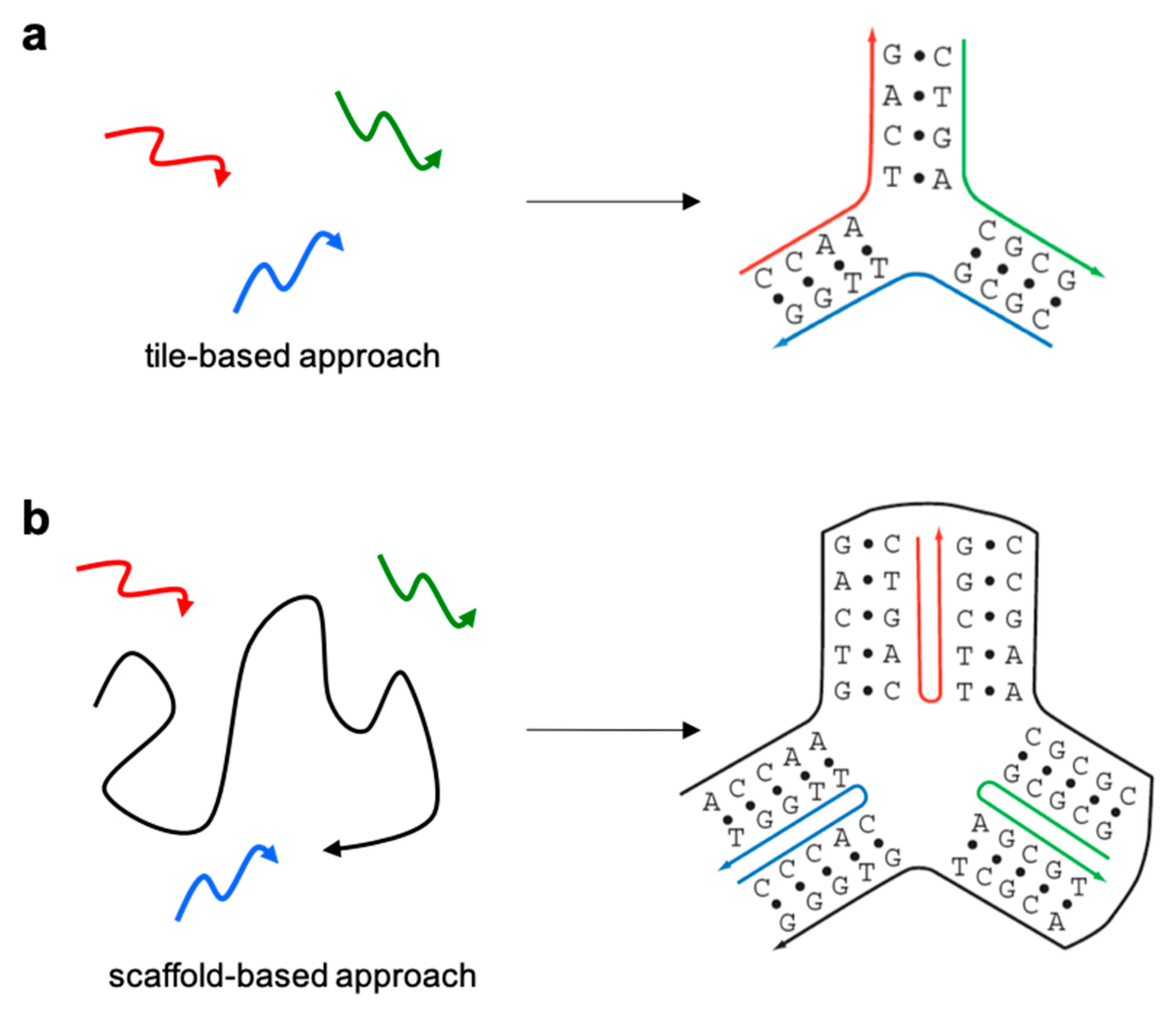
3.1. Mono- and Two-Dimensional Nanostructures
3.2. Small DNA Tiles
3.3. Large Three-Dimensional Nanostructures
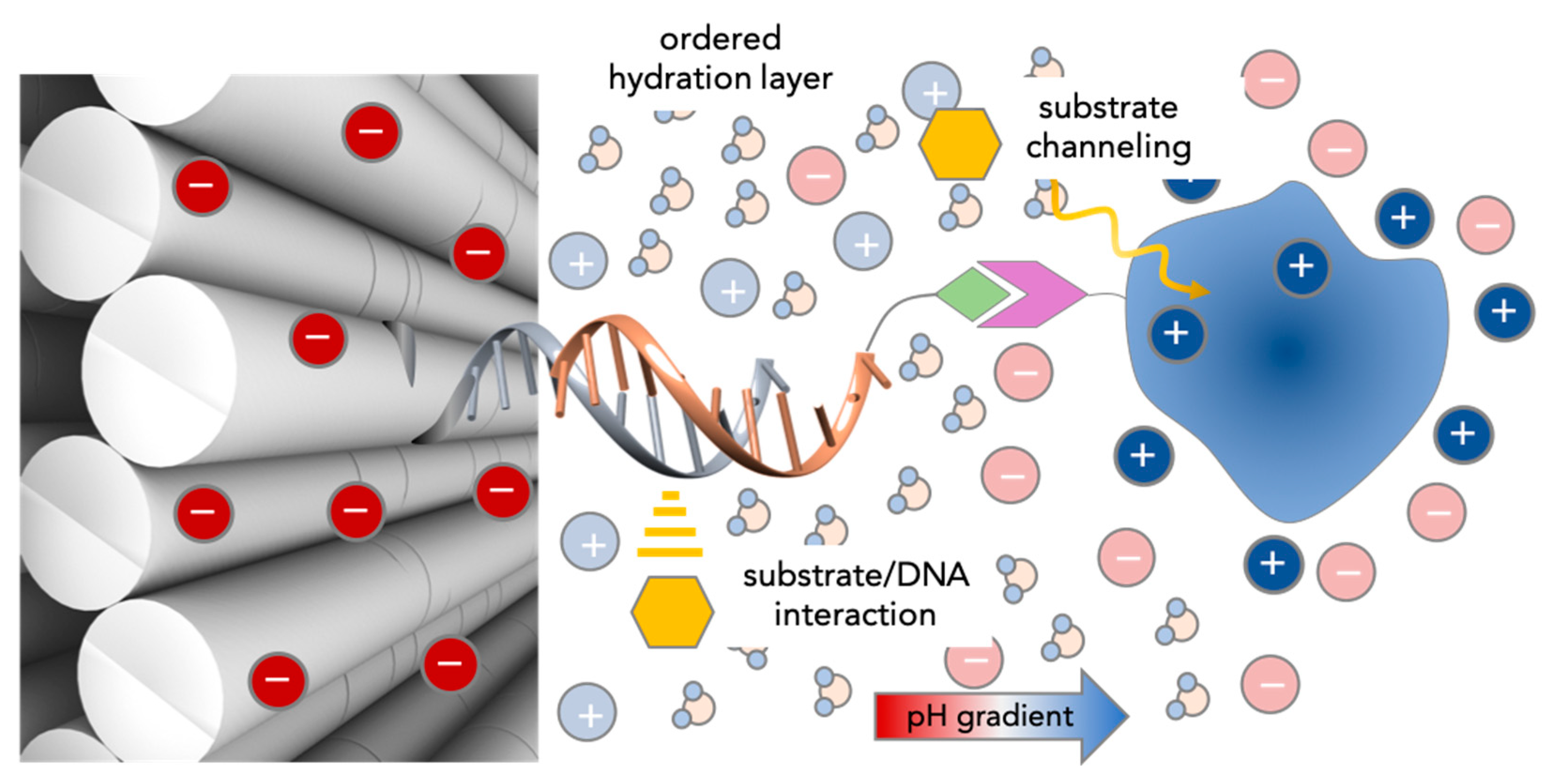
4. DNA-Nanostructures for Confined Multivalency: A Thermodynamic Model of Entropic Avidity
5. Catalytic Activity of DNA-Enzyme Systems: Experimental Observations and Proposed Models
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Seeman, N.C. Nucleic acid junctions and lattices. J. Theor. Biol. 1982, 99, 237–247. [Google Scholar] [CrossRef]
- Seeman, N.C. Macromolecular design, nucleic acid junctions, and crystal formation. J. Biomol. Struct. Dyn. 1985, 3, 11–34. [Google Scholar] [CrossRef]
- Du, S.M.; Zhang, S.; Seeman, N.C. DNA junctions, antijunctions, and mesojunctions. Biochemistry 1992, 31, 10955–10963. [Google Scholar] [CrossRef]
- Zheng, J. From molecular to macroscopic via the rational design of a self-assembled 3D DNA crystal. Nature 2009, 461, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.Y.; Seelig, G. Dynamic DNA nanotechnology using strand-displacement reactions. Nat. Chem. 2011, 3, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Mao, C. A nanomechanical device based on the B-Z transition of DNA. Nature 1999, 397, 144–146. [Google Scholar] [CrossRef]
- Endo, M. Single-molecule visualization of the hybridization and dissociation of photoresponsive oligonucleotides and their reversible switching behavior in a DNA nanostructure. Angew. Chem. Int. Ed. 2012, 51, 10518–10522. [Google Scholar] [CrossRef] [PubMed]
- Liedl, T.; Simmel, F.C. Switching the conformation of a DNA molecule with a chemical oscillator. Nano Lett. 2005, 5, 1894–1898. [Google Scholar] [CrossRef]
- Liu, D. A reversible pH-driven DNA nanoswitch array. J. Am. Chem. Soc. 2006, 128, 2067–2071. [Google Scholar] [CrossRef]
- Yurke, B. A DNA-fuelled molecular machine made of DNA. Nature 2000, 406, 605–608. [Google Scholar] [CrossRef]
- Sacca, B. Nanolattices of switchable DNA-based motors. Small 2012, 8, 3000–3008. [Google Scholar] [CrossRef] [PubMed]
- Sannohe, Y. Visualization of dynamic conformational switching of the G-quadruplex in a DNA nanostructure. J. Am. Chem. Soc. 2010, 132, 16311–16313. [Google Scholar] [CrossRef] [PubMed]
- Seeman, N.C. Nanomaterials based on DNA. Annu. Rev. Biochem. 2010, 79, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Rothemund, P.W. Folding DNA to create nanoscale shapes and patterns. Nature 2006, 440, 297–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeman, N.C.; Kallenbach, N.R. DNA branched junctions. Annu. Rev. Biophys. Biomol. Struct. 1994, 23, 53–86. [Google Scholar] [CrossRef]
