
Multi-Target Chemometric Modelling, Fragment Analysis and Virtual Screening with ERK Inhibitors as Potential Anticancer Agents



Abstract
1. Introduction
2. Results and Discussions
2.1. Linear mt-QSAR Model
2.2. Linear Interpretable mt-QSAR-LDA Model
+ 18.180 D[Tssq11(CH)_MN]me − 0.027 D[Tssq5(POL)_MX]me
+ 0.005 D[Tnsq1(PSA)_GM]me + 0.003 D[Tnsq13(VDW)_N2]bt
− 0.111 D[Tssq2(HYD)_N1]me
2.3. Interpretation of Molecular Descriptors
2.4. Quantitative Contributions of the Molecular Fragments
2.5. Non-Linear mt-QSAR-RF Model
2.6. Virtual Screening with Kinase Database
2.7. Molecular Docking Analysis
2.8. Molecular Dynamics Analysis
2.9. Assessment of Drug-Likeness
3. Materials and Methods
3.1. Dataset Curation and Descriptor Calculation
3.2. Box–Jenkins Approach
3.3. Model Development and Validation
3.4. Molecular Docking Analysis
3.5. Molecular Dynamics Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cicenas, J.; Zalyte, E.; Rimkus, A.; Dapkus, D.; Noreika, R.; Urbonavicius, S. JNK, p38, ERK, and SGK1 Inhibitors in Cancer. Cancers 2017, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Seger, R.; Krebs, E.G. The MAPK signaling cascade. FASEB 1995, 9, 726–735. [Google Scholar] [CrossRef]
- Schubbert, S.; Shannon, K.; Bollag, G. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 2007, 7, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef] [PubMed]
- Smalley, I.; Smalley, K.S.M. ERK inhibition: A new front in the war against MAPK pathway-driven cancers? Cancer Disc. 2018, 8, 140–142. [Google Scholar] [CrossRef] [PubMed]
- Hatzivassiliou, G.; Liu, B.N.; O’Brien, C.; Spoerke, J.M.; Hoeflich, K.P.; Haverty, P.M.; Soriano, R.; Forrest, W.F.; Heldens, S.; Chen, H.; et al. ERK inhibition overcomes acquired resistance to MEK inhibitors. Mol. Cancer Ther. 2012, 11, 1143–1154. [Google Scholar] [CrossRef]
- Carlino, M.S.; Todd, J.R.; Gowrishankar, K.; Mijatov, B.; Pupo, G.M.; Fung, C.; Snoyman, S.; Hersey, P.; Long, G.V.; Kefford, R.F.; et al. Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma. Mol. Oncol. 2014, 8, 544–554. [Google Scholar] [CrossRef]
- Liu, F.F.; Yang, X.T.; Geng, M.Y.; Huang, M. Targeting ERK, an Achilles’ heel of the MAPK pathway, in cancer therapy. Acta Pharm. Sin. B 2018, 8, 552–562. [Google Scholar] [CrossRef]
- Kidger, A.M.; Sipthorp, J.; Cook, S.J. ERK1/2 inhibitors: New weapons to inhibit the RAS-regulated RAF-MEK1/2-ERK1/2 pathway. Pharmacol. Ther. 2018, 187, 45–60. [Google Scholar] [CrossRef]
- Woodson, E.N.; Kedes, D.H. Distinct roles for extracellular signal-regulated kinase 1 (ERK1) and ERK2 in the structure and production of a primate gammaherpesvirus. J. Vir. 2012, 86, 9721–9736. [Google Scholar] [CrossRef]
- Shin, M.; Franks, C.E.; Hsu, K.L. Isoform-selective activity-based profiling of ERK signaling. Chem. Sci. 2018, 9, 2419–2431. [Google Scholar] [CrossRef] [PubMed]
- Busca, R.; Pouyssegur, J.; Lenormand, P. ERK1 and ERK2 map kinases: Specific roles or functional redundancy? Front. Cell Dev. Biol. 2016, 4, 53. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, B.S.; Durinck, S.; Stawiski, E.W.; Yin, J.; Wang, W.; Lin, E.; Moffat, J.; Martin, S.E.; Modrusan, Z.; Seshagiri, S. ERK mutations and amplification confer resistance to ERK-inhibitor therapy. Clin. Cancer Res. 2018, 24, 4044–4055. [Google Scholar] [CrossRef] [PubMed]
