1. Introduction
Although the biotechnology of Izumoring [
1,
2] and related approaches [
3] have revolutionized the availability of large quantities of many rare sugars, the practical conversion of
d-sorbose to
d-idose
6 by xylose isomerase [
4] is beset by the 97:3 equilibrium in favor of
d-sorbose and cannot easily produce significant quantities of
d-idose
6 [
5].
d-Idose, the rarest of all rare hexoses [
6,
7,
8], is the least stable to acid, base, or heat; idose is the only aldohexose to never be crystallized. The easy preparation of a stable precursor that could be converted to
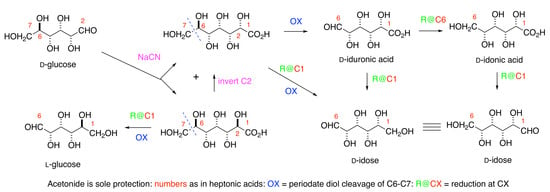
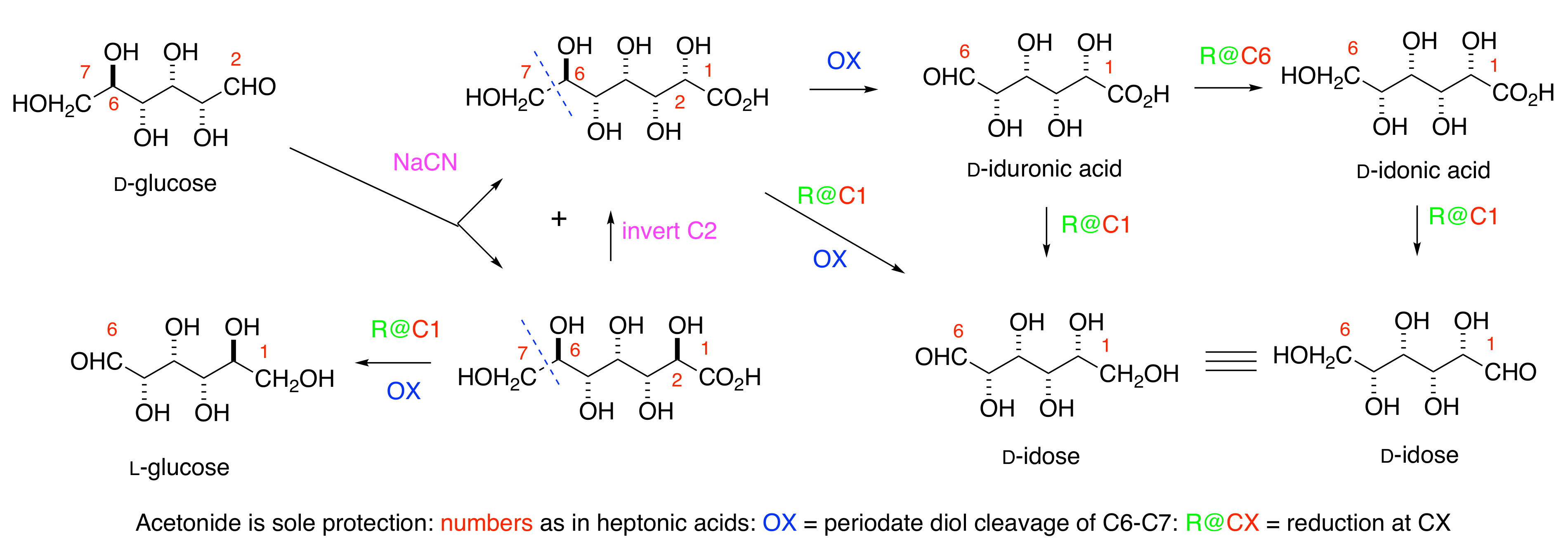
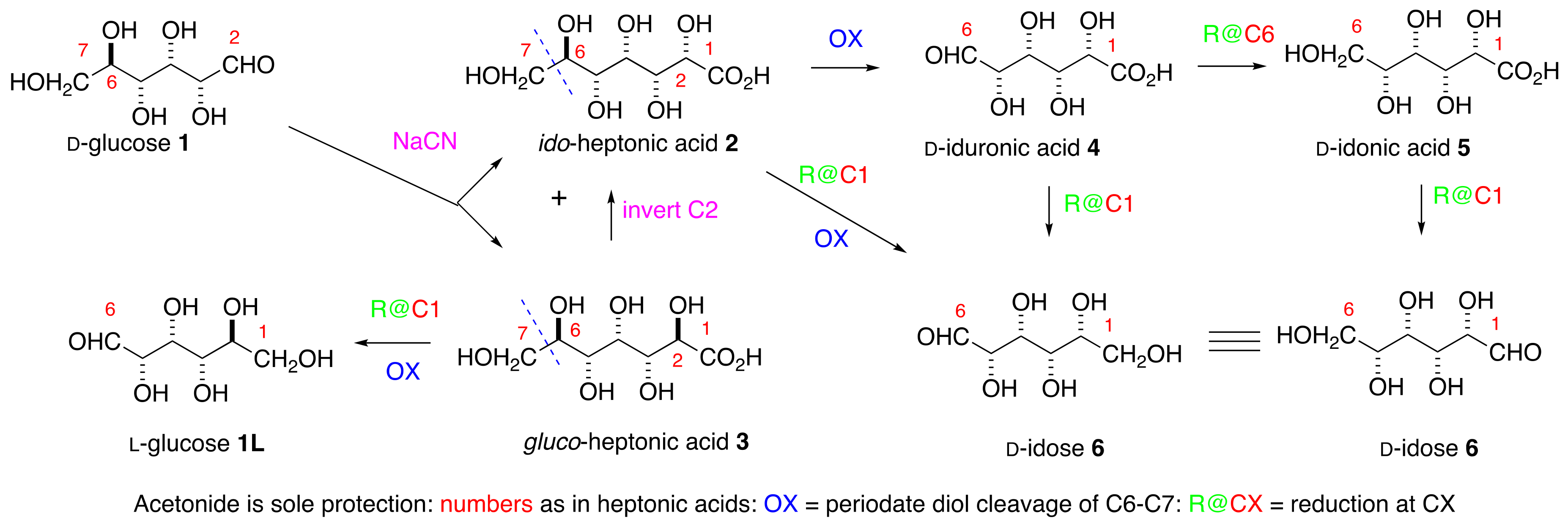
d-idose under very mild conditions, would simplify both the investigation of its biological properties and its incorporation into oligosaccharides and aglycone pharmacophores. The Kiliani reaction [
9] of
d-glucose
1 with sodium cyanide, to give the sodium salts of the epimeric
d-
ido (
2) and
l-
gluco- (
3) heptonic acids in a ratio of approximately 1:4, has been a massive industrial process for many decades (
Scheme 1) [
10].
Salts of the
gluco-acid
3, available cheaply [
11] in >99% purity, are used as chelating agents, detergents, and for many other purposes. The mother liquors after the first crystallization contain more of the