- Seeman, N.C.; Kallenbach, N.R. Design of immobile nucleic acid junctions. Biophys. J. 1983, 44, 201–209. [Google Scholar] [CrossRef] [Green Version]
- Feldkamp, U.; Niemeyer, C.M. Rational engineering of dynamic DNA systems. Angew. Chem. Int. Ed. 2008, 47, 3871–3873. [Google Scholar] [CrossRef]
- Simmel, F.C. Programming the dynamics of biochemical reaction networks. ACS Nano 2013, 7, 6–10. [Google Scholar] [CrossRef]
- Zhang, D.Y. Engineering entropy-driven reactions and networks catalyzed by DNA. Science 2007, 318, 1121–1125. [Google Scholar] [CrossRef]
- Kosinski, R. Sites of high local frustration in DNA origami. Nat. Commun. 2019, 10, 1061. [Google Scholar] [CrossRef]
- Lee, A.J.; Walti, C. DNA nanostructures: A versatile lab-bench for interrogating biological reactions. Comput. Struct. Biotechnol. J. 2019, 17, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F. Structural DNA Nanotechnology: State of the Art and Future Perspective. J. Am. Chem. Soc. 2014, 136, 11198–11211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Niemeyer, C.M. From DNA Nanotechnology to Material Systems Engineering. Adv. Mater. 2019, 31, e1806294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, A.A.; de Silva, C. Beyond the smiley face: applications of structural DNA nanotechnology. Nano Rev. Exp. 2018, 9, 1430976. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q. Rationally Designed DNA-Origami Nanomaterials for Drug Delivery In Vivo. Adv. Mater. 2018, e1804785. [Google Scholar] [CrossRef]
- Zhou, K. Toward Precise Manipulation of DNA-Protein Hybrid Nanoarchitectures. Small 2019, 15, e1804044. [Google Scholar] [CrossRef]
- Yang, Y.R.; Liu, Y.; Yan, H. DNA Nanostructures as Programmable Biomolecular Scaffolds. Bioconj. Chem. 2015, 26, 1381–1395. [Google Scholar] [CrossRef]
- Madsen, M.; Gothelf, K.V. Chemistries for DNA Nanotechnology. Chem. Rev. 2019, 6384–6458. [Google Scholar] [CrossRef]
- Trads, J.B.; Tørring, T.; Gothelf, K.V. Site-Selective Conjugation of Native Proteins with DNA. Acc. Chem. Res. 2017, 50, 1367–1374. [Google Scholar] [CrossRef]
- Chen, X.; Wu, Y.-W. Selective chemical labeling of proteins. Org. Biomol. Chem. 2016, 14, 5417–5439. [Google Scholar] [CrossRef] [Green Version]
- Niemeyer, C.M. Semisynthetic DNA–Protein Conjugates for Biosensing and Nanofabrication. Angew. Chem. Int. Ed. 2010, 49, 1200–1216. [Google Scholar] [CrossRef] [PubMed]
- Sacca, B.; Niemeyer, C.M. Functionalization of DNA nanostructures with proteins. Chem. Soc. Rev. 2011, 40, 5910–5921. [Google Scholar] [CrossRef] [PubMed]
