Comparative Proteomic Analysis of Molecular Differences between Leaves of Wild-Type Upland Cotton and Its Fuzzless-Lintless Mutant


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
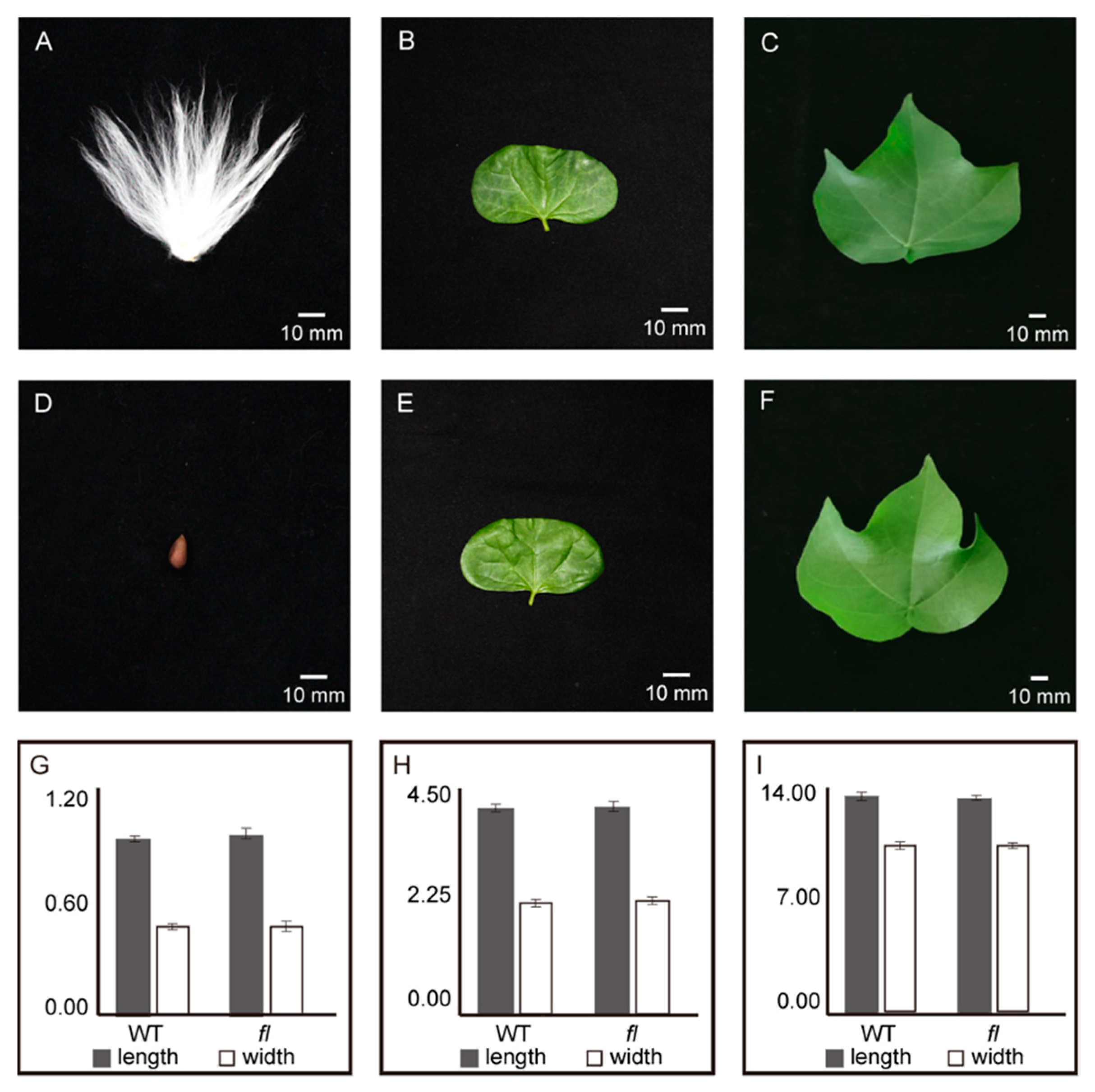
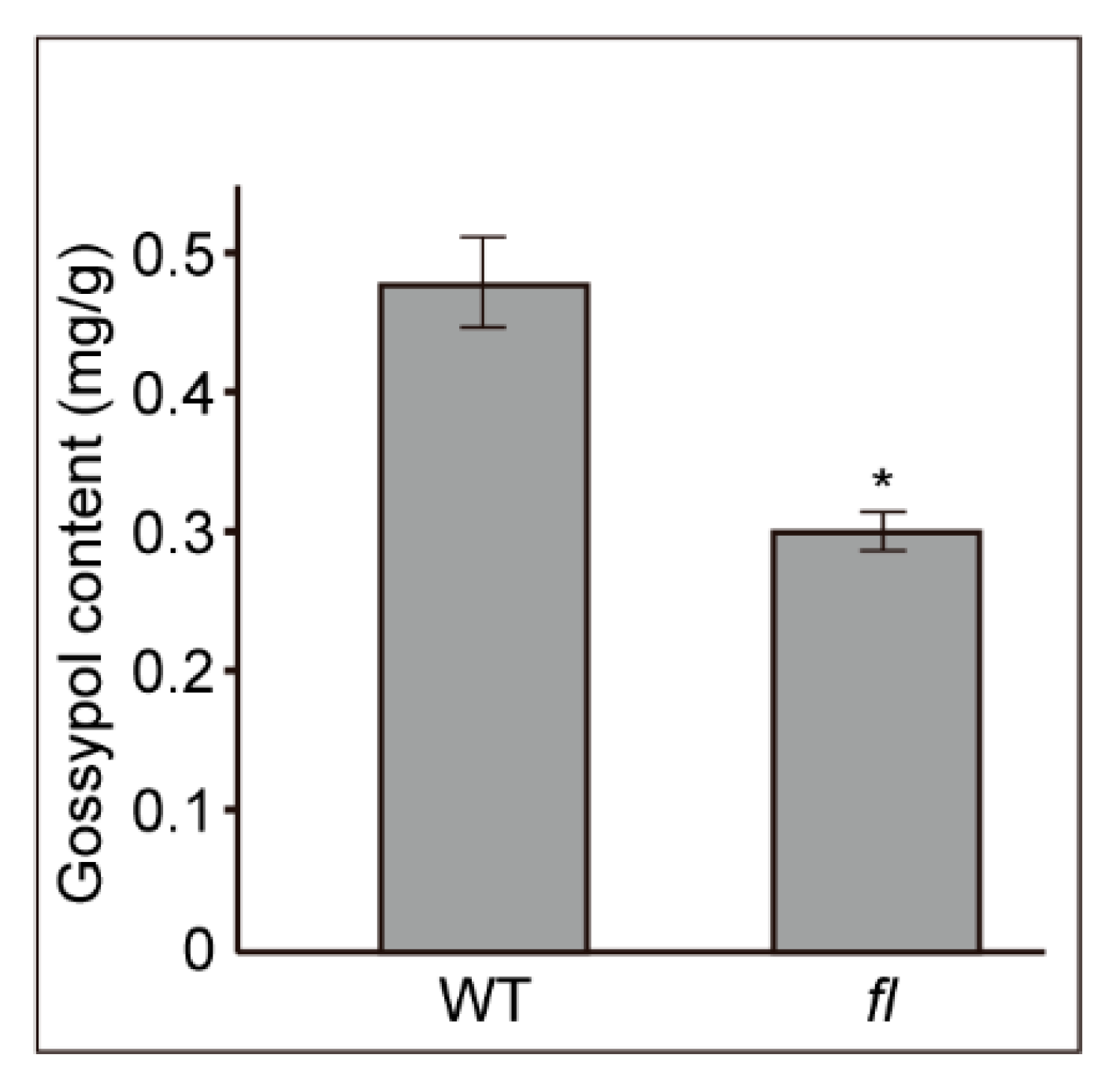
2.1. Phenotypes and Gossypol Contents of Leaves from Xu142 WT and fl Mutant
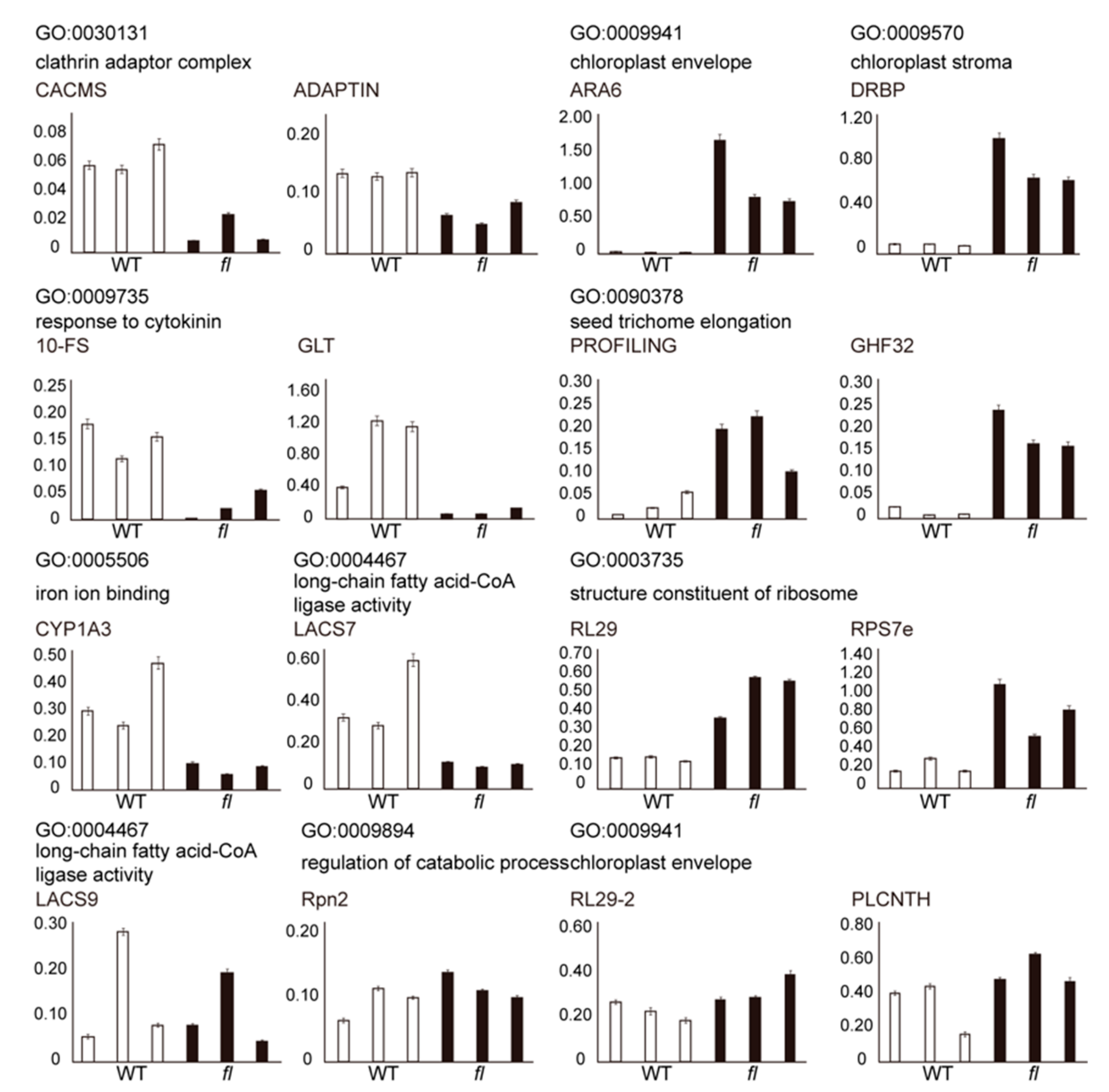
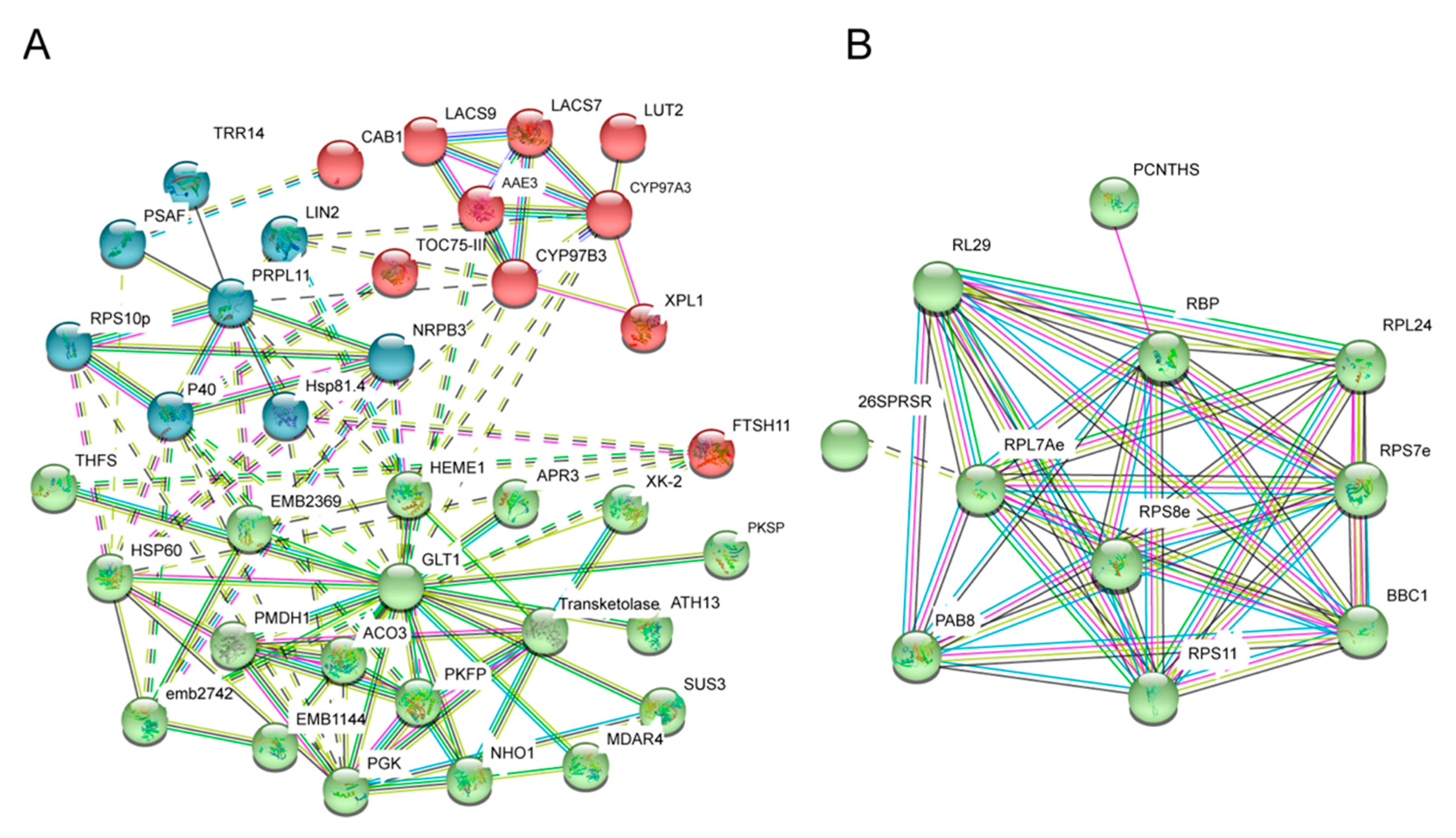
2.2. Identification and Validation of WT- and fl-Specific Proteins
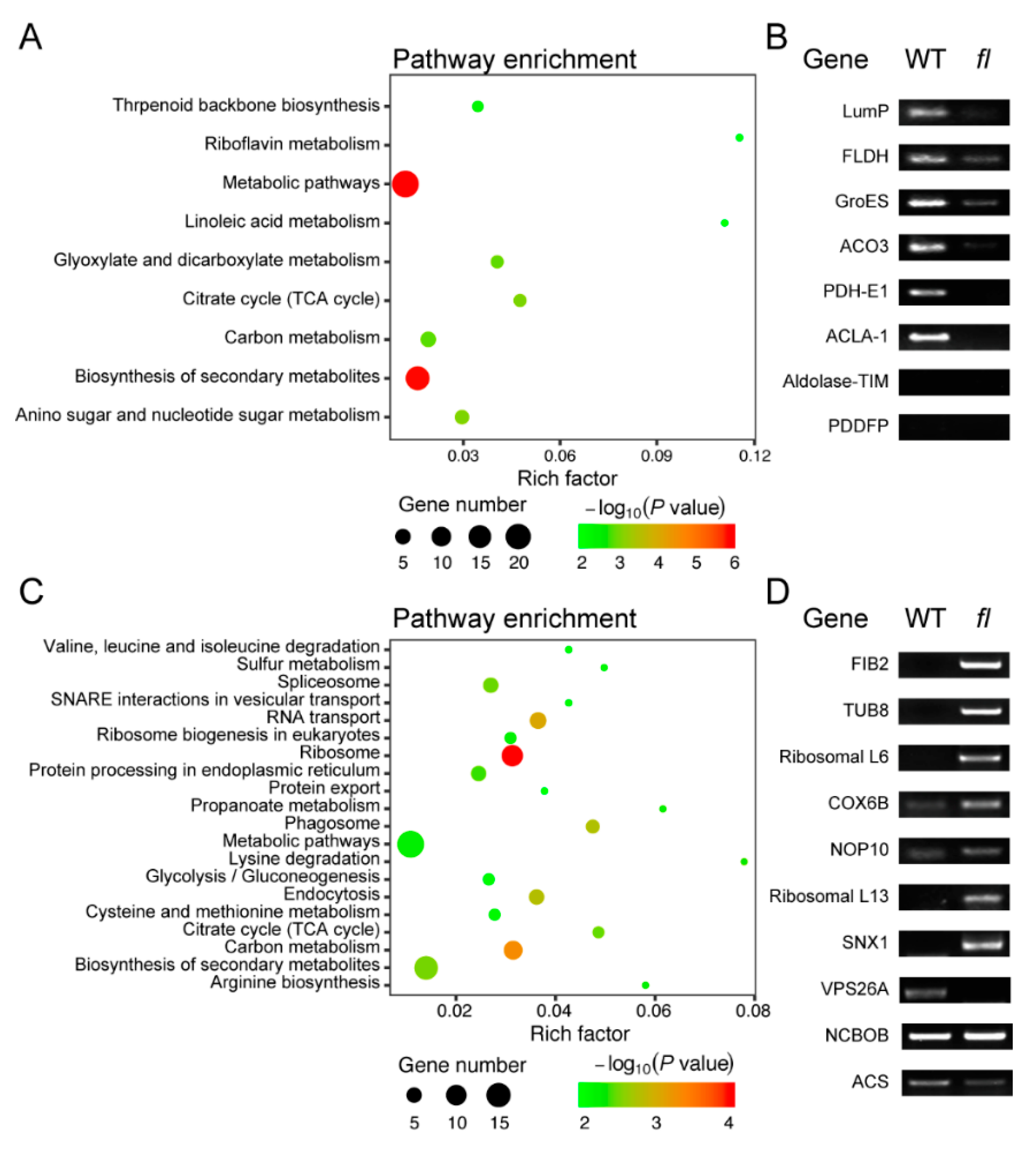
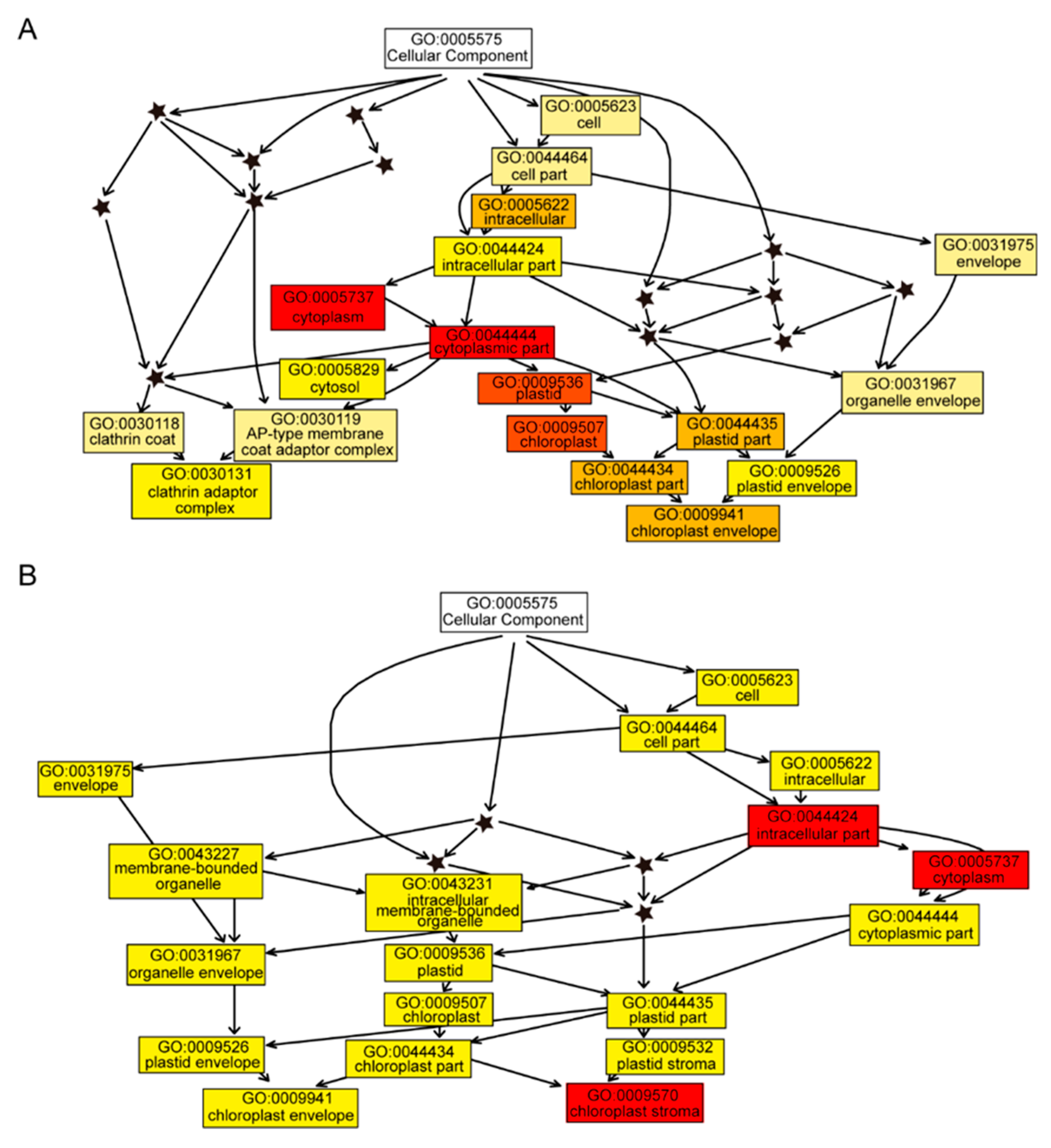
2.3. Analysis and Validation of Differential Abundant Proteins (DAPs)
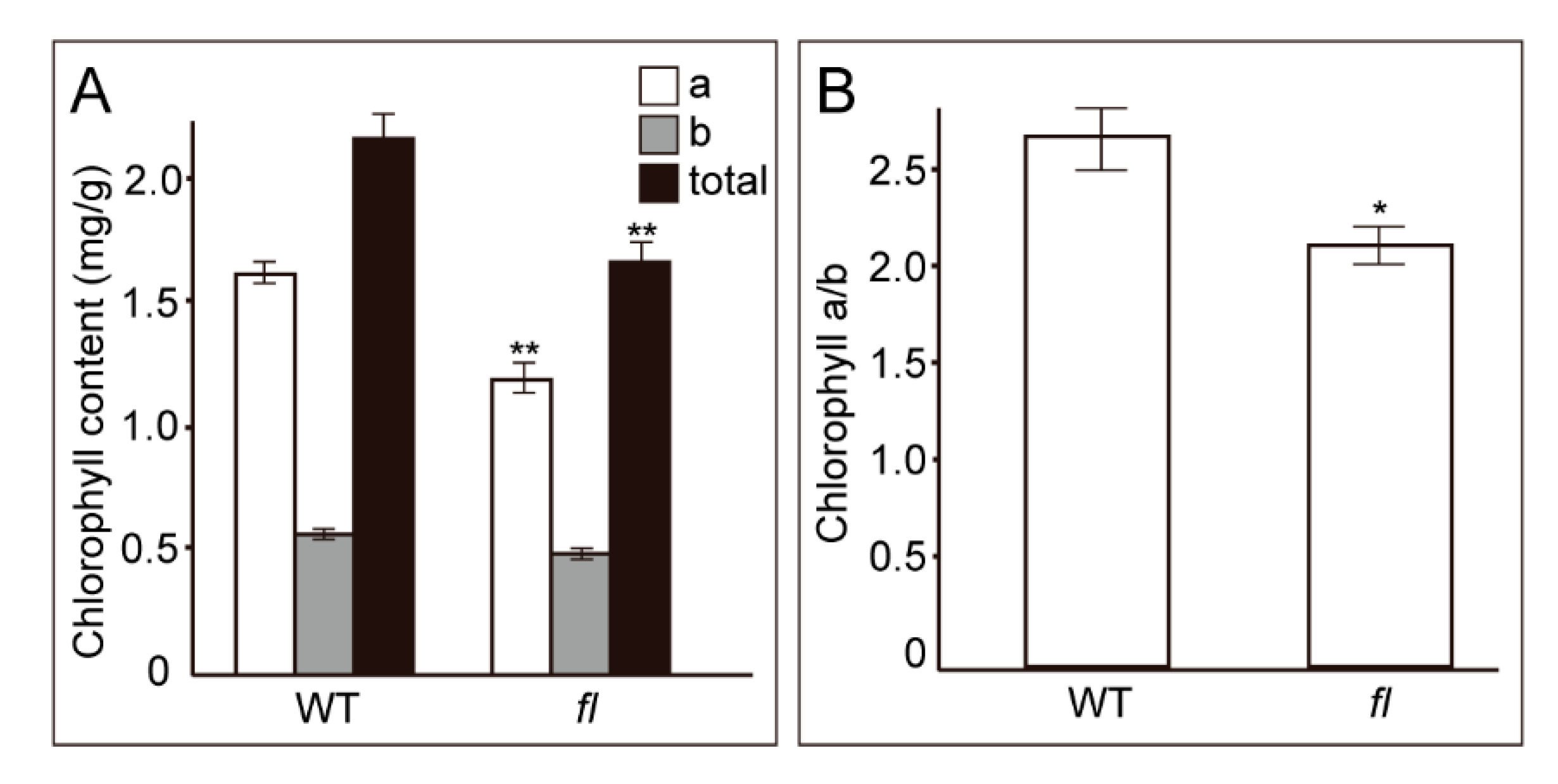
2.4. Determination of Chlorophyll Contents
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Morphological Observation
4.2. Gossypol Contents Determination
4.3. Protein Extraction
4.4. Protein Identification and Bioinformatic Analysis
4.5. RNA Extraction and PCR/qPCR Validation
4.6. Chlorophyll Contents Measurement
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Qin, Y.M.; Zhu, Y.X. How cotton fibers elongate: A tale of linear cell-growth mode. Curr. Opin. Plant Biol. 2011, 14, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Woodward, A.W.; Chen, Z.J. Gene expression changes and early events in cotton fibre development. Ann. Bot. 2007, 100, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Arpat, A.B.; Waugh, M.; Sullivan, J.P.; Gonzales, M.; Frisch, D.; Main, D.; Wood, T.; Leslie, A.; Wing, R.A.; Wilkins, T.A. Functional genomics of cell elongation in developing cotton fibers. Plant Mol. Biol. 2004, 54, 911–929. [Google Scholar] [CrossRef]
