Aromatic vs. Aliphatic Hyperbranched Polyphosphoesters as Flame Retardants in Epoxy Resins

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Thermal Characterization of FRs
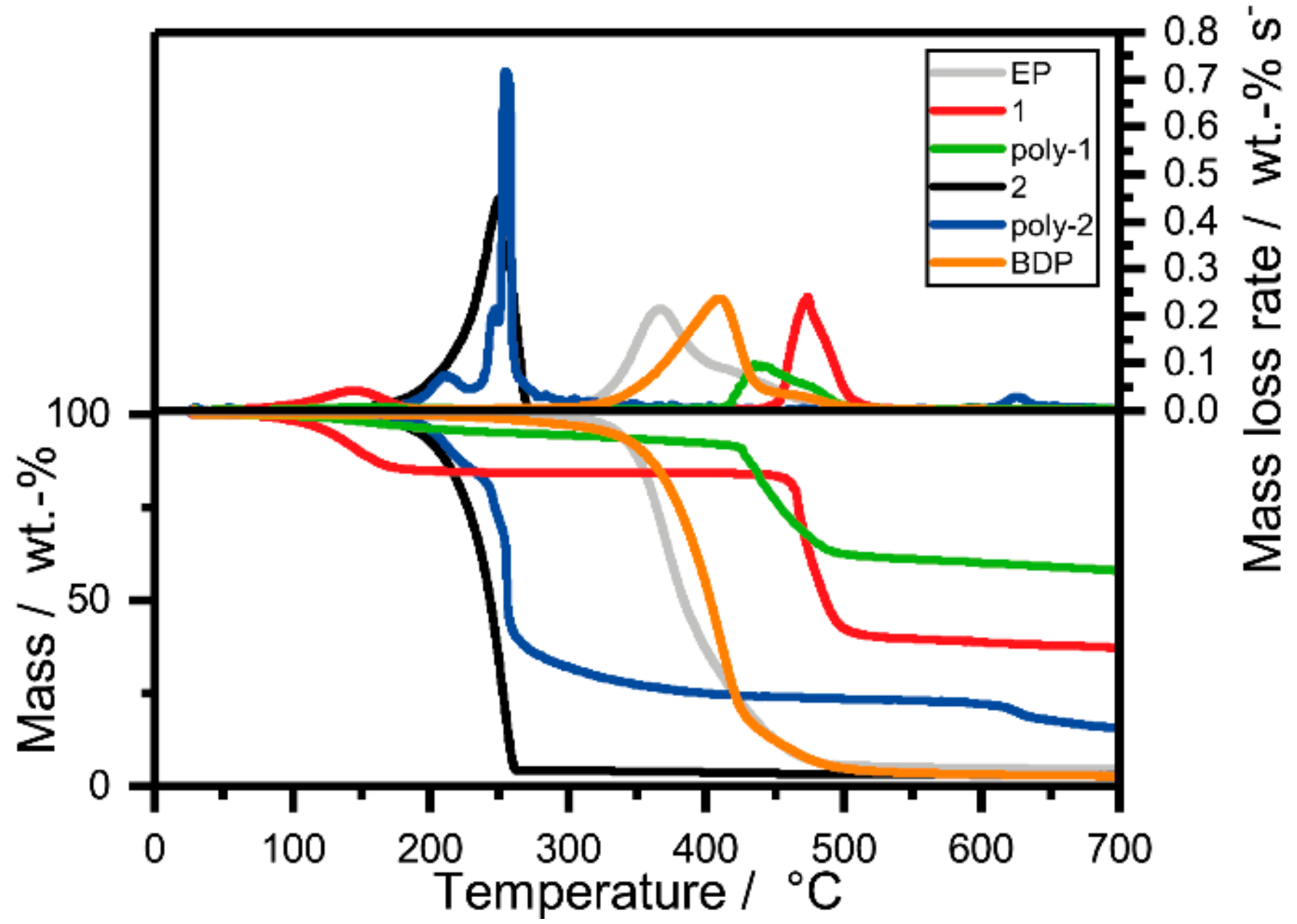
2.2. Pyrolysis: Thermal Decomposition via Thermogravimetric Analysis
2.3. Pyrolysis: Evolved Gas Analysis via TGA-FTIR
2.4. Thermal Characterization of FRs in Epoxy Resins
2.5. Pyrolysis: Evolved-Gas Analysis via TGA/TGA-FTIR
2.6. Pyrolysis: Condensed Phase Analysis via Hot-Stage FTIR
2.7. Fire Behavior: Cone Calorimeter
3. Materials and Methods
3.1. Materials
3.2. Instrumentation and Characterization Techniques
3.2.1. Size Exclusion Chromatography (SEC)
3.2.2. Nuclear Magnetic Resonance (NMR)
3.2.3. Electrospray Ionization Mass Spectrometry (ESI-MS)
3.2.4. Differential Scanning Calorimetry (DSC)
3.2.5. TGA-FTIR
3.2.6. Hot Stage FTIR
3.2.7. Cone Calorimeter
3.3. Synthetic Procedures
3.3.1. Synthesis of the 4-Vinylphenol
3.3.2. Synthesis of the Tris(p-vinylphenyl)phosphate (1)
3.3.3. ATMET Polymerization to Poly(1)
3.3.4. Tri(hex-5-en-1-yl)phosphate (2)
3.3.5. Poly(tri(hex-5-en-1-yl)phosphate) (poly-2) by ATMET Polymerization
3.3.6. Epoxy Resins
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alexander, B.; Jens, C.M.; Frederik, R.W.; Bernhard, S. Matrix matters: Hyperbranched flame retardants in aliphatic and aromatic epoxy resins. Polym. Degrad. Stab. 2019, 108986. [Google Scholar] [CrossRef]
- Perret, B.; Pawlowski, K.; Schartel, B. Fire retardancy mechanisms of arylphosphates in polycarbonate (PC) and PC/acrylonitrile-butadiene-styrene. J. Therm. Anal. Calorim. 2009, 97, 949–958. [Google Scholar] [CrossRef]
- Despinasse, M.-C.; Schartel, B. Influence of the structure of aryl phosphates on the flame retardancy of polycarbonate/acrylonitrile–butadiene–styrene. Polym. Degrad. Stab. 2012, 97, 2571–2580. [Google Scholar] [CrossRef]
- Schartel, B.; Perret, B.; Dittrich, B.; Ciesielski, M.; Krämer, J.; Müller, P.; Altstädt, V.; Zang, L.; Döring, M. Flame retardancy of polymers: The role of specific reactions in the condensed phase. Macromol. Mater. Eng. 2016, 301, 9–35. [Google Scholar] [CrossRef]
- Morgan, A.B.; Wilkie, C.A. Flame Retardant Polymer Nanocomposites; Wiley: Hoboken, NJ, USA, 2007. [Google Scholar]
