Abstract
A series of new ferrocene- and ruthenocene-containing iridium(III) heteroleptic complexes of the type [(ppy)2Ir(RCOCHCOR′)], with ppy = 2-pyridylphenyl, R = Fc = FeII(η5-C5H4)(η5-C5H5) and R′ = CH3 (1) or Fc (2), as well as R = Rc = RuII(η5-C5H4)(η5-C5H5) and R′ = CH3 (3), Rc (4) or Fc (5) was synthesized via the reaction of appropriate metallocene-containing β-diketonato ligands with [(ppy)2(μ-Cl)Ir]2. The single crystal structure of 3 (monoclinic, P21/n, Z = 4) is described. Complexes 1–5 absorb light strongly in the region 280−480 nm the metallocenyl β-diketonato substituents quench phosphorescence in 1–5. Cyclic and square wave voltammetric studies in CH2Cl2/[N(nBu)4][B(C6F5)4] allowed observation of a reversible IrIII/IV redox couple as well as well-resolved ferrocenyl (Fc) and ruthenocenyl (Rc) one-electron transfer steps in 1−5. The sequence of redox events is in the order Fc oxidation, then IrIII oxidation and finally ruthenocene oxidation, all in one-electron transfer steps. Generation of IrIV quenched phosphorescence in 6, [(ppy)2Ir(H3CCOCHCOCH3)]. This study made it possible to predict the IrIII/IV formal reduction potential from Gordy scale group electronegativities, χR and/or ΣχR′ of β-diketonato pendent side groups as well as from DFT-calculated energies of the highest occupied molecular orbital of the species involved in the IrIII/IV oxidation at a 98% accuracy level.
1. Introduction
Despite the scarcity and high price of iridium, an important commercial use of this metal and its complexes is the CativaTM process for acetic acid generation by carbonylation of methanol [1,2]. However, large scale commercial iridium-catalysed generation of hydrogen by water electrolysis is hindered by the scarceness and high cost of the metal. During electrolysis, iridium catalyses the oxygen evolution half reaction [3,4]. Although homoleptic tris(β-diketonato)iridium(III) complexes are almost unheard of in literature with only a handful of reports available [5,6], heteroleptic Ir(III) complexes containing a chelating β-diketonato ligand in addition to N- and C- bonding ligands have been extensively studied [7]. Complexes incorporating heavy transition metals such as platinum [8], osmium [9] and iridium [10,11] have been employed as emitters in electroluminescence devices, of which the iridium complexes are generally the most effective. The extensively studied organic light emitting iridium complex, fac-tris(2-pyridylphenyl-N,C2′)iridium(III), [Ir(ppy)3], has C∧N cyclometalate ligands [12]. This anionic ligand offers a strong Ir-C covalent interaction and, consequently, exhibits a highly stabilized ligand-field strength. Metallocene-containing β-diketonato complexes of Ir(III) are hitherto unknown.
Ferrocene-containing β-diketones, FcCOCH2COR, have been synthesized and characterized [13] before, as well as tested for cytotoxic activity [14] The ruthenocene analogues of these ligands have also been described [15] and tested for cytotoxicity [16]. While there are many homometallic β-diketonato complexes known as precursors for the preparation of oxide materials [17], heterometallic β-diketonato complexes are less common [18,19] although they are known since the seventies [20]. Especially 1-ferrocenyl-1,3-butanedione have been used to generate bi-, tri- and multi nuclear complexes of Re [21], Ni, Pd [22], Pr, Eu, Gd, Tb, Dy, Ho [23], Sm, Er and Yb [24]. These multi nuclear complexes often shows reversible electrochemistry with resolved ferrocenyl redox processes as shown for Hf and Zr phthalocyaninato complexes [25] as well as for octahedral tris(β-diketonato)aluminium(III) [26] and tris(β-diketonato)-manganese(III) [27] complexes. Cyclic voltammetric measurements showed these [M(FcCOCHCOR)3] complexes (M = Al or Mn) also display improved electronic communication between pendent β-diketonato substituents compared to [Cu(FcCOCHCOR)2] complexes [28]. Strong electronic communication between β-diketonato R substituents and the metal to which they are co-ordinated even influence kinetic parameters: square planar rhodium β-diketonato complexes showed their reactivity towards oxidative addition or substitution respectively is enhanced several orders of magnitude respectively by electron-donating β-diketonato R side groups [29,30]. Similarly, density functional theory (DFT) studies on the substitution of β-diketonato-1,5-cyclooctadieneiridium(I) complexes with 1,10-phenanthroline showed linear relationships between calculated orbital energies and charges of [Ir(β-diketonato)(cod)] complexes as well as the experimental second order substitution rate constant of the reaction [31].