- Kim, H.J.; Triplett, B.A. Cotton fiber growth in planta and in vitro. Models for plant cell elongation and cell wall biogenesis. Plant Physiol. 2001, 127, 1361–1366. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.L.; Chourey, P.S. A fiberless seed mutation in cotton is associated with lack of fiber cell initiation in ovule epidermis and alterations in sucrose synthase expression and carbon partitioning in developing seeds. Plant Physiol. 1998, 118, 399–406. [Google Scholar] [CrossRef]
- Thyssen, G.N.; Fang, D.D.; Turley, R.B.; Florane, C.B.; Li, P.; Mattison, C.P.; Naoumkina, M. A Gly65Val substitution in an actin, GhACT_LI1, disrupts cell polarity and F-actin organization resulting in dwarf, lintless cotton plants. Plant J. 2017, 90, 111–121. [Google Scholar] [CrossRef]
- Han, X.; Xu, X.; Fang, D.D.; Zhang, T.; Guo, W. Cloning and expression analysis of novel Aux/IAA family genes in Gossypium hirsutum. Gene 2012, 503, 83–91. [Google Scholar] [CrossRef]
- Wan, Q.; Zhang, H.; Ye, W.; Wu, H.; Zhang, T. Genome-wide transcriptome profiling revealed cotton fuzz fiber development having a similar molecular model as Arabidopsis trichome. PLoS ONE 2014, 9, e97313. [Google Scholar] [CrossRef]
- Padmalatha, K.V.; Patil, D.P.; Kumar, K.; Dhandapani, G.; Kanakachari, M.; Phanindra, M.L.; Kumar, S.; Mohan, T.C.; Jain, N.; Prakash, A.H.; et al. Functional genomics of fuzzless-lintless mutant of Gossypium hirsutum L. cv. MCU5 reveal key genes and pathways involved in cotton fibre initiation and elongation. BMC Genom. 2012, 13, 624. [Google Scholar] [CrossRef]