- Gimenez, N.; Martinez-Trillos, A.; Montraveta, A.; Lopez-Guerra, M.; Rosich, L.; Nadeu, F.; Valero, J.G.; Aymerich, M.; Magnano, L.; Rozman, M.; et al. Mutations in the RAS-BRAF-MAPK-ERK pathway define a specific subgroup of patients with adverse clinical features and provide new therapeutic options in chronic lymphocytic leukemia. Haematologica 2019, 104, 576–586. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Targeting ERK1/2 protein-serine/threonine kinases in human cancers. Pharmacol. Res. 2019, 142, 151–168. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Infante, J.R.; Janku, F.; Wong, D.J.L.; Sosman, J.A.; Keedy, V.; Patel, M.R.; Shapiro, G.I.; Mier, J.W.; Tolcher, A.W.; et al. First-in-class ERK1/2 inhibitor ulixertinib (BVD-523) in patients with MAPK mutant advanced solid tumors: Results of a phase I dose-escalation and expansion study. Cancer Discov. 2018, 8, 184–195. [Google Scholar] [CrossRef]
- Cao, Z.; Liao, Q.; Su, M.; Huang, K.; Jin, J.; Cao, D. AKT and ERK dual inhibitors: The way forward? Cancer Lett. 2019, 459, 30–40. [Google Scholar] [CrossRef]
- Searls, D.B. Data integration: Challenges for drug discovery. Nat. Rev. Drug Discov. 2005, 4, 45–58. [Google Scholar] [CrossRef]
- Lo, Y.C.; Rensi, S.E.; Torng, W.; Altman, R.B. Machine learning in chemoinformatics and drug discovery. Drug Discov. Today 2018, 23, 1538–1546. [Google Scholar] [CrossRef]
- Speck-Planche, A. Recent advances in fragment-based computational drug design: Tackling simultaneous targets/biological effects. Futur. Med. Chem. 2018, 10, 2021–2024. [Google Scholar] [CrossRef]
- Larif, S.; Ben Salem, C.; Hmouda, H.; Bouraoui, K. In silico screening and study of novel ERK2 inhibitors using 3D QSAR, docking and molecular dynamics. J. Mol. Graph. Model. 2014, 53, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Chung, J.Y.; Ryu, J.S.; Hah, J.M. Structure tuning of pyrazolylpyrrole derivatives as ERK inhibitors utilizing dual tools; 3D-QSAR and side-chain hopping. Bioorganic Med. Chem. Lett. 2011, 21, 4900–4904. [Google Scholar] [CrossRef] [PubMed]
- Aronov, A.M.; Baker, C.; Bemis, G.W.; Cao, J.R.; Chen, G.J.; Ford, P.J.; Germann, U.A.; Green, J.; Hale, M.R.; Jacobs, M.; et al. Flipped out: Structure-guided design of selective pyrazolylpyrrole ERK inhibitors. J. Med. Chem. 2007, 50, 1280–1287. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Luan, F.; Cordeiro, M.N.D.S. Role of ligand-based drug design methodologies toward the discovery of new anti-Alzheimer agents: Futures perspectives in fragment-based ligand design. Curr. Med. Chem. 2012, 19, 1635–1645. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A.; Kleandrova, V.V.; Luan, F.; Cordeiro, M.N.D.S. Multi-target drug discovery in anti-cancer therapy: Fragment-based approach toward the design of potent and versatile anti-prostate cancer agents. Bioorganic Med. Chem. 2011, 19, 6239–6244. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A.; Kleandrova, V.V.; Luan, F.; Cordeiro, M.N.D.S. Fragment-based QSAR model toward the selection of versatile anti-sarcoma leads. Eur. J. Med. Chem. 2011, 46, 5910–5916. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Cordeiro, M.N.D.S. Fragment-based in silico modeling of multi-target inhibitors against breast cancer-related proteins. Mol. Div. 2017, 21, 511–523. [Google Scholar] [CrossRef]
- Halder, A.K.; Cordeiro, M.N.D.S. Development of multi-target chemometric models for the inhibition of class I PI3K enzyme isoforms: A case study using QSAR-Co tool. Int. J. Mol. Sci. 2019, 20, 4191. [Google Scholar] [CrossRef]
- Anderson, A.C. The process of structure-based drug design. Chem. Biol. 2003, 10, 787–797. [Google Scholar] [CrossRef]
- Software QUBILs-MAS v1.0. Available online: http://tomocomd.com/qubils-mas (accessed on 30 August 2019).