We report here the synthesis, spectroscopy and electrochemical characterization of new ferrocene- and ruthenocene-containing iridium(III) heteroleptic complexes [(ppy)2Ir(RCOCHCOR′)], with R = Fc = FeII(η5–C5H4)(η5–C5H5) and Rʹ = CH3 (1), Fc (2), as well as R = Rc = RuII(η5–C5H4)(η5–C5H5) and R′ = CH3 (3), Rc (4) and Fc (5) and also R = R′ = CH3 (6); (ppy = 2-pyridylphenyl). Iridium(III) oxidation potentials are also related to DFT-calculated HOMO energy levels of the species involved.
2. Results and Discussion
2.1. Synthesis
The metallocene-containing iridium(III) heteroleptic complexes [(ppy)2Ir(RCOCHCOR′)], 1–6, were prepared as shown in Scheme 1 via adaption of published methods [32] to be applicable to synthesis of the present new complexes. The chloride-bridged dimer [(ppy)2(μ-Cl)Ir]2 was readily converted to the monomeric complexes by reacting it with the bidentate, monoanionic metallocene-containing β-diketonato ligands RCOCH2COR′ where R = Fc and R′ = CH3 (7), Fc (8), as well as R = Rc and R′ = CH3 (9), Rc (10) and Fc (11) and also R = R′ = CH3 (12).
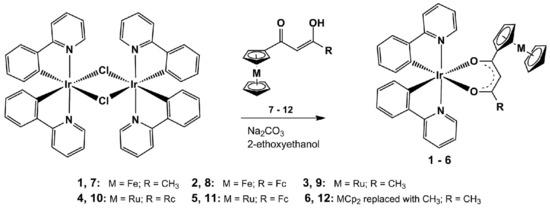
Scheme 1.
Synthesis of 1–6 from the chloro-bridged dimeric iridium(III) complex di-μ-chlorotetrakis[2-(2-pyridinyl-kN)phenyl-kC]diiridium(III). Trans isomers with respect to N-atoms of the ppy-ligands of 1–6 are shown as the single crystal X-ray structure determination, see below, confirms this geometric orientation. Fc = ferrocenyl, Rc = ruthenocenyl and Cp = cyclopentadienyl = C5H5−.
After work-up, which included column chromatography and recrystallization, the new air-stable heteroleptic iridium(III) complexes 1–5 could be isolated as solids, ranging from yellow (compound 1) to orange-red (compounds 2, 5), in moderate (30–70%) yields. Complexes 1–5 are soluble in most organic solvents and crystallographic quality needle-like crystals were obtained from dichloromethane:n-heptane = 1:1. For comparison purposes the known acetylacetonato derivative [(ppy)Ir(CH3COCHCOCH3)] (6) was also synthesized.
2.2. Single Crystal X-Ray Structure of 3
The structure of [(ppy)2Ir(RcCOCHCOCH3)] (3), was solved to determine the orientation of the ppy ligands relative to each other. Complex 3 crystallizes as yellow plates from CH2Cl2/n-heptane in the monoclinic space group P21/n, with one molecule of 3 and a disordered CH2Cl2 (33% occupation) contained in the asymmetric unit cell. A molecular plot of 3 is shown in Figure 1 and crystallographic data are given in Table 1.