ido-acid
2 mixtures, such as Seqlene-50 [
12] consisting of a 1:1 ratio of
2 and
3 at 50% concentration, are very cheap industrial cleaners.
Acetonation is the only protection needed for the synthesis of
d-idose
6,
d-iduronic
4, and
d-idonic
5 acids. The key steps include (i) the Shing [
13] periodate cleavage of the C6-C7 bond in heptonic acids
2 and
3, and the (ii) adjustment of the oxidation levels at C1 and C6 in the heptonic acid [
Scheme 1]. The C
2 symmetry of the
ido-motif means that the aldehyde in
d-idose may derived from either C1 or C6 of the
ido-heptonic acid
2. Efficient inversion at C2 of
gluco-acid
3 allows the unambiguous synthesis of the
d-
ido targets in the paper without any separation of diastereomers. Preliminary studies of the use of Seqlene to access acetonides of
2 are reported.
A similar strategy has been used for the scalable synthesis of
l-glucose
1L from
gluco-acid
3 with no column chromatography [
14]. Treatment of the sodium salt of
3 with 2,2-dimethoxypropane in methanolic HCl formed the triacetonide
8 (54%) (
Scheme 2); selective hydrolysis of
8 gave diol
7 (86%) and, in four steps,
l-glucose
1L in 80% yield. The triacetonide
8 was also efficiently converted to many other rare sugars with differing oxidation levels at C1 and C6 [
15].