- Valdes-Martini, J.R.; Marrero-Ponce, Y.; Garcia-Jacas, C.R.; Martinez-Mayorga, K.; Barigye, S.J.; Vaz d’Almeida, Y.S.; Pham-The, H.; Perez-Gimenez, F.; Morell, C.A. QuBiLS-MAS, open source multi-platform software for atom- and bond-based topological (2D) and chiral (2.5D) algebraic molecular descriptors computations. J. Cheminform. 2017, 9, 35. [Google Scholar] [CrossRef]
- Medina Marrero, R.; Marrero-Ponce, Y.; Barigye, S.J.; Echeverria Diaz, Y.; Acevedo-Barrios, R.; Casanola-Martin, G.M.; Garcia Bernal, M.; Torrens, F.; Perez-Gimenez, F. QuBiLs-MAS method in early drug discovery and rational drug identification of antifungal agents. SAR QSAR Environ. Res. 2015, 26, 943–958. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A. Combining ensemble learning with a fragment-based topological approach to generate new molecular diversity in drug discovery: In silico design of Hsp90 inhibitors. ACS Omega 2018, 3, 14704–14716. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A.; Scotti, M.T. BET bromodomain inhibitors: Fragment-based in silico design using multi-target QSAR models. Mol. Div. 2019, 23, 555–572. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A.; Cordeiro, M.N.D.S. De novo computational design of compounds virtually displaying potent antibacterial activity and desirable in vitro ADMET profiles. Med. Chem. Res. 2017, 26, 2345–2356. [Google Scholar] [CrossRef]
- Ambure, P.; Aher, R.B.; Gajewicz, A.; Puzyn, T.; Roy, K. “NanoBRIDGES” software: Open access tools to perform QSAR and nano-QSAR modeling. Chemom. Intell. Lab. Syst. 2015, 147, 1–13. [Google Scholar] [CrossRef]
- Rogers, D.; Hopfinger, A.J. Application of genetic function approximation to quantitative structure-activity-relationships and quantitative structure-property relationships. J. Chem. Inf. Comp. Sci. 1994, 34, 854–866. [Google Scholar] [CrossRef]
- Ambure, P.; Halder, A.K.; Gonzalez Diaz, H.; Cordeiro, M.N.D.S. QSAR-Co: An open source software for developing robust multitasking or multitarget classification-based QSAR models. J. Chem. Inf. Model. 2019, 59, 2538–2544. [Google Scholar] [CrossRef]
- QSAR-Co Tool. Available online: https://sites.google.com/view/qsar-co (accessed on 4 August 2019).