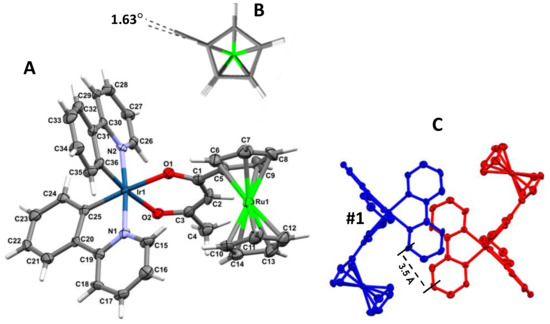
Figure 1.
Left, A: Molecular structure of [(ppy)2Ir(RcCOCHCOCH3)] (3) showing atom labeling and, B, the ruthenocenyl group from above highlighting the near-eclipse configuration. The thermal ellipsoids represent a 50% probability limit. For clarity, a co-crystallized molecule of CH2Cl2 have been omitted. Right: C shows the crystal packing for 3, with the foreground molecule labelled #1 in blue and viewed perpendicular to the ppy ligand plane of the molecule (red) in the background. The perpendicular π–π spacing between the ppy ligands of adjacent molecules is ca. 3.5 Å. Selected bond distances (angstroms) are: Ir(1)–N(1) 2.046(7), Ir(1)–N(2) 2.045(7), Ir(1)–C(36) 1.987(8), Ir(1)–C(25) 1.992(9), Ir(1)–O(1) 2.112(6), Ir(1)–O(2) 2.158(6), O(1)–C(1) 1.262(10), O(2)–C(3) 1.250(11), C(1)–C(2) 1.402(14), C(2)–C(3) 1.393(14), C(1)–C(5) 1.500(14), C(3)–C(4) 1.528(13); Selected bond angles (degrees): N(1)–Ir(1)–C(25) 80.4(3), N(2)–Ir(1)–C(36) 80.8(3), O(1)–Ir(1)–O(2) 87.8(2), Ir(1)–O(1)–C(1) 126.1(6), Ir(1)–O(2)–C(3) 124.8(6), N(1)–Ir(1)–N(2) 174.3(3), O(1)–Ir(1)–C(25) 172.9(3), O(2)–Ir(1)–C(36) 172.3(3), O(1)–C(1)–C(2) 126.6(9), O(2)–C(3)–C(2) 127.4(8), C(1)–C(2)–C(3) 126.9(9), O(1)–C(1)–C(5) 113.9(9), O(2)–C(3)–C(4) 114.6(9). Other bond lengths and angles are available in the Supporting Information. Symmetry transformations used to generate equivalent atoms: #1 −x, −y, −z.

Table 1.
Crystal Data and Structural Refinement for 3.
Complex 3 displayed a slightly distorted octahedral coordination geometry around the central Ir atom. Similar to [(ppy)2Ir(CH3COCHCOCH3)], 6, [32], it retains the cis-C,C′ trans-N,N′ chelate disposition of the chloride-bridged precursor dimer, [(ppy)2(μ-Cl)Ir]2 [33].
The average Ir–C bonds of (Ir–Cavg = 1.990(9) Å) are shorter than the Ir–N bonds (Ir–Navg = 2.046(7) Å). These Ir–C bond lengths are comparable to values reported for the precursor dimer [(ppy)2(μ-Cl)Ir]2 (Ir–C = 1.986(3) − 2.008(7) Å) [33] and complex 6 (Ir−Cavg = 2.003(8) Å) [32]. Similarly, the Ir–N bond lengths (Ir–Navg = 2.046(7) Å) also fall within the typical range of values reported for iridium complexes with ppy ligands, including 6 (Ir–Navg = 2.021(7) Å), but shorter than the average length of 2.140 (5) Å observed in fac-Ir(ppy)3 [34].
The Ir(1)–O(1) and Ir(1)–O(2) bond lengths of 2.112(6) and 2.158(6) Å respectively are notably longer than the mean Ir–O value of 2.088 Å reported for related iridium compounds in the Cambridge Crystallographic Database [35]. Since the Ir–O bond lengths are within 4σ(I) Å the same, coordination of the β-diketonato ligand to the Ir core is considered to approach a symmetric coordination sphere. All other bond lengths and angles within the chelate ligands are typical for cyclometalated ppy and acetylacetonato ligands bound to Ir(III).