3. Experimental
3.1. General Experimental
All commercial reagents were used as supplied. Solvents were used as supplied (analytical or HPLC grade), without prior purification. Thin-layer chromatography (TLC) was performed on aluminium sheets coated with 60 F
254 silica. Plates were visualized using a 0.2%
w/
v cerium (IV) sulfate and 5% ammonium molybdate solution in 2 M sulfuric acid. Melting points were recorded on a Kofler hot block and are uncorrected. Optical rotations of the protected sugars were recorded on a Perkin-Elmer 241 polarimeter with a path length of 1 dm; concentrations are quoted in g 100 mL
−1. Optical rotations were recorded on a Jasco R1030 polarimeter, Na
+ lamp, (Jasco, Tokyo, Japan) at 20 °C, polarimeter with a path length of 1 dm. Infrared spectra were recorded on a Perkin-Elmer 1750 IR Fourier transform spectrophotometer using thin films on a diamond ATR surface (thin film). Only the characteristic peaks are quoted. Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker AMX 500 (
1H: 500 MHz and
13C: 125.7 MHz) or Bruker AVIII 400 HD nanobay and Bruker DQX 400 (
1H: 400 MHz,
13C: 100.6 MHz and
19F: 375 MHz) or Bruker DPX 250 (
1H: 250 MHz and
13C: 62.5 MHz) or Varian Mercury 300 (
1H: 300 MHz,
13C: 75 MHz) spectrometers in the deuterated solvent stated.
1H and
13C NMR spectra were assigned by utilizing 2D COSY, HSQC and HMBC spectra. All chemical shifts (δ) are quoted in ppm and coupling constants (
J) in Hz. Residual signals from the solvents were used as an internal reference. For solutions in D
2O acetonitrile was used as an internal reference. HRMS measurements were made using a micro-time-of-flight (TOF) mass analyzer using electrospray ionization (ESI) or an HP 5988A mass spectrometer using chemical ionization (CI). The purity of
d-idose by high-performance liquid chromatography (Hitachi GL-611 column, Tokyo, Japan, and Shimadzu RID-6A refractive index detector, Kyoto, Japan) at 60 °C, eluted with 10
−4 M NaOH at a flow rate of 1.0 mL/min. Seqlene-50 was obtained from Hallstar Engineering of Chicago, IL,USA (see
Supplementary Materials for
1H and
13C NMR spectra).
3.2. Methyl 2,3:4,5:6,7-Tri-O-Isopropylidene-d-glycero-d-ido-Heptonate 11
Method 1 from Ido-Heptono-1,4-Lactone 10
A methanolic solution of hydrogen chloride [prepared by dropwise addition of acetyl chloride (0.8 mL, 11.2 mmol) to methanol (6.6 mL) under argon at 0 °C] was added to a solution of the ido-diacetonide 10 (3.45 g, 12.0 mmol) in 2,2-dimethoxypropane (50 mL). The reaction mixture was then refluxed for 2 h when TLC (cyclohexane/ethyl acetate, 1:1) showed the formation of a major product (Rf 0.70). Sodium carbonate (5 g) was added into reaction mixture to neutralize the pH to 7 (the color of reaction mixture turned from brown to light yellow). After the solids were removed by filtration, the solvent was removed in vacuo to give a residue that was dissolved into cyclohexane (50 mL). The solution was washed with distilled water (3 × 50 mL), dried (MgSO4) and solvent was removed in vacuo to yield pure triacetonide 11 (3.10 g, 8.6 mmol, yield 71%). Further extraction of the aqueous layer by ethyl acetate (3 × 50 mL) gave partially acetonated products; removal of solvent in vacuo, afforded a residue (~2.0 g), which was dissolved into acetone (20 mL), and the procedures above were repeated to obtain more 11 (4.10 g in total, 95%) as a syrup, which used in the next step without further purification.
Method 2 from Seqlene without Recrystallization
A solution of Seqlene (2 mL) was fully dried in vacuo to give a dark brown residue (~1.3 g). A methanolic solution of hydrogen chloride (prepared by dropwise addition of acetyl chloride (0.4 mL, 5.6 mmol) to methanol (3.3 mL) under argon at 0 °C) was added to a solution of the residue in 2,2-dimethoxypropane (25 mL). The reaction mixture was then refluxed for 5 h when TLC (cyclohexane/ethyl acetate, 1:1) showed the formation of a major product (Rf 0.70). Sodium carbonate (2 g) was added to the reaction mixture to neutralize the pH to 7 (the color of reaction mixture turned from brown to light yellow). After the solids were removed by filtration, the solvent was removed in vacuo to give a residue that was dissolved into cyclohexane (20 mL). The solution was washed with distilled water (3 × 10 mL), dried (MgSO4) and solvent was removed in vacuo to yield a mixture of 8 and 11 (680 mg in total, 36%) as a NMR ratio of 1:1 (some impurities were also detected).