- Müller, P.; Schartel, B. Melamine poly(metal phosphates) as flame retardant in epoxy resin: Performance, modes of action, and synergy. J. Appl. Polym. Sci. 2016, 133. [Google Scholar] [CrossRef]
- Velencoso, M.M.; Battig, A.; Markwart, J.C.; Schartel, B.; Wurm, F.R. Molecular firefighting-how modern phosphorus chemistry can help solve the challenge of flame retardancy. Angew. Chem. Int. Ed. Engl. 2018, 57, 10450–10467. [Google Scholar] [CrossRef]
- Schartel, B. Phosphorus-Based flame retardancy mechanisms—Old hat or a starting point for future development? Mater. 2010, 3, 4710–4745. [Google Scholar] [CrossRef]
- Lu, S.-Y.; Hamerton, I. Recent developments in the chemistry of halogen-free flame retardant polymers. Prog. Polym. Sci. 2002, 27, 1661–1712. [Google Scholar] [CrossRef]
- Bauer, K.N.; Liu, L.; Andrienko, D.; Wagner, M.; Macdonald, E.K.; Shaver, M.P.; Wurm, F.R. Polymerizing phostones: A fast way to in-chain poly(phosphonate)s with adjustable hydrophilicity. Macromol. 2018, 51, 1272–1279. [Google Scholar] [CrossRef]
- Steinbach, T.; Wurm, F.R. Poly(phosphoester)s: A new platform for degradable polymers. Angew. Chem. Int. Ed. 2015, 54, 6098–6108. [Google Scholar] [CrossRef]
- Monge, S.; Canniccioni, B.; Graillot, A.; Robin, J.-J. Phosphorus-containing polymers: A great opportunity for the biomedical field. Biomacromolecules 2011, 12, 1973–1982. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Liang, G.; Gu, A.; Zhang, Z.; Han, J.; Yuan, L. Novel phosphorus-containing hyperbranched polysiloxane and its high performance flame retardant cyanate ester resins. Polym. Degrad. Stab. 2013, 98, 597–608. [Google Scholar] [CrossRef]
- Kim, Y.H. Hyperbranched polymers 10 years after. J. Polym. Sci. Part. A Polym. Chem. 1998, 36, 1685–1698. [Google Scholar] [CrossRef]
- Täuber, K.; Marsico, F.; Wurm, F.R.; Schartel, B. Hyperbranched poly(phosphoester)s as flame retardants for technical and high performance polymers. Polym. Chem. 2014, 5, 7042–7053. [Google Scholar] [CrossRef]
- Gao, C.; Yan, D. Hyperbranched polymers: From synthesis to applications. Prog. Polym. Sci. 2004, 29, 183–275. [Google Scholar] [CrossRef]