Unconjugated C=O bond lengths in β-diketones are typically 1.206 Å, while C−O bond lengths are 1.300 Å [36,37]. For 3, the C−O bond lengths are between these C=O and alkoxy bond length extremes with C(1)−O(1) = 1.262(10) Å and C(3)−O(2) = 1.250(11) Å. For 3 the difference between lengths of the C−O bonds is 0.012(15) Å, while the difference between unconjugated C=O and C−O bonds in β-diketones is 0.094(10) Å. The C−O bonds encountered in 3 are thus indicative of significant delocalized character in the β-diketonato fragment. Bond lengths C(1)–C(2) and C(2)–C(3) are within 1σ(I) the same (Figure 1) implying the β-diketone is symmetric.
Regarding the ruthenocenyl groups, the average C−C bond length within the ruthenocenyl group is 1.414(16) Å for the unsubstituted cyclopentadienyl (Cp) rings and 1.420(17) Å for substituted cyclopentadienyl rings. The largest deviations from this average are +0.016(23) Å for the C(12)−C(13) bond and −0.016(22) Å for the C(5)−C(9) bond. Bond angles in the unsubstituted and substituted cyclopentadienyl rings averaged 108°, which is the ideal theoretical value. The largest deviation from the average values was for the C(10)−C(14)−C(13) angle (+1.70910)o) on the unsubstituted Cp ring. The ruthenocenyl group thus exhibit the expected normal delocalized bond lengths and angles. Cp-rings of the ruthenocenyl group were found to exist almost exactly in the eclipsed configuration. The deviation from a complete eclipsed conformation, as measured with the dihedral angle C(14)−cent(Cp-ring)−cent(subst-Cp-ring)−C(5) was only ca. 1.63° (cent = centroid), Figure 1. Deviation of the two Cp-rings from parallel was measured at ca. 1.15° while deviation of the substituted Cp-ring from the β-diketonato chelate plane Ir-O(1)-C(1)-C(2)-C(3)-O(2) was measured at ca. 18.97o.
The crystal packing of 3 shows similar ppy-ligand positioning to that of 6, with a nearest neighbour molecule related by a C2 rotation that positions a ppy-ligand staggered relative to that of adjacent molecules, as shown in Figure 1 right. For both 3 and 6, the distance between adjacent ring planes of the aromatic ring systems was measured as ca. 3.5 Å face-to-face separation, and a centroid(arene)-centroid(pyridyl) distance of 4.53 Å (at an angle of 5.65°), falls within the range for classification of weak π–π interactions [34].
2.3. Spectroscopic Studies
The IR spectra of 1–6 showed the characteristic strong ν(C=O) vibrations found between 1510–1562 cm−1 (Materials and Methods section) which is typical for chelate-bonded β-diketonato ligands in transition metal complexes [38,39]. Generally, a shift to lower wave numbers is observed for 1–6 compared to the free β-diketones 7–12 [40,41], which allows monitoring of the reaction progress.
The UV–Vis absorption spectra of complexes 1–5 in CH2Cl2 solutions at room temperature are shown in Figure 2 and peak maxima are summarized in Table 2. Each absorption spectrum is composed of an intense absorption band in ultraviolet region and a weaker absorption band in visible region. In analogy to other studies, the higher energy absorption bands below 300 nm are assigned to spin-allowed intraligand π-π* transitions (corresponding to ligand centred states, 1LC), while the lower energy absorption bands between 340 and 540 nm are assigned to a mixture of metal to ligand charge transfers (1MLCT and 3MLCT) [10,11,12,42]. A linear relationship between absorbance and concentration (insert Figure 2) indicated all complexes followed the Beer-Lambert law, A = εCl.