Method 3 (from Seqlene with Recrystallization)
A solution of Seqlene (10 mL, ~7.2 g solid) was diluted with water (60 mL) and passed through a column containing Amberlite IR-120 (H+) cation-exchange resin column (~40 mL). The eluent was stirred at 100 °C for 5 h with cadmium carbonate (2.7 g). Then, cadmium chloride (2.9 g) was added. After filtration by active carbon, the solution was concentrated in vacuo to half of the volume. Ethanol (~40 mL) was added until the solution became muddy. The solution was left to recrystallize at rt overnight to obtain a cadmium salt (1.5 g) of lactones that was subjected to the protection conditions as shown above to form a mixture of triacetonide 8 and 11 (2.0 g) as a ratio of 3:10 according to 1H NMR.
HRMS m/z (ESI + ve): found 383.1673 [M + Na]+, C17H28O8Na+ requires 383.1676; [α]D20 + 18.9 (c 1.25, MeOH); νmax (thin film): 1764 (s, C=O); δH (CD3OD, 400MHz): 1.22 (3H, s, CH3), 1.28 (3H, s, CH3), 1.29 (6H, s, 2 x CH3), 1.35 (3H, s, CH3), 1.36 (3H, s, CH3), 3.69 (3H, s, OCH3), 3.82–3.85 (1H, m, H7), 3.92–3.94 (1H, m, H5), 4.00 (1H, dd, H4, J4,3 2.1, J4,5 7.5), 4.01–4.04 (2H, m, H6, H7′), 4.17 (1H, dd, H3, J3,4 = 2.1, J3,2 7.8), 4.53 (1H, d, H2, J2,3 7.8), δC (CD3OD, 100MHz) 24.1 (CH3), 24.8 (CH3), 25.5 (CH3), 25.7 (CH3), 26.2 (CH3), 26.6 (CH3), 51.5 (OCH3), 67.1 (C7), 75.3 (C2), 77.0 (C5), 77.2 (C6), 77.9 (C4), 78.6 (C3), 109.5 (C(CH3)2), 109.6 (C(CH3)2), 111.5 (C(CH3)2), 171.3 (C1); m/z (ESI + ve): 383 ([M + Na]+, 100%).
3.3. Methyl 2,3:4,5-Di-O-Isopropylidene-d-glycero-d-ido-Heptonate 12
A solution of triacetonide 11 (4.10 g, 11.4 mmol) in acetic acid:water:methanol (15 mL, 2:1:3) was stirred at 40 °C for 5.5 h until TLC (ethyl acetate) showed the formation of one major product (Rf 0.60). The reaction mixture was concentrate in vacuo to ~2 mL and then stirred with NaHCO3 (sat. aq, 40 mL). Cyclohexane (3 × 40 mL) was used to extract the unreacted starting material 11 (~2.2 g) on which the process was repeated. The aqueous layer was then washed with dichloromethane (3 × 40 mL), the combined extracts dried (MgSO4), and the solvent removed in vacuo to obtain 12 (1.20 g, 32%) as a clear oil. The unreacted 11 from cyclohexane was recycled by the hydrolysis protocol to obtain more 12 (2.21 g, 61% based on recovered 12).
HRMS m/z (ESI + ve): found 343.1360 [M + Na]+, C14H24O8Na+ requires 343.1363; [α]D20 + 63 (c 0.46, MeOH); νmax (thin film): 3469 (br, OH), 1760 (s, C=O); δH (CDCl3, 400MHz) 1.44 (3H, s, CH3), 1.45 (3H, s, CH3), 1.47 (3H, s, CH3), 1.51 (3H, s, CH3), 2.42 (1H, t, OH7, JOH,H7 = JOH,H7′ 5.5), 2.98 (1H, d, OH6, JOH, H6 5.3), 3.72–3.79 (2H, m, H6, H7), 3.83 (3H, s, OCH3), 3.84–3.87 (1H, m, H7′), 4.12 (1H, t, H5, J5,6 = J5,4 7.3), 4.26 (1H, dd, H4, J4,3 3.1, J4,5 7.3), 4.33 (1H, dd, H3, J3,4 3.1, J3,2 7.6), 4.70 (1H, d, H2, J2,3 7.6); δC (CDCl3, 100MHz) 26.0 (CH3), 26.6 (CH3), 26.7 (CH3), 27.4 (CH3), 52.6 (OCH3), 63.9 (C7), 74.7 (C4), 73.0 (C6), 75.5 (C2), 76.8 (C5), 78.4, 78.6 (C3 and C4), 110.0 (C(CH3)2), 111.6 (C(CH3)2), 171.4 (C1); m/z (ESI + ve): 343 ([M + Na]+, 100%).