- Kwak, P.; Wang, Q.; Chen, X.; Qiu, C.; Yang, Z. Enrichment of a set of microRNAs during the cotton fiber development. BMC Genom. 2009, 10, 457. [Google Scholar] [CrossRef]
- Wang, M.; Yuan, D.; Tu, L.; Gao, W.; He, Y.; Hu, H.; Wang, P.; Liu, N.; Lindsey, K.; Zhang, X. Long noncoding RNAs and their proposed functions in fibre development of cotton (Gossypium spp.). New Phytol. 2015, 207, 1181–1197. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Li, C.; Zhang, J.; Li, F.; Ma, L.; Tan, Y.; Wang, Q.; Zhang, B. Differential expression of microRNAs during fiber development between fuzzless-lintless mutant and its wild-type allotetraploid cotton. Sci. Rep. 2017, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.Z.; Pan, J.J. Genetic analysis of a fuzzless-lintless mutant in Gossypium hirsutum L. Jiangsu J. Agric. Sci. 1991, 7, 13–16. [Google Scholar]
- Yu, X.; Zhu, Y.; Lu, S.; Zhang, T.; Chen, X.; Xu, Z. A comparative analysis of a fuzzless-lintless mutant of Gossypium hirsutum L. cv. Xu-142. Sci. China C Life Sci. 2000, 43, 623–630. [Google Scholar] [CrossRef]
- Shi, Y.H.; Zhu, S.W.; Mao, X.Z.; Feng, J.X.; Qin, Y.M.; Zhang, L.; Cheng, J.; Wei, L.P.; Wang, Z.Y.; Zhu, Y.X. Transcriptome profiling, molecular biological, and physiological studies reveal a major role for ethylene in cotton fiber cell elongation. Plant Cell 2006, 18, 651–664. [Google Scholar] [CrossRef]
- Pang, C.Y.; Wang, H.; Pang, Y.; Xu, C.; Jiao, Y.; Qin, Y.M.; Western, T.L.; Yu, S.X.; Zhu, Y.X. Comparative proteomics indicates that biosynthesis of pectic precursors is important for cotton fiber and Arabidopsis root hair elongation. Mol. Cell Proteom. 2010, 9, 2019–2033. [Google Scholar] [CrossRef]