- Corey, D.R.; Schultz, P.G. Generation of a hybrid sequence-specific single-stranded deoxyribonuclease. Science 1987, 238, 1401–1403. [Google Scholar] [CrossRef] [PubMed]
- Duckworth, B.P. A Universal Method for the Preparation of Covalent Protein–DNA Conjugates for Use in Creating Protein Nanostructures. Angew. Chem. Int. Ed. 2007, 46, 8819–8822. [Google Scholar] [CrossRef] [PubMed]
- Debets, M.F. Aza-dibenzocyclooctynes for fast and efficient enzyme PEGylation via copper-free (3+2) cycloaddition. Chem. Commun. 2010, 46, 97–99. [Google Scholar] [CrossRef]
- Kohn, M.; Breinbauer, R. The Staudinger ligation-a gift to chemical biology. Angew. Chem. Int. Ed. Engl. 2004, 43, 3106–3116. [Google Scholar] [CrossRef]
- Keppler, A. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 2003, 21, 86. [Google Scholar] [CrossRef]
- Los, G.V. HaloTag: A Novel Protein Labeling Technology for Cell Imaging and Protein Analysis. ACS Chem. Biol. 2008, 3, 373–382. [Google Scholar] [CrossRef]
- Takeda, S.; Tsukiji, S.; Nagamune, T. Site-specific conjugation of oligonucleotides to the C-terminus of recombinant protein by expressed protein ligation. Bioorg. Med. Chem. Lett. 2004, 14, 2407–2410. [Google Scholar] [CrossRef]
- Takeda, S. Covalent split protein fragment-DNA hybrids generated through N-terminus-specific modification of proteins by oligonucleotides. Org. Biomol. Chem. 2008, 6, 2187–2194. [Google Scholar] [CrossRef]
- Tominaga, J. An enzymatic method for site-specific labeling of recombinant proteins with oligonucleotides. Chem. Comm. 2007, 401–403. [Google Scholar] [CrossRef] [PubMed]
- Theile, C.S. Site-specific N-terminal labeling of proteins using sortase-mediated reactions. Nat. Protoc. 2013, 8, 1800–1807. [Google Scholar] [CrossRef] [PubMed]
- Koussa, M.A.; Sotomayor, M.; Wong, W.P. Protocol for sortase-mediated construction of DNA-protein hybrids and functional nanostructures. Methods 2014, 67, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, R.S. Enzymatic ligation of large biomolecules to DNA. ACS Nano 2013, 7, 8098–8104. [Google Scholar] [CrossRef] [PubMed]
- Rosen, C.B. Template-directed covalent conjugation of DNA to native antibodies, transferrin and other metal-binding proteins. Nat. Chem. 2014, 6, 804–809. [Google Scholar] [CrossRef]
- Zhang, Y. Recent progress in enzymatic protein labelling techniques and their applications. Chem. Soc. Rev. 2018, 47, 9106–9136. [Google Scholar] [CrossRef]
- Sprengel, A. Tailored protein encapsulation into a DNA host using geometrically organized supramolecular interactions. Nat. Commun. 2017, 8, 14472. [Google Scholar] [CrossRef] [Green Version]