- Gore, P.A. 11-Cluster Analysis. In Handbook of Applied Multivariate Statistics and Mathematical Modeling; Tinsley, H.E.A., Brown, S.D., Eds.; Academic Press: San Diego, CA, USA, 2000; pp. 297–321. [Google Scholar]
- Nizami, B.; Tetko, I.V.; Koorbanally, N.A.; Honarparvar, B. QSAR models and scaffold-based analysis of non-nucleoside HIV RT inhibitors. Chemom. Intell. Lab. Syst. 2015, 148, 134–144. [Google Scholar] [CrossRef]
- Halder, A.K. Finding the structural requirements of diverse HIV-1 protease inhibitors using multiple QSAR modelling for lead identification. SAR QSAR Environ. Res. 2018, 29, 911–933. [Google Scholar] [CrossRef]
- Wilks, S.S. Certain generalizations in the analysis of variance. Biometrika 1932, 24, 471–494. [Google Scholar] [CrossRef]
- Brown, M.T.; Wicker, L.R. 8-Discriminant Analysis. In Handbook of Applied Multivariate Statistics and Mathematical Modeling; Tinsley, H.E.A., Brown, S.D., Eds.; Academic Press: San Diego, CA, USA, 2000; pp. 209–235. [Google Scholar]
- Boughorbel, S.; Jarray, F.; El-Anbari, M. Optimal classifier for imbalanced data using Matthews correlation coefficient metric. PLoS ONE 2017, 12, e0177678. [Google Scholar] [CrossRef] [PubMed]
- Hanczar, B.; Hua, J.; Sima, C.; Weinstein, J.; Bittner, M.; Dougherty, E.R. Small-sample precision of ROC-related estimates. Bioinformatics 2010, 26, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Halder, A.K.; Cordeiro, M.N.D.S. Probing the environmental toxicity of deep eutectic solvents and their components: An in silico modeling approach. ACS. Sustain. Chem. Eng. 2019, 7, 10649–10660. [Google Scholar] [CrossRef]
- Rucker, C.; Rucker, G.; Meringer, M. Y-randomization and its variants in QSPR/QSAR. J. Chem. Inf. Model. 2007, 47, 2345–2357. [Google Scholar] [CrossRef]
- Roy, K.; Kar, S.; Ambure, P. On a simple approach for determining applicability domain of QSAR models. Chemom. Intell. Lab. Syst. 2015, 145, 22–29. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Cordeiro, M.N.D.S. Speeding up early drug discovery in antiviral research: A fragment-based in silico approach for the design of virtual anti-hepatitis C leads. ACS Comb. Sci. 2017, 19, 501–512. [Google Scholar] [CrossRef]
- Ghose, A.K.; Crippen, G.M. Atomic physicochemical parameters for three-dimensional-structure-directed quantitative structure-activity relationships. 2. Modeling dispersive and hydrophobic interactions. J. Chem. Inf. Comp. Sci. 1987, 27, 21–35. [Google Scholar] [CrossRef]
- Bemis, G.W.; Murcko, M.A. The properties of known drugs. 1. Molecular frameworks. J. Med. Chem. 1996, 39, 2887–2893. [Google Scholar] [CrossRef]
- Sushko, I.; Novotarskyi, S.; Korner, R.; Pandey, A.K.; Rupp, M.; Teetz, W.; Brandmaier, S.; Abdelaziz, A.; Prokopenko, V.V.; Tanchuk, V.Y.; et al. Online chemical modeling environment (OCHEM): Web platform for data storage, model development and publishing of chemical information. J. Comput. Aided Mol. Des. 2011, 25, 533–554. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Cordeiro, M.N.D.S. Advanced in silico approaches for drug discovery: Mining information from multiple biological and chemical data through mtk- QSBER and pt-QSPR Strategies. Curr. Med. Chem. 2017, 24, 1687–1704. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Fawagreh, K.; Gaber, M.M.; Elyan, E. Random forests: From early developments to recent advancements. Syst. Sci. Control Eng. 2014, 2, 602–609. [Google Scholar] [CrossRef]
- Marchese Robinson, R.L.; Palczewska, A.; Palczewski, J.; Kidley, N. Comparison of the predictive performance and interpretability of random forest and linear models on benchmark data sets. J. Chem. Inf. Model. 2017, 57, 1773–1792. [Google Scholar] [CrossRef] [PubMed]
- Guha, R. On the interpretation and interpretability of quantitative structure-activity relationship models. J. Comput. Aid. Mol. Des. 2008, 22, 857–871. [Google Scholar] [CrossRef]
- Nawar, S.; Mouazen, A.M. Comparison between random forests, artificial neural networks and gradient boosted machines methods of on-line Vis-NIR spectroscopy measurements of soil total nitrogen and total carbon. Sensors 2017, 17, 2428. [Google Scholar] [CrossRef]
- Asinex Kinase Library. Available online: http://www.asinex.com/focus_kinases/ (accessed on 30 August 2019).