- Li, H.B.; Qin, Y.M.; Pang, Y.; Song, W.Q.; Mei, W.Q.; Zhu, Y.X. A cotton ascorbate peroxidase is involved in hydrogen peroxide homeostasis during fibre cell development. New Phytol. 2007, 175, 462–471. [Google Scholar] [CrossRef]
- Tao, C.; Jin, X.; Zhu, L.; Li, H. Two-dimensional gel electrophoresis-based proteomic analysis reveals N-terminal truncation of the Hsc70 protein in cotton fibers in vivo. Sci. Rep. 2016, 6, 36961. [Google Scholar] [CrossRef]
- Liu, G.J.; Xiao, G.H.; Liu, N.J.; Liu, D.; Chen, P.S.; Qin, Y.M.; Zhu, Y.X. Targeted lipidomics studies reveal that linolenic acid promotes cotton fiber elongation by activating phosphatidylinositol and phosphatidylinositol monophosphate biosynthesis. Mol. Plant 2015, 8, 911–921. [Google Scholar] [CrossRef]
- Tang, W.; Tu, L.; Yang, X.; Tan, J.; Deng, F.; Hao, J.; Guo, K.; Lindsey, K.; Zhang, X. The calcium sensor GhCaM7 promotes cotton fiber elongation by modulating reactive oxygen species (ROS) production. New Phytol. 2014, 202, 509–520. [Google Scholar] [CrossRef]
- Liu, K.; Han, M.; Zhang, C.; Yao, L.; Sun, J.; Zhang, T. Comparative proteomic analysis reveals the mechanisms governing cotton fiber differentiation and initiation. J. Proteom. 2012, 75, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Wang, M.; Ding, Y.; Zhu, S.; Zhao, G.; Tu, L.; Zhang, X. Transcriptomic repertoires depict the initiation of lint and fuzz fibres in cotton (Gossypium hirsutum L.). Plant Biotechnol. J. 2018, 16, 1002–1012. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zhang, B.; Dong, C.J.; Du, Y.; Jiang, L.; Liu, J.Y. Comparative proteomic and biochemical analyses reveal different molecular events occurring in the process of fiber initiation between wild-type allotetraploid cotton and its fuzzless-lintless mutant. PLoS ONE 2015, 10, e0117049. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.M.; Wang, L.L.; Han, L.B.; Wang, J.; Yao, Y.; Wang, H.Y.; Du, X.M.; Luo, Y.M.; Xia, G.X. Proteomic identification of differentially expressed proteins in the Ligon lintless mutant of upland cotton (Gossypium hirsutum L.). J. Proteome Res. 2010, 9, 1076–1087. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, X.; Jin, X.; Jia, R.; Huang, Q.; Tan, Y.; Guo, A. Comparative proteomics of Bt-transgenic and non-transgenic cotton leaves. Proteome Sci. 2015, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Xu, F.; Sun, J.; Peng, Z.; He, S.; Pan, Z.; Du, X. iTRAQ-based comparative proteomic analysis of seedling leaves of two upland cotton genotypes differing in salt tolerance. Front. Plant Sci. 2017, 8, 2113. [Google Scholar] [CrossRef] [PubMed]
- Daud, M.K.; Quiling, H.; Lei, M.; Ali, B.; Zhu, S.J. Ultrastructural, metabolic and proteomic changes in leaves of upland cotton in response to cadmium stress. Chemosphere 2015, 120, 309–320. [Google Scholar] [CrossRef]
- Fan, S.; Meng, Y.; Song, M.; Pang, C.; Wei, H.; Liu, J.; Zhan, X.; Lan, J.; Feng, C.; Zhang, S.; et al. Quantitative phosphoproteomics analysis of nitric oxide-responsive phosphoproteins in cotton leaf. PLoS ONE 2014, 9, e94261. [Google Scholar] [CrossRef]
- Zheng, X.; Fan, S.; Wei, H.; Tao, C.; Ma, Q.; Ma, Q.; Zhang, S.; Li, H.; Pang, C.; Yu, S. iTRAQ-based quantitative proteomic analysis reveals cold responsive proteins involved in leaf senescence in upland cotton (Gossypium hirsutum L.). Int. J. Mol. Sci. 2017, 18, 1984. [Google Scholar] [CrossRef]