- Samanta, K. Protein Surface Recognition by Synthetic Molecules. In Comprehensive Supramolecular Chemistry II; Wilson, A., Ed.; Elsevier: Amsterdam, The Netherland, 2017. [Google Scholar]
- Hatai, J.; Schmuck, C. Diverse Properties of Guanidiniocarbonyl Pyrrole-Based Molecules: Artificial Analogues of Arginine. Acc. Chem. Res. 2019, 52, 1709–1720. [Google Scholar] [CrossRef]
- Schrader, T.; Bitan, G.; Klarner, F.G. Molecular tweezers for lysine and arginine - powerful inhibitors of pathologic protein aggregation. Chem. Commun. 2016, 52, 11318–11334. [Google Scholar] [CrossRef]
- Klarner, F.G.; Schrader, T. Aromatic interactions by molecular tweezers and clips in chemical and biological systems. Acc. Chem. Res. 2013, 46, 967–978. [Google Scholar] [CrossRef]
- Barrow, S.J. Cucurbituril-Based Molecular Recognition. Chem. Rev. 2015, 115, 12320–12406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Pathak, P.; Jayawickramarajah, J. Design, synthesis, and applications of DNA-macrocyclic host conjugates. Chem. Commun. 2018, 54, 11668–11680. [Google Scholar] [CrossRef] [PubMed]
- Li, H. DNA-templated self-assembly of protein and nanoparticle linear arrays. J. Am. Chem. Soc. 2004, 126, 418–419. [Google Scholar] [CrossRef] [PubMed]
- Shen, W. NTA directed protein nanopatterning on DNA Origami nanoconstructs. J. Am. Chem. Soc. 2009, 131, 6660–6661. [Google Scholar] [CrossRef]
- Williams, B.A. Creating protein affinity reagents by combining peptide ligands on synthetic DNA scaffolds. J. Am. Chem. Soc. 2009, 131, 17233–17241. [Google Scholar] [CrossRef]
- He, Y. Antibody nanoarrays with a pitch of approximately 20 nanometers. J. Am. Chem. Soc. 2006, 128, 12664–12665. [Google Scholar] [CrossRef]
- Fruk, L.; Müller, J.; Niemeyer, C.M. Kinetic Analysis of Semisynthetic Peroxidase Enzymes Containing a Covalent DNA–Heme Adduct as the Cofactor. Chem. Eur. J. 2006, 12, 7448–7457. [Google Scholar] [CrossRef]
- Fruk, L.; Niemeyer, C.M. Covalent Hemin–DNA Adducts for Generating a Novel Class of Artificial Heme Enzymes. Angew. Chem. Int. Ed. 2005, 44, 2603–2606. [Google Scholar] [CrossRef]
- Laity, J.H.; Lee, B.M.; Wright, P.E. Zinc finger proteins: New insights into structural and functional diversity. Curr. Opin. Struct. Biol. 2001, 11, 39–46. [Google Scholar] [CrossRef]
- Xin, L. Regulation of an enzyme cascade reaction by a DNA machine. Small 2013, 9, 3088–3091. [Google Scholar] [CrossRef]
- Goodman, R.P. A facile method for reversibly linking a recombinant protein to DNA. Chembiochem 2009, 10, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z. Simple and efficient method to purify DNA-protein conjugates and its sensing applications. Anal. Chem. 2014, 86, 3869–3875. [Google Scholar] [CrossRef] [PubMed]