- Chaikuad, A.; Tacconi, E.M.C.; Zimmer, J.; Liang, Y.K.; Gray, N.S.; Tarsounas, M.; Knapp, S. A unique inhibitor binding site in ERK1/2 is associated with slow binding kinetics. Nat. Chem. Biol. 2014, 10, 853–860. [Google Scholar] [CrossRef]
- Morris, E.J.; Jha, S.; Restaino, C.R.; Dayananth, P.; Zhu, H.; Cooper, A.; Carr, D.; Deng, Y.; Jin, W.; Black, S.; et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Disc. 2013, 3, 742–750. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. Software News and Update AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Heightman, T.D.; Berdini, V.; Braithwaite, H.; Buck, I.M.; Cassidy, M.; Castro, J.; Courtin, A.; Day, J.E.H.; East, C.; Fazal, L.; et al. Fragment-based discovery of a potent, orally bioavailable inhibitor that modulates the phosphorylation and catalytic activity of ERK1/2. J. Med. Chem. 2018, 61, 4978–4992. [Google Scholar] [CrossRef]
- Uehling, D.E.; Harris, P.A. Recent progress on MAP kinase pathway inhibitors. Bioorganic Med. Chem. Lett. 2015, 25, 4047–4056. [Google Scholar] [CrossRef] [PubMed]
- Mauri, A.; Consonni, V.; Pavan, M.; Todeschini, R. Dragon software: An easy approach to molecular descriptor calculations. MATCH Commun. Math. Comput. Chem. 2006, 56, 237–248. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Alonso, N.; Caamano, O.; Romero-Duran, F.J.; Luan, F.; Cordeiro, M.N.D.S.; Yanez, M.; Gonzalez-Diaz, H.; Garcia-Mera, X. Model for high-throughput screening of multitarget drugs in chemical neurosciences: Synthesis, assay, and theoretic study of rasagiline carbamates. ACS Chem. Neurosci. 2013, 4, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- STATISTICA, version 6.0; Statsoft-Team: Tulsa, OK, USA, 2001.
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.; Meyer, E.F., Jr.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The Protein Data Bank. A computer-based archival file for macromolecular structures. Eur. J. Biochem. 1977, 80, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Protein Data Bank. Available online: www.rcsb.org (accessed on 30 August 2019).
- Fiser, A.; Do, R.K.G.; Sali, A. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Bas, D.C.; Rogers, D.M.; Jensen, J.H. Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins 2008, 73, 765–783. [Google Scholar] [CrossRef]
- Gasteiger, J.; Marsili, M. New model for calculating atomic charges in molecules. Tetrahedron Lett. 1978, 3181–3184. [Google Scholar] [CrossRef]