3.4. 2,3:4,5-Di-O-Isopropylidene-d-glycero-d-ido-Heptitol 13
Sodium borohydride (334 mg, 8.80 mmol) was added into a solution of 12 (1.41 g, 4.40 mmol) in methanol (20 mL) at 0 °C, and the solution was stirred for 3 h at rt until TLC (ethyl acetate) showed the consumption of starting material (Rf 0.57) and the formation of a new product (Rf 0.37). Acetic acid (~0.5 mL) was added into the solution to adjust the pH to 7, and the solvent was removed in vacuo to obtain a crude product which was further purified by flash column chromatography (ethyl acetate/methanol, 50:1) to obtain the triol 13 as a white solid (1.20 g, 95%).
HRMS m/z (ESI + ve): found 315.1415 [M + Na]+, C13H24O7Na+ requires 315.1414; m.p. 92 °C–94 °C; [α]D20 + 64 (c, 0.68 in MeOH); νmax (thin film): 3389 (broad, OH); δH (CD3OD, 400MHz) 1.38 (3H, s, CH3), 1.40 (9H, s, 3 x CH3), 3.55 (1H, dd, H7, J7,6 6.3, Jgem 11.0), 3.60–3.53 (1H, m, H6), 3.65 (1H, dd, H1, J1,2 5.2, Jgem 11.9), 3.71 (1H, dd, H1′, J1′,2 4.0, Jgem 11.9), 3.73 (1H, dd, H7′, J7′,6 2.9, Jgem 11.0), 3.99 (1H, dd, H3, J3,4 1.5, J3,2 8.5), 4.03–4.05 (2H, m, H4, H5), 4.18 (1H, ddd, H2, J2,1′ 4.1, J2,1 5.2, J2,3 8.5); δC (CD3OD, 100MHz) 25.8 (2 x CH3), 26.2 (CH3), 26.3 (CH3), 61.7 (C1), 63.6 (C7), 73.8 (C6), 76.6, 78.5 (C4, C5), 77.4 (C3), 77.9 (C2), 108.9 (C(CH3)3), 109.2 (C(CH3)2); m/z (ESI + ve): 315 ([M + Na]+, 100%).
3.5. Methyl 2,3:4,5-Di-O-Isopropylidene-d-glycero-d-Iduronate 15 and Methyl 2,3:4,5-Di-O-Isopropylidene-d-glycero-d-Idonate 16
Silica gel-supported NaIO4 (4.50 g) was added portionwise to a vigorously stirred solution of 12 (791 mg, 2.47 mmol) in dichloromethane (30 mL). After 1 h, TLC analysis (cyclohexane/ethyl acetate, 1:1) showed no remaining starting material (Rf 0.19) and formation of a single product (Rf 0.70). The mixture was filtered and the silica gel was thoroughly washed with CH2Cl2 (4 × 30 mL). The solvents were removed in vacuo to afford the crude aldehyde 15 (630 mg, 89%). Then, sodium borohydride (20.9 mg, 1.36 mmol) was added to a solution of the crude aldehyde 15 in methanol (5 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 1 h until TLC (cyclohexane/ethyl acetate, 1:1) indicated the formation of one major product (Rf 0.53) and a minor product (0.30). After acetic acid (~0.2 mL) was added into the reaction mixture to adjust pH to 7, ethyl acetate (3 × 10 mL) was used to extract the product and organic layer was dried (MgSO4), filtered and the solvent was removed to obtain a residue which was further purified by flash column chromatography (cyclohexane/ethyl acetate, 1:3) to obtain the major product 16 as a clear oil (410 mg, 57% 2 steps).