- Liang, W.; Fang, L.; Xiang, D.; Hu, Y.; Feng, H.; Chang, L.; Zhang, T. Transcriptome analysis of short fiber mutant Ligon lintless-1 (Li1) reveals critical genes and key pathways in cotton fiber elongation and leaf development. PLoS ONE 2015, 10, e0143503. [Google Scholar] [CrossRef]
- Tian, X.; Ruan, J.X.; Huang, J.Q.; Yang, C.Q.; Fang, X.; Chen, Z.W.; Hong, H.; Wang, L.J.; Mao, Y.B.; Lu, S.; et al. Characterization of gossypol biosynthetic pathway. Proc. Natl. Acad. Sci. USA 2018, 115, 5410–5418. [Google Scholar] [CrossRef]
- Gao, W.; Long, L.; Zhu, L.F.; Xu, L.; Gao, W.H.; Sun, L.Q.; Liu, L.L.; Zhang, X.L. Proteomic and virus-induced gene silencing (VIGS) analyses reveal that gossypol, brassinosteroids, and jasmonic acid contribute to the resistance of cotton to Verticillium dahliae. Mol. Cell. Proteom. 2013, 12, 3690–3703. [Google Scholar] [CrossRef]
- Chen, M. Chlorophyll modifications and their spectral extension in oxygenic photosynthesis. Annu. Rev. Biochem. 2014, 83, 317–340. [Google Scholar] [CrossRef]
- Liu, J.; Meng, Y.; Lv, F.; Chen, J.; Ma, Y.; Wang, Y.; Chen, B.; Zhang, L.; Zhou, Z. Photosynthetic characteristics of the subtending leaf of cotton boll at different fruiting branch nodes and their relationships with lint yield and fiber quality. Front. Plant Sci. 2015, 6, 747. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Ni, Z.; Zhang, H.; Chen, Q.; Gao, W.; Cai, Y.; Li, M.; Sun, G.; Qu, Y. The gene encoding subunit A of the vacuolar H+-ATPase from cotton plays an important role in conferring tolerance to water deficit. Front. Plant Sci. 2018, 9, 758. [Google Scholar] [CrossRef]