- Discovery Studio Visualizer 2017 R2. Available online: https://www.3dsbiovia.com/products/ (accessed on 25 August 2019).
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Vanderspoel, D.; Vandrunen, R. Gromacs—A message-passing parallel molecular-dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Schuttelkopf, A.W.; van Aalten, D.M.F. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- PRODRG Server. Available online: http://prodrg1.dyndns.org/ (accessed on 6 September 2019).
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Giri, A.K.; Spohr, E. Cluster formation of NaCl in bulk solutions: Arithmetic vs. geometric combination rules. J. Mol. Liq. 2017, 228, 63–70. [Google Scholar] [CrossRef]
- Giri, A.K.; Teixeira, F.; Cordeiro, M.N.D.S. Structure and kinetics of water in highly confined conditions: A molecular dynamics simulation study. J. Mol. Liq. 2018, 268, 625–636. [Google Scholar] [CrossRef]
- Giri, A.K.; Teixeira, F.; Cordeiro, M.N.D.S. Salt separation from water using graphene oxide nanochannels: A molecular dynamics simulation study. Desalination 2019, 460, 1–14. [Google Scholar] [CrossRef]
- Hockney, R.W.; Eastwood, J.W. Computer Simulation Using Particles; CRC Press: Boca Raton, FL, USA, 1988. [Google Scholar]
- Parrinello, M.; Rahman, A. polymorphic transitions in single-crystals—A new molecular-dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald—An N.Log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification a | Sub-Training Set | Test Set |
|---|---|---|
| NDTotal b | 3585 | 896 |
| NDactive b | 1306 | 316 |
| CCDactive c | 1256 | 310 |
| Sensitivity(%) | 96.17 | 98.10 |
| NDinactive b | 2279 | 580 |
| CCDinactive c | 1779 | 510 |
| Specificity (%) | 89.03 | 87.93 |
| F-measure | 0.893 | 0.891 |
| Accuracy (%) | 91.63 | 91.52 |
| MCC | 0.831 | 0.832 |
| Descriptors | D[Tnsq5(CH)N2]me | D[Tnsq3(CH)MN]bt | D[Tssq11(CH)MN]me | D[Tssq5(POL)MX]me | D[Tnsq1(PSA)GM]me | D[Tnsq13(VDW)N2]bt | D[Tnsq2(HYD)N1]bt |
|---|---|---|---|---|---|---|---|
| D[Tnsq5(CH)N2]me | 1.000 | −0.779 | −0.613 | 0.225 | 0.218 | −0.015 | 0.059 |
| D[Tnsq3(CH)MN]bt | −0.779 | 1.000 | 0.606 | −0.092 | −0.277 | 0.093 | 0.073 |
| D[Tssq11(CH)MN]me | −0.613 | 0.606 | 1.000 | 0.097 | 0.015 | 0.176 | −0.130 |
| D[Tssq5(POL)MX]me | 0.225 | −0.092 | 0.097 | 1.000 | 0.127 | 0.461 | 0.139 |
| D[Tnsq1(PSA)GM]me | 0.218 | −0.277 | 0.015 | 0.127 | 1.000 | 0.158 | −0.259 |
| D[Tnsq13(VDW)N2]bt | −0.015 | 0.093 | 0.176 | 0.461 | 0.158 | 1.000 | 0.451 |