- Yeo, J.E. Synthesis of site-specific DNA-protein conjugates and their effects on DNA replication. ACS Chem. Biol. 2014, 9, 1860–1868. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.; Benson, E.; Hogberg, B. Purification of functionalized DNA origami nanostructures. ACS Nano 2015, 9, 4968–4975. [Google Scholar] [CrossRef] [PubMed]
- Muller, J.; Niemeyer, C.M. DNA-directed assembly of artificial multienzyme complexes. Biochem. Biophys. Res. Commun. 2008, 377, 62–67. [Google Scholar] [CrossRef]
- Conrado, R.J. DNA-guided assembly of biosynthetic pathways promotes improved catalytic efficiency. Nucleic Acids Res. 2012, 40, 1879–1889. [Google Scholar] [CrossRef]
- Wilner, O.I. Enzyme cascades activated on topologically programmed DNA scaffolds. Nat. Nanotechnol. 2009, 4, 249–254. [Google Scholar] [CrossRef]
- Wilner, O.I. Self-assembly of enzymes on DNA scaffolds: En route to biocatalytic cascades and the synthesis of metallic nanowires. Nano Lett. 2009, 9, 2040–2043. [Google Scholar] [CrossRef]
- Ke, G. Directional Regulation of Enzyme Pathways through the Control of Substrate Channeling on a DNA Origami Scaffold. Angew. Chem. Int. Ed. 2016, 55, 7483–7486. [Google Scholar] [CrossRef]
- Fu, J. Interenzyme Substrate Diffusion for an Enzyme Cascade Organized on Spatially Addressable DNA Nanostructures. J. Am. Chem. Soc. 2012, 134, 5516–5519. [Google Scholar] [CrossRef] [Green Version]
- Rinker, S. Self-assembled DNA nanostructures for distance-dependent multivalent ligand-protein binding. Nat. Nanotechnol. 2008, 3, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Fu, J. Multi-enzyme complexes on DNA scaffolds capable of substrate channelling with an artificial swinging arm. Nat. Nanotechnol. 2014, 9, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Liu, M. A DNA tweezer-actuated enzyme nanoreactor. Nat. Commun. 2013, 4, 2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linko, V.; Eerikainen, M.; Kostiainen, M.A. A modular DNA origami-based enzyme cascade nanoreactor. Chem. Commun. 2015, 51, 5351–5354. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z. Nanocaged enzymes with enhanced catalytic activity and increased stability against protease digestion. Nat. Commun. 2016, 7, 10619. [Google Scholar] [CrossRef] [Green Version]
- Grossi, G. Control of enzyme reactions by a reconfigurable DNA nanovault. Nat. Commun. 2017, 8, 992. [Google Scholar] [CrossRef]
- Zhou, C.; Yang, Z.; Liu, D. Reversible regulation of protein binding affinity by a DNA machine. J. Am. Chem. Soc. 2012, 134, 1416–1418. [Google Scholar] [CrossRef]