- Jin, X.; Zhu, L.; Tao, C.; Xie, Q.; Xu, X.; Chang, L.; Tan, Y.; Ding, G.; Li, H.; Wang, X. An improved protein extraction method applied to cotton leaves is compatible with 2-DE and LC-MS. BMC Genom. 2019, 20, 285. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Jin, J.P.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.C.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, 1040–1045. [Google Scholar] [CrossRef]
- Wang, X.C.; Li, Q.; Jin, X.; Xiao, G.H.; Liu, G.J.; Liu, N.J.; Qin, Y.M. Quantitative proteomics and transcriptomics reveal key metabolic processes associated with cotton fiber initiation. J. Proteom. 2015, 114, 16–27. [Google Scholar] [CrossRef]
- Ma, D.; Hu, Y.; Yang, C.; Liu, B.; Fang, L.; Wan, Q.; Liang, W.; Mei, G.; Wang, L.; Wang, H.; et al. Genetic basis for glandular trichome formation in cotton. Nat. Commun. 2016, 7, 10456. [Google Scholar] [CrossRef]
- Wu, G. Amino acids: Metabolism, functions, and nutrition. Amino Acids 2009, 37, 1–17. [Google Scholar] [CrossRef]
- Liaqat, A.; Shahid, M.; Saeed, N.A. Present status and future prospects of mechanized production of oilseed crops in Pakistan—A review. Pak. J. Agric. Res. 2010, 23, 83–93. [Google Scholar]
- Ali, M.; Arifullah, S.; Memon, M.H. Edible oil deficit and its impact on food expenditure in Pakistan. Pak. Dev. Rev. 2008, 47, 531–546. [Google Scholar] [CrossRef]
- Ai, C.; Kong, L. CGPS: A machine learning-based approach integrating multiple gene set analysis tools for better prioritization of biologically relevant pathways. J. Genet. Genom. 2018, 45, 489–504. [Google Scholar] [CrossRef]