| D[Tnsq2(HYD)N1]me | 0.059 | 0.073 | −0.130 | 0.139 | −0.259 | 0.451 | 1.000 |
| Descriptor | Description |
|---|---|
| D[Tnsq13(VDW)N2]bt | Total atom-based non-stochastic quadratic index of order 13 weighted by the van der Waals volume, modified by the Euclidean distance as mathematical operator, and depending on the chemical structure and the target |
| D[Tnsq3(CH)MN]bt | Total atom-based non-stochastic quadratic index of order 3 weighted by the charge, modified by the minimum value as mathematical operator, and depending on the chemical structure and the target |
| D[Tssq5(POL)MX]me | Total atom-based stochastic quadratic index of order 5 weighted by the polarizability, modified by the maximum value as mathematical operator, and depending on the chemical structure and the measure of effect |
| D[Tnsq2(HYD)N1]me | Total atom-based non-stochastic quadratic index of order 2 weighted by the hydrophobicity, modified by the Manhattan distance as mathematical operator, and depending on the chemical structure and the measure of effect |
| D[Tnsq5(CH)N2]me | Total atom-based non-stochastic quadratic index of order 5 weighted by the charge, modified by the Euclidean distance as mathematical operator, and depending on the chemical structure and the measure of effect |
| D[Tnsq1(PSA)GM]me | Total atom-based non-stochastic quadratic index of order 1 weighted by the polar surface area, modified by the geometric mean as mathematical operator, and depending on the chemical structure and the measure of effect |
| D[Tssq11(CH)MN]me | Total atom-based stochastic quadratic index of order 11 weighted by the charge, modified by the minimum value as mathematical operator, and depending on the chemical structure and the measure of effect |
| Classification a | Sub-Training Set (10-Fold CV) | Test Set |
|---|---|---|
| NDTotal b | 3585 | 896 |
| NDactive b | 1306 | 316 |
| CCDactive c | 1239 | 304 |
| Sensitivity(%) | 94.87 | 96.20 |
| NDinactive b | 2279 | 580 |
| CCDinactive c | 2209 | 559 |
| Specificity (%) | 96.93 | 96.38 |
| F-measure | 0.962 | 0.948 |
| Accuracy (%) | 96.18 | 96.32 |
| MCC | 0.918 | 0.920 |
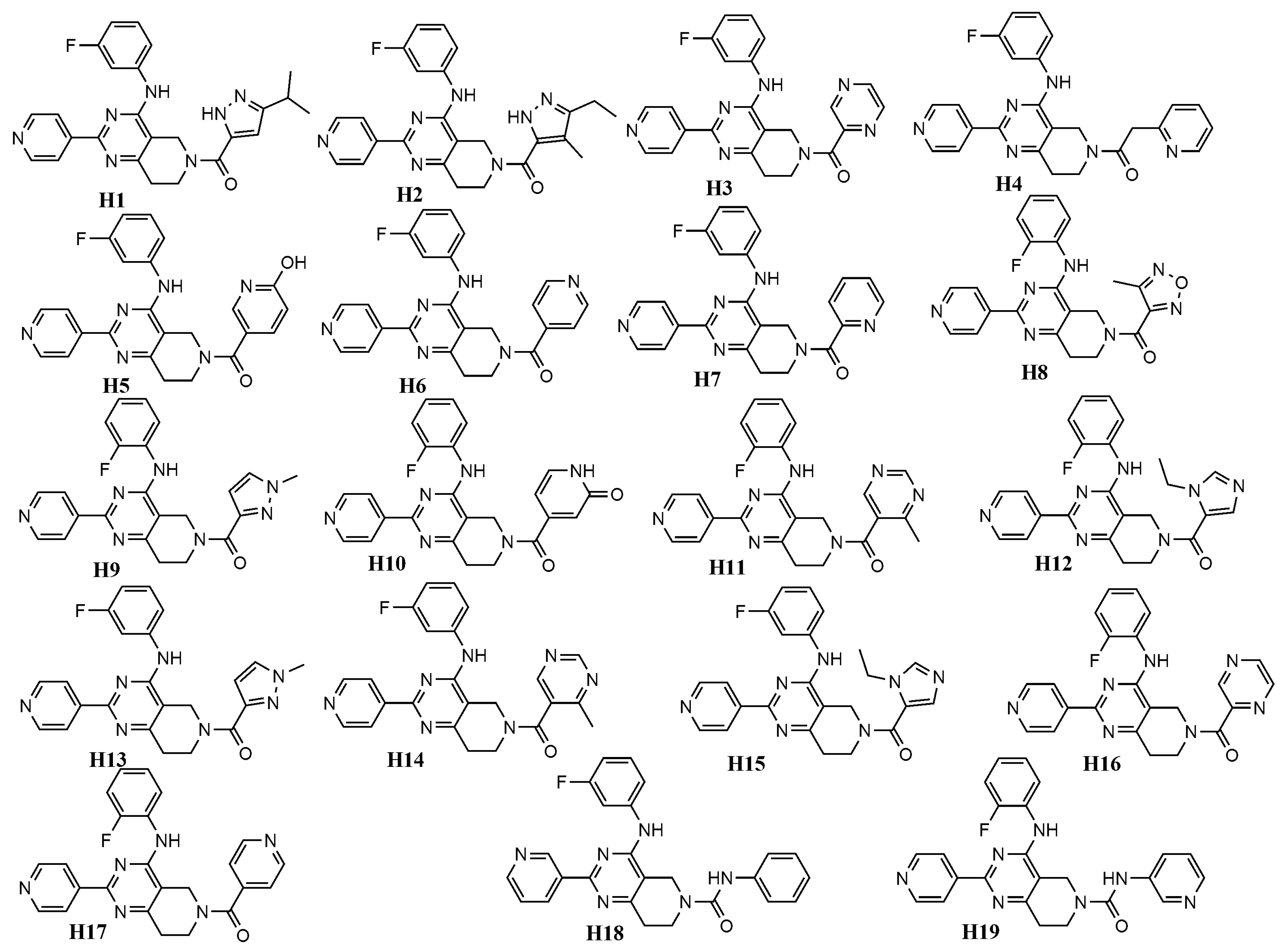
| Cpd | Rigid Docking | Flexible Docking | ||
|---|---|---|---|---|
| ERK-1 (4QTB) | ERK-2 (4QTA) | ERK-1 (4QTB) | ERK-2 (4QTA) | |
| H1 | −9.48 | −10.22 | −10.79 | −10.87 |
| H2 | −9.64 | −9.25 | −10.21 | −10.39 |
| H3 | −9.36 | −9.7 | −10.27 | −9.87 |
| H4 | −8.99 | −8.96 | −10.74 | −9.72 |
| H5 | −9.68 | −10.69 | −9.84 | −11.23 |