- Henke, H.; Brüggemann, O.; Teasdale, I. Branched macromolecular architectures for degradable, multifunctional phosphorus-based polymers. Macromol. Rapid Commun. 2017, 38, 1600644. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.F.; Shi, W.F. Synthesis and thermal decomposition of a novel hyperbranched polyphosphate ester used for flame retardant systems. Polym. Degrad. Stab. 2006, 91, 1289–1294. [Google Scholar] [CrossRef]
- Deng, J.; Zhu, S.W.; Shi, W.F. Effect of molecular chain structure of the cured epoxy resin containing hyperbranched (3-hydroxyphenyl) phosphate on expansion and flame retardance. J. Appl. Polym. Sci. 2004, 94, 2065–2070. [Google Scholar] [CrossRef]
- Deng, J.; Shi, W.F. Synthesis and effect of hyperbranched (3-hydroxyphenyl) phosphate as a curing agent on the thermal and combustion behaviours of novolac epoxy resin. Eur. Polym. J. 2004, 40, 1137–1143. [Google Scholar] [CrossRef]
- Chen, X.; Jiao, C.; Li, S.; Sun, J. Flame retardant epoxy resins from bisphenol-A epoxy cured with hyperbranched polyphosphate ester. J. Polym. Res. 2011, 18, 2229–2237. [Google Scholar] [CrossRef]
- Chen, X.; Zhuo, J.; Jiao, C. Thermal degradation characteristics of flame retardant polylactide using TG-IR. Polym. Degrad. Stab. 2012, 97, 2143–2147. [Google Scholar] [CrossRef]
- Pawlowski, K.H.; Schartel, B. Flame retardancy mechanisms of triphenyl phosphate, resorcinol bis(diphenyl phosphate) and bisphenol A bis(diphenyl phosphate) in polycarbonate/acrylonitrile–butadiene–styrene blends. Polym. Int. 2007, 56, 1404–1414. [Google Scholar] [CrossRef]
- Weil, E.D. Fire-Protective and flame-retardant coatings-A State-of-the-Art Review. J. Fire Sci. 2011, 29, 259–296. [Google Scholar] [CrossRef]
- Weil, E.D.; Levchik, S.V. Flame Retardants for Plastics and Textiles: Practical Applications; Carl Hanser Verlag GmbH Co KG: München, Germany, 2015. [Google Scholar] [CrossRef]
- Marsico, F.; Wagner, M.; Landfester, K.; Wurm, F.R. Unsaturated polyphosphoesters via acyclic diene metathesis polymerization. Macromol. 2012, 45, 8511–8518. [Google Scholar] [CrossRef]
- Steinbach, T.; Alexandrino, E.M.; Wahlen, C.; Landfester, K.; Wurm, F.R. Poly(phosphonate)s via olefin metathesis: Adjusting hydrophobicity and morphology. Macromol. 2014, 47, 4884–4893. [Google Scholar] [CrossRef]