- Engelen, W.; Janssen, B.M.G.; Merkx, M. DNA-based control of protein activity. Chem. Comm. 2016, 52, 3598–3610. [Google Scholar] [CrossRef] [Green Version]
- Kitov, P.I.; Bundle, D.R. On the nature of the multivalency effect: A thermodynamic model. J. Am. Chem. Soc. 2003, 125, 16271–16284. [Google Scholar] [CrossRef]
- Jencks, W.P. On the attribution and additivity of binding energies. Proc. Natl. Acad. Sci. USA 1981, 78, 4046–4050. [Google Scholar] [CrossRef] [Green Version]
- Glettenberg, M.; Niemeyer, C.M. Tuning of peroxidase activity by covalently tethered DNA oligonucleotides. Bioconjug. Chem. 2009, 20, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.N. Tuning Enzyme Kinetics through Designed Intermolecular Interactions Far from the Active Site. ACS Catal. 2015, 5, 2149–2153. [Google Scholar] [CrossRef]
- Lin, J.-L.; Wheeldon, I. Kinetic Enhancements in DNA–Enzyme Nanostructures Mimic the Sabatier Principle. ACS Catal. 2013, 3, 560–564. [Google Scholar] [CrossRef]
- Idan, O.; Hess, H. Origins of activity enhancement in enzyme cascades on scaffolds. ACS Nano 2013, 7, 8658–8665. [Google Scholar] [CrossRef]
- Zhang, Y.; Tsitkov, S.; Hess, H. Proximity does not contribute to activity enhancement in the glucose oxidase–horseradish peroxidase cascade. Nat. Commun. 2016, 7, 13982. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
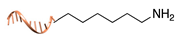
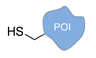
| C+R- (Covalent/non Regioselective Methods) | ||
|---|---|---|
| DNA | Protein | DNA-Protein Conjugate |
| sulfo- SMCC crosslinking [33] | ||
 | 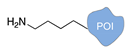 | 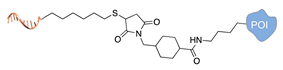 |
 |  |  |
| C+R+ (Covalent/Regioselective Methods) | ||
|---|---|---|
| DNA | Protein | DNA-Protein Conjugate |
| Cu(I)-catalyzed azide-alkyne cycloaddition [34] | ||
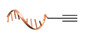 | 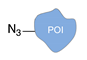 |  |
| Cu(I)-free azide-alkyne cycloaddition [35] | ||
 |  | 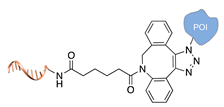 |
| Staudinger ligation [36] | ||
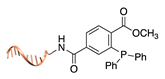 | 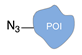 | 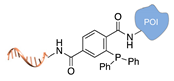 |
| SNAP tag [37] | ||
 | 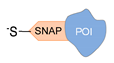 | 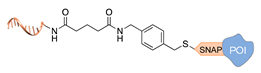 |
| Halo-tag [38] | ||
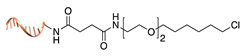 | 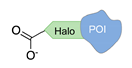 |  |
| Expressed protein ligation [39,40] | ||
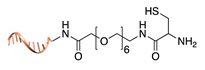 | 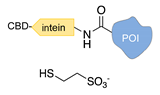 | 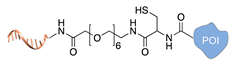 |
| Microbial transglutaminase [41] | ||
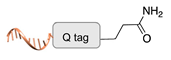 |  |  |