| H6 | −9.63 | −9.35 | −10.31 | −10.8 |
| H7 | −8.92 | −9.03 | −10.48 | −10.97 |
| H8 | −9.65 | −10.52 | −10.83 | −9.78 |
| H9 | −9.28 | −9.51 | −10.19 | −10.21 |
| H10 | −9.63 | −10.19 | −10.55 | −9.62 |
| H11 | −9.46 | −10.12 | −10.06 | −10.27 |
| H12 | −9.21 | −9.56 | −10.51 | −9.47 |
| H13 | −9.21 | −9.39 | −10.3 | −10.63 |
| H14 | −9.24 | −10.06 | −10.02 | −9.59 |
| H15 | −9.19 | −9.51 | −9.62 | −10.06 |
| H16 | −9.16 | −9.75 | −9.92 | −9.93 |
| H17 | −9.63 | −10.15 | −10.43 | −10.54 |
| H18 | −10.07 | −9.9 | −10.76 | −10.58 |
| H19 | −9.16 | −9.24 | −10.86 | −11.01 |
| Ulixertinib | −8.77 | −8.38 | −9.97 | −9.73 |
| Complexes | ΔGBind |
|---|---|
| ERK2-H1 | −33.46 |
| ERK1-H1 | −23.28 |
| ERK2-ULX | −27.44 |
| ERK1-ULX | −21.38 |
| NAME | MW | nHDon | nHAcc | ALOGP |
|---|---|---|---|---|
| H1 | 457.56 | 2 | 8 | 4.18 |
| H2 | 457.56 | 2 | 8 | 4.20 |
| H3 | 427.48 | 1 | 9 | 2.16 |
| H4 | 440.52 | 1 | 8 | 3.34 |
| H5 | 442.49 | 2 | 9 | 3.15 |
| H6 | 426.49 | 1 | 8 | 2.88 |
| H7 | 426.49 | 1 | 8 | 3.31 |
| H8 | 431.47 | 1 | 10 | 2.55 |
| H9 | 429.50 | 1 | 8 | 3.11 |
| H10 | 442.49 | 2 | 9 | 1.84 |
| H11 | 441.51 | 1 | 9 | 2.53 |
| H12 | 443.53 | 1 | 8 | 2.76 |
| H13 | 429.50 | 1 | 8 | 3.11 |
| H14 | 441.51 | 1 | 9 | 2.53 |
| H15 | 443.53 | 1 | 8 | 2.76 |
| H16 | 427.48 | 1 | 9 | 2.16 |
| H17 | 426.49 | 1 | 8 | 2.88 |
| H18 | 440.52 | 2 | 8 | 3.94 |
| H19 | 441.51 | 2 | 9 | 2.79 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halder, A.K.; Giri, A.K.; Cordeiro, M.N.D.S. Multi-Target Chemometric Modelling, Fragment Analysis and Virtual Screening with ERK Inhibitors as Potential Anticancer Agents. Molecules 2019, 24, 3909. https://doi.org/10.3390/molecules24213909
Halder AK, Giri AK, Cordeiro MNDS. Multi-Target Chemometric Modelling, Fragment Analysis and Virtual Screening with ERK Inhibitors as Potential Anticancer Agents. Molecules. 2019; 24(21):3909. https://doi.org/10.3390/molecules24213909
Chicago/Turabian StyleHalder, Amit Kumar, Amal Kanta Giri, and Maria Natália Dias Soeiro Cordeiro. 2019. "Multi-Target Chemometric Modelling, Fragment Analysis and Virtual Screening with ERK Inhibitors as Potential Anticancer Agents" Molecules 24, no. 21: 3909. https://doi.org/10.3390/molecules24213909
APA StyleHalder, A. K., Giri, A. K., & Cordeiro, M. N. D. S. (2019). Multi-Target Chemometric Modelling, Fragment Analysis and Virtual Screening with ERK Inhibitors as Potential Anticancer Agents. Molecules, 24(21), 3909. https://doi.org/10.3390/molecules24213909