- Steinbach, T.; Alexandrino, E.M.; Wurm, F.R. Unsaturated poly(phosphoester)s via ring-opening metathesis polymerization. Polym. Chem. 2013, 4, 3800–3806. [Google Scholar] [CrossRef] [Green Version]
- Steinmann, M.; Markwart, J.; Wurm, F.R. Poly(alkylidene chlorophosphate)s via acyclic diene metathesis polymerization: A general platform for the postpolymerization modification of poly(phosphoester)s. Macromolecules 2014, 47, 8506–8513. [Google Scholar] [CrossRef]
- Mutlu, H.; de Espinosa, L.M.; Meier, M.A.R. Acyclic diene metathesis: A versatile tool for the construction of defined polymer architectures. Chem. Soc. Rev. 2011, 40, 1404–1445. [Google Scholar] [CrossRef]
- Cowie, J.M.G.; Arrighi, V. Polymers: Chemistry and Physics of Modern Materials, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Schartel, B.; Wilkie, C.A.; Camino, G. Recommendations on the scientific approach to polymer flame retardancy: Part 1—Scientific terms and methods. J. Fire Sci. 2016, 34, 447–467. [Google Scholar] [CrossRef]
- Rakotomalala, M.; Wagner, S.; Doring, M. Recent developments in halogen free flame retardants for epoxy resins for electrical and electronic applications. Materials 2010, 3, 4300–4327. [Google Scholar] [CrossRef]
- Ciesielski, M.; Schäfer, A.; Döring, M. Novel efficient DOPO-based flame-retardants for PWB relevant epoxy resins with high glass transition temperatures. Polym. Adv. Technol. 2008, 19, 507–515. [Google Scholar] [CrossRef]
- Braun, U.; Balabanovich, A.I.; Schartel, B.; Knoll, U.; Artner, J.; Ciesielski, M.; Döring, M.; Perez, R.; Sandler, J.K.W.; Altstädt, V.; et al. Influence of the oxidation state of phosphorus on the decomposition and fire behaviour of flame-retarded epoxy resin composites. Polymer 2006, 47, 8495–8508. [Google Scholar] [CrossRef]
- Markwart, J.C.; Battig, A.; Zimmermann, L.; Wagner, M.; Fischer, J.; Schartel, B.; Wurm, F.R. Systematically controlled decomposition mechanism in phosphorus flame retardants by precise molecular architecture: P–O vs. P–N. ACS Appl. Polym. Mater. 2019, 1, 1118–1128. [Google Scholar] [CrossRef]
- Linstrom, P.J.; Mallard, W.G. NIST Chemistry WebBook, NIST Standard Reference Database Number 69. Available online: https://doi.org/10.18434/T4D303 (accessed on 27 August 2019).