| Sortase A [42,43] | ||
 | 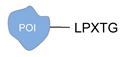 | 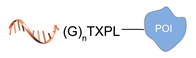 |
| Terminal deoxynucleotidyl transferase [44] | ||
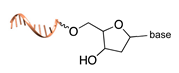 | 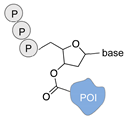 | 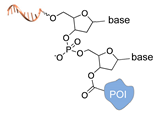 |
| DNA-templated conjugation [45] | ||
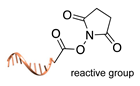 | 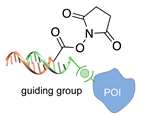 | 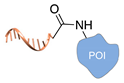 |
| C-R- (Non Covalent/Non-Regioselective Methods) | ||
|---|---|---|
| DNA | Protein | DNA-Protein Conjugate |
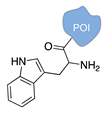
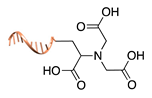
| Asp/Glu-selective GCP [48,49] | ||
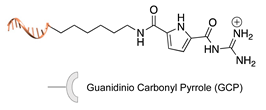 | 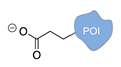 | 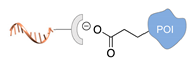 |
| Lys/Arg-selective molecular tweezers+ [50,51] | ||
 | 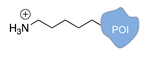 | 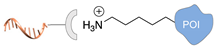 |
| cucurbit(8)uril-mediated dimerization [52,53] | ||
 |  | 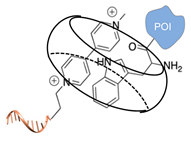 |
| C-R+ (Non Covalent/Regioselective Methods) | ||
|---|---|---|
| DNA | Protein | DNA-Protein Conjugate |
| biotin/monoavidin [54] | ||
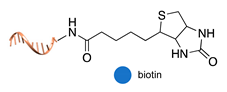 | 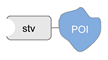 |  |
| NTA/6His tag/Ni2+ [55] | ||
 | 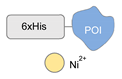 | 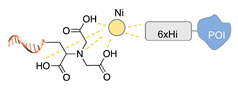 |
| DNA aptamer/protein [56] | ||
 |  |  |
| peptide ligand/protein [47] | ||
 |  | 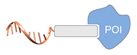 |
| antigen/antibody-protein [57] | ||
 | 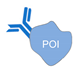 | 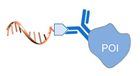 |
| cofactor/apoenzyme [58,59] | ||
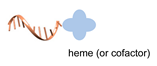 |  |  |
| DNA binding protein [60] | ||
 | 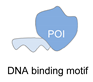 | 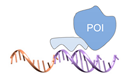 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaekel, A.; Stegemann, P.; Saccà, B. Manipulating Enzymes Properties with DNA Nanostructures. Molecules 2019, 24, 3694. https://doi.org/10.3390/molecules24203694
Jaekel A, Stegemann P, Saccà B. Manipulating Enzymes Properties with DNA Nanostructures. Molecules. 2019; 24(20):3694. https://doi.org/10.3390/molecules24203694
Chicago/Turabian StyleJaekel, Andreas, Pierre Stegemann, and Barbara Saccà. 2019. "Manipulating Enzymes Properties with DNA Nanostructures" Molecules 24, no. 20: 3694. https://doi.org/10.3390/molecules24203694
APA StyleJaekel, A., Stegemann, P., & Saccà, B. (2019). Manipulating Enzymes Properties with DNA Nanostructures. Molecules, 24(20), 3694. https://doi.org/10.3390/molecules24203694