- Jin, X.; Zhu, L.; Yao, Q.; Meng, X.; Ding, G.; Wang, D.; Xie, Q.; Tong, Z.; Tao, C.; Yu, L.; et al. Expression profiling of mitogen-activated protein kinase genes reveals their evolutionary and functional diversity in different rubber tree (Hevea brasiliensis) cultivars. Genes 2017, 8, 261. [Google Scholar] [CrossRef]
- Purdy, S.J.; Cunniff, J.; Maddison, A.L.; Jones, L.E.; Barraclough, T.; Castle, M.; Davey, C.L.; Jones, C.M.; Shield, L.; Gallagher, J.; et al. Seasonal carbohydrate dynamics and climatic regulation of senescence in the perennial grass, miscanthus. BioEnergy Res. 2015, 8, 28–41. [Google Scholar] [CrossRef]
Sample Availability: Samples of the Xu142 and fuzzless-lintless mutant are available from the corresponding authors. |

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, L.; Zheng, B.; Song, W.; Tao, C.; Jin, X.; Li, H. Comparative Proteomic Analysis of Molecular Differences between Leaves of Wild-Type Upland Cotton and Its Fuzzless-Lintless Mutant. Molecules 2019, 24, 3769. https://doi.org/10.3390/molecules24203769
Zhu L, Zheng B, Song W, Tao C, Jin X, Li H. Comparative Proteomic Analysis of Molecular Differences between Leaves of Wild-Type Upland Cotton and Its Fuzzless-Lintless Mutant. Molecules. 2019; 24(20):3769. https://doi.org/10.3390/molecules24203769
Chicago/Turabian StyleZhu, Liping, Bowen Zheng, Wangyang Song, Chengcheng Tao, Xiang Jin, and Hongbin Li. 2019. "Comparative Proteomic Analysis of Molecular Differences between Leaves of Wild-Type Upland Cotton and Its Fuzzless-Lintless Mutant" Molecules 24, no. 20: 3769. https://doi.org/10.3390/molecules24203769