HRMS m/z (ESI + ve): found 343.1362 [M + Na]+, C14H24O8Na+ requires 343.1363; [α]D20 + 33 (c 1.02, CHCl3); νmax (thin film): 3495 (br, OH), 1759 (s, C=O); δH (CDCl3, 400MHz) 1.46 (3H, s, CH3), 1.47 (3H, s, CH3), 1.48 (3H, s, CH3), 1.50 (3H, s, CH3), 2.11 (1H, br-dd, OH6, JOH,H6′ 5.0, JOH,6 7.0), 3.71 (1H, ddd, H6, J6,5 4.0, J6,OH 7.3, Jgem 12.0), 3.82 (3H, s, OCH3), 3.88 (1H, dt, H6′, J6′,5 = J6′,OH 4.0, Jgem 12.0), 4.15 (1H, dd, H4, JH4,H3 3.2, JH4,H5 8.2), 4.21 (1H, dd, H3, J3,4 3.2, J3,2 7.5), 4.24 (1H, q, H5, J5,6 = J5,6′ 4.0, J5.4 8.2), 4.62 (1H, d, H2, J2,3 7.5), δC (CDCl3, 100MHz) 26.0 (CH3), 26.5 (CH3), 26.7 (CH3), 27.3 (CH3), 52.6 (OCH3), 61.7 (C6), 75.5 (C2), 76.2 (C4), 77.3, 77.7 (C3 and C5), 109.8 (C(CH3)2), 111.7 (C(CH3)2), 171.0 (C1); m/z (ESI + ve): 343 ([M + Na]+, 100%).
3.6. d-Idose 6
3.6.1. Method 1 (from 13)
Silica gel-supported NaIO4 (4.50 g) was added portionwise to a vigorously stirred solution of the triol 13 (668 mg, 2.29 mmol) in dichloromethane (30 mL). After 2 h, TLC analysis (ethyl acetate) showed no remaining starting material (Rf 0.31) and formation of one elongated spot (Rf 0.56–0.65). The reaction mixture was filtered and the silica gel was thoroughly washed with dichloromethane (4 × 30 mL). The solvents were removed in vacuo to afford the crude aldehyde 14 (600 mg, 100%), which was dissolved in water (20 mL) and treated with DOWEX® 50WX8-200 (~400 mg, prewashed with water). After stirring at rt for 24 h, TLC analysis (ethyl acetate) showed no remaining starting material and formation of a single product (baseline). The resin was filtered and washed with water. Removal of water in vacuo afforded D-idose 6 (400 mg, 97% from 13; 53% from 11) as a colorless syrup.
[α]
D20 + 10.7 (
c 0.55, water) (authentic sample: [α]
D20 + 10.0 (
c 0.80, water), [
1] [α]
D17 + 13.7 (
c 2.47, water)],
1H, and
13C NMR are identical with those of authentic sample; HPLC showed purity 85%; After HPLC purification, purity reached to 100%;
m/
z (ESI + ve): 203 ([M + Na]
+, 100%) HRMS
m/
z (ESI + ve): found 203.0524 ([M + Na]
+), C
6H
12O
6Na
+ requires 203.0526.
3.6.2. Method 2 (from 16)
Diisobutylaluminum hydride (1.0 M in toluene, 4.05 mL, 4.05 mmol) was added dropwise to a solution of 16 (391 mg, 1.35 mmol) in dichloromethane (5 mL) at −78 °C. The reaction mixture was stirred at −78 °C for 2 h until TLC analysis (ethyl acetate) showed no remaining starting material (Rf 0.31) and the formation of one spot (Rf 0.56–0.65). Mass spectrometry also showed the formation of desired product peak ([M + MeOH + Na]+ 315) and disappearance of starting material peak ([M + Na]+ 313). The mixture was diluted with ethyl acetate (10 mL) and potassium sodium tartrate (sat., aq., 2 mL) was added. After stirring for 8 h, the reaction mixture was diluted with water (10 mL) and extracted with ethyl acetate (3 × 10 mL). The organic phase was dried (MgSO4) and filtered; then, the solvent was removed in vacuo to obtain an oil that was further purified by flash chromatography (cyclohexane/ethyl acetate 7:1 to 3:1) to yield crude aldehyde 14 as a syrup (267 mg, 76%). The crude aldehyde 14 was dissolved in water (20 mL) was treated with DOWEX® 50WX8-200 (~300 mg, prewashed with water). After 24 h, TLC analysis (ethyl acetate) showed no remaining starting material and formation of a single product (baseline). The resin was filtered and washed with water. Removal of water in vacuo afforded d-idose 6 (185 mg, 76% from 16; 21% from 11) as a colorless syrup.
[α]D20 + 11.7 (c 0.65, water, eq), 1H and 13C NMR were identical to those produced by Method 1 above.



{kind=link}
{kind=link}
{kind=link}
{kind=link}