- Socrates, G. Infrared and Raman Characteristic Group Frequencies: Tables and Charts; Wiley: Hoboken, NJ, USA, 2004. [Google Scholar]
- Hesse, M.; Meier, H.; Zeeh, B. Spektroskopische Methoden in der organischen Chemie; Thieme Verlag: Stuttgart, Germany, 2005. [Google Scholar]
- Levchik, S.V.; Weil, E.D. Thermal decomposition, combustion and flame-retardancy of epoxy resins—a review of the recent literature. Polym. Int. 2004, 53, 1901–1929. [Google Scholar] [CrossRef]
- Levchik, S.V.; Camino, G.; Luda, M.P.; Costa, L.; Muller, G.; Costes, B. Epoxy resins cured with aminophenylmethylphosphine oxide—II. Mechanism of thermal decomposition. Polym. Degrad. Stab. 1998, 60, 169–183. [Google Scholar] [CrossRef]
- Bishop, D.P.; Smith, D.A. Combined pyrolysis and radiochemical gas chromatography for studying the thermal degradation of epoxy resins and polyimides. I. The degradation of epoxy resins in nitrogen between 400 °C and 700 °C. J. Appl. Polym. Sci. 1970, 14, 205–223. [Google Scholar] [CrossRef]
- Lee, L.-H. Mechanisms of thermal degradation of phenolic condensation polymers. II. Thermal stability and degradation schemes of epoxy resins. J. Polym. Sci. Part. A Gen. Pap. 1965, 3, 859–882. [Google Scholar] [CrossRef]
- Ciesielski, M.; Burk, B.; Heinzmann, C.; Döring, M. 2-Fire-retardant High-performance Epoxy-based Materials. In Novel Fire Retardant Polymers and Composite Materials; Wang, D.-Y., Ed.; Woodhead Publishing: Cambridge, UK, 2017; pp. 3–51. [Google Scholar] [CrossRef]
- Thomas, L.C. Interpretation of the Infrared Spectra of Organophosphorus Compounds; Heydon: London, UK, 1974. [Google Scholar]
- Battig, A.; Markwart, J.C.; Wurm, F.R.; Schartel, B. Hyperbranched phosphorus flame retardants: Multifunctional additives for epoxy resins. Polym. Chem. 2019, 10, 4346–4358. [Google Scholar] [CrossRef]
- Schartel, B.; Hull, T.R. Development of fire-retarded materials—Interpretation of cone calorimeter data. Fire Mater. 2007, 31, 327–354. [Google Scholar] [CrossRef]
- Ricks-Laskoski, H.L.; Chaloux, B.L.; Deese, S.M.; Laskoski, M.; Miller, J.B.; Buckley, M.A.; Baldwin, J.W.; Hickner, M.A.; Saunders, K.M.; Christensen, C.M. Tetrazolation of side chains and anhydrous conductivity in a hydrophobic polymer. Macromol. 2014, 47, 4243–4250. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markwart, J.C.; Battig, A.; Velencoso, M.M.; Pollok, D.; Schartel, B.; Wurm, F.R. Aromatic vs. Aliphatic Hyperbranched Polyphosphoesters as Flame Retardants in Epoxy Resins. Molecules 2019, 24, 3901. https://doi.org/10.3390/molecules24213901
Markwart JC, Battig A, Velencoso MM, Pollok D, Schartel B, Wurm FR. Aromatic vs. Aliphatic Hyperbranched Polyphosphoesters as Flame Retardants in Epoxy Resins. Molecules. 2019; 24(21):3901. https://doi.org/10.3390/molecules24213901
Chicago/Turabian StyleMarkwart, Jens C., Alexander Battig, Maria M. Velencoso, Dennis Pollok, Bernhard Schartel, and Frederik R. Wurm. 2019. "Aromatic vs. Aliphatic Hyperbranched Polyphosphoesters as Flame Retardants in Epoxy Resins" Molecules 24, no. 21: 3901. https://doi.org/10.3390/molecules24213901
APA StyleMarkwart, J. C., Battig, A., Velencoso, M. M., Pollok, D., Schartel, B., & Wurm, F. R. (2019). Aromatic vs. Aliphatic Hyperbranched Polyphosphoesters as Flame Retardants in Epoxy Resins. Molecules, 24(21), 3901. https://doi.org/10.3390/molecules24213901