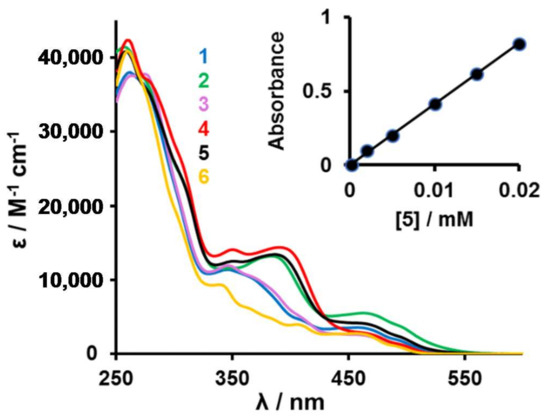
Figure 2.
UV–Vis spectra of [(ppy)2Ir(RCOCHCOR′)] complexes 1 (R = Fc, R′ = CH3, blue), 2 (R = R′ = Fc, green), 3 (R = Rc, R′ = CH3, purple), 4 (R = R′ = Rc, red), 5 (R = Fc, R′ = Rc, black) and 6 (R = R′ = CH3, orange) in CH2Cl2. Insert: The linear relationship between absorbance and concentration for complex 5 at λ = 262 nm.
Table 2.
Wavelengths of absorption maxima (λmax) and extinction coefficients in brackets at these wavelengths, ε, for [(ppy)2Ir(RCOCHCORʹ)] complexes 1–6 in CH2Cl2 and Gordy scale group electronegativities, χR, for R and Rʹ groups.
In the wavelength region 355 ≤ λ ≤ 430 nm, the influence of the number of metallocenyl substituents on absorption bands is observable. Each of the complexes 2, 4 and 5 has two metallocenyl groups in the β-diketonato ligand coordinated to the iridium centre. These complexes exhibit relatively strong absorbance bands with λmax at 384, 390 and 386 nm (ε > 13200 M−1·cm−1) grouped closely together. The same absorption bands in complexes 1 and 3, which have only one metallocenyl substituent, became much weaker and was shifted to shorter wave lengths. It is only observable as a shoulder at ca. 371 nm (ε = 9010 M−1·cm−1). For complex 6, which has no metallocene in its structure, the comparable absorption band was barely detectable as a weak shoulder at 363 nm (ε = 6330 M−1·cm−1).
2.4. Electrochemistry
Cyclic voltammetry (CV), linear sweep voltammetry (LSV), and Osteryoung square wave voltammetry (OSW) were conducted on 0.5 mM solutions of iridium(III) complexes 1–6 in dry CH2Cl2 utilizing 0.1 mol·dm−3 [(nBu)4N][B(C6F5)4] as supporting electrolyte. Data for cyclic voltammetry experiments are summarized in Table 3, and CVs of 5 at different scan rates are shown in Figure 3 (left) while Figure 3 (right) enables comparison of CV’s of 1−6 at a scan rate of 100 mV·s−1 with each other. CH2Cl2 was used as solvent because it minimizes solvent−compound interactions, while the chosen supporting electrolyte, [(nBu)4N][B(C6F5)4], minimizes ionic interactions of the type (cations)n+···−[B(C6F5)4] [15,43,44].
Table 3.
Cyclic voltammetry data of 0.5 mmol·dm−3 solutions of 1–5 in CH2Cl2 containing 0.1 mol·dm−3 [(nBu)4N][B(C6F5)4] as supporting electrolyte at 20 °C. Scan rate = 100 mV·s−1, potentials are vs. FcH/FcH+.
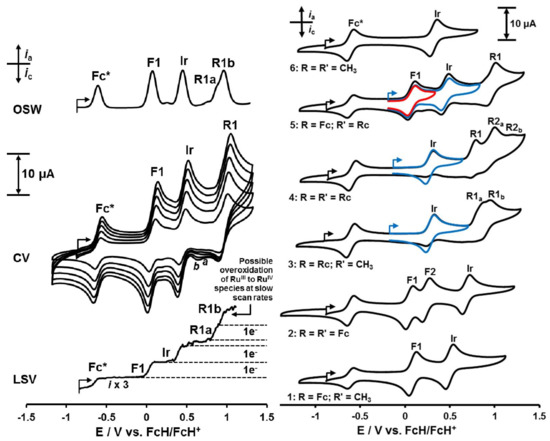
Figure 3.
Left, Top: OSW (Osteryoung Square Wave) voltammogram of 0.5 mmol·dm−3 solutions of [(ppy)2Ir(FcCOCHCORc)], 5, in CH2Cl2 (20 oC) at 10 Hz in the presence of Fc* = decamethylferrocene as internal standard. Left, Middle: Cyclic voltammograms at scan rates 0.1 (smallest currents), 0.2, 0.3, 0.4 and 0.5 V·s−1 (largest current). Left, Bottom: LSV (Linear Sweep Voltammogram) at 2 mV·s−1. The ferrocenyl-, iridium- and ruthenocenyl-based redox processes are labelled as F1, Ir and R1 respectively. The small CV reduction peaks labeled “a” and “b” may be associated with either the presence of a RuIV species, or more likely a dimeric RuIII ruthenocenium species [43]. Likewise, the LSV peak R1b counting 1 electron is consistent with RuIV formation on LSV timescale. Right: Cyclic voltammograms of ca. 0.5 mmol·dm−3 solutions of [(ppy)2Ir(RCOCHCORʹ)] complexes 1–6, scanned at 100 mV·s−1 in CH2Cl2 at 20 oC in the presence of 0.1 mol·dm−3 [N(nBu)4][B(C6F5)4] as supporting electrolyte. Decamethylferrocene, Fc*, was used as internal reference, it is identified by the wave at -0.608 V vs. FvH/FcH+. Blue and red inserts: CV scans of 3, 4 and 5 where scan directions are reversed before ruthenocene oxidation commenced.
In this study, all studied iridium complexes 1–6 showed electrochemically reversible oxidation waves corresponding to the Fc0/+, IrIII/IV and Rc0/+ redox couples, although the Rc0/+ couple was not chemically reversible. One-electron electrochemical reversible redox processes are theoretically characterized by CV peak separations of ΔEp = Epa − Epc = 59 mV [45,46]. Chemical reversibility is exhibited by ipc/ipa current ratios approaching unity.
The first redox process observed in the electrochemical sequence of redox events of 1–6 is the ferrocenyl oxidation wave labeled F1 in the CV’s of 1, 2 and 5 (Figure 3). ΔE for all these redox processes was 82 mV or less. As the internal standard decamethylferrocene, Fc*, showed ΔE = 77 mV (Table 3), waves F1 in all cases were regarded as electrochemical reversible as the Fc* redox process at slow (100 mV·s−1) scan rates. Current ratios for wave F1 of 1 and 2 approached unity (Table 3) but for 5 it was only 0.55 at 100 mV s−1 scan rate. This indicated that, unlike for 1 and 2, wave F1 for 5 was not chemically reversible. However, if the reversing potential was set to exclude redox activity of the ruthenocenyl couple, (blue and red inserts Figure 3, right), the ferrocenyl wave current ratio also approached 1.
It is concluded that the unstable and highly reactive ruthenocenium species, Rc+ that is generated when the ruthenocenyl group is oxidized [43], probably interacts with the ppy ligands of 5 in such a way that the molecule degrades, thereby interfering with the reversibility of the Fc/Fc+ couple. Formal reduction potentials, Eo′ = ½(Epa + Epc), could therefore be calculated [47,48] for wave F1 of 1, 2 and 5 and are summarized in Table 3. F1 Eoʹ values for 1, 2 and 5 deviate from 0 mV vs. FcH/FcH+ in the range 50 ≤ Eo′Fc of Ir complex ≤ 86 mV, Table 3. This is a smaller deviation than for the Mn(FcCOCHCOR)3 [27], Cu(FcCOCHCOR)2 [28] or the free β-diketones FcCOCH2COR [13] which exhibited 52 ≤ Eo′Fc group of Mn ≤ 97 mV, 98 ≤ Eo′Fc group of Cu ≤ 233 mV and 188 ≤ Eo′Fc group free β-diketone ≤ 236 mV, respectively. Al(FcCOCHCOR)3 complexes exhibited a ferrocenyl reduction potential range of 33 ≤ Eo′Fc group of Al ≤ 86 mV [26]. These comparisons indicate that the electron-withdrawing effect that the β-diketone C=O group has on F1 ferrocenyl oxidation, moving it to larger (more cathodic) potentials, are much more negated by coordination to IrIII than coordination to MnIII or CuII, but it is about the same as in AlIII coordination.
Complex 2 has two ferrocenyl groups and the oxidation of the second ferrocenyl group is associated with wave F2 (Figure 3). This process is also electrochemically and chemically reversible at Eo′ = 0.235 V vs FcH/FcH+. Upon oxidation of the ferrocenyl groups in complexes 1, 2 and 5, the electron-donating ferrocenyl group with group electronegativity [49,50], χFc = 1.86 converts to an electron-withdrawing ferrocenium group with χFc+ = 2.82 (Table 2). The quantity “group electronegativity” also takes into account charge effects. The ferrocenium group is therefore almost as electron-withdrawing as the CF3 group which exhibits χCF3 = 3.01. This strong electron-withdrawing property of Fc+ is the reason why wave F2 of complex 2 exhibits Eo′ at a potential 185 mV larger than wave F1. The first-oxidized ferricenium group withdraws electron density via the conjugated β-diketonato backbone from the second still-to-be oxidized ferrocenyl group in 2 making this ferrocenyl group much more difficult to oxidize than the first one. The strong electron-withdrawing property of the Fc+ group also has a profound effect on the potential at which the IrIII/IV couple is observed.
The next general redox process observed in the electrochemical sequence of redox events of 1–6 is the iridium wave labeled Ir in Figure 3 and Table 3 in the potential range Eo′ = 0.252 − 0.681 V versus FcH/FcH+. For complex 6, similar to that of the internal reference decamethylferrocene (Fc*), the iridium-based redox process was chemically and electrochemically reversible, displaying ΔEp = 0.081 V and ipc/ipa ratios approaching 1 at slow (100 mV·s−1) scan rates. A value of Eo′ = 0.318 V was then calculated for 6 which is notably different than the published Eo′ value [32] of 0.870 V versus Ag/AgCl (in DCM) or by subtracting 0.450 V [51] at 0.42V vs. FcH/FcH+ for 6.
The Ir wave of complexes 1–6 displayed comparable ΔE values in the range 82–88 mV at 100 mV·s−1 scan rate, implying all IrIII/IV couples approached electrochemical reversibility at slow (100 mV·s−1) scan rates. Only complexes 1 and 2 exhibited IrIII/IV chemical reversibility with ipc/ipa approaching 1 though, Table 3. However, when one sets the switching potential on anodic scans of 3, 4 and 5 in such a way that the ruthenocenyl moiety is not oxidized at wave R, see blue inserts in Figure 3, right, then the IrIII/IV couple of complexes 3, 4 and 5 also becomes chemically reversible with current ratios approaching 1. It is clear that the highly reactive oxidized Rc+ fragments interact with the oxidized molecules as a whole and decomposes it to the extent that ipc for wave F1 of 5 and the Ir wave of 3, 4 and 5 are smaller than stoichiometric reductions require. In the process, as explained before [15,43], a RuIV species probably irreversibly forms. In addition, all ruthenocene-containing complexes showed ipc/ipa ratios also much less than one for the ruthenocenyl-based redox process implying the ipc of the ruthenocenyl group waves is also much less than expected. This contrasts the electrochemistry of neat ruthenocene which do show ipc/ipa current ratios of one in CH2Cl2/[(nBu)4N][B(C6F5)4] [15]. The high reactivity of Rc+ is highlighted with the fact that even [(nBu)4N][PF6] destroys it, generating a RuIV species [15,43]. The smaller-than-expected ipc currents of waves F1, Ir and R in 3, 4 and 5 are consistent with the unstable and highly reactive 17 electron ruthenicenium cation, Rc+, which forms in the anodic sweeps of compounds 3, 4 and 5 at wave R, interacts with the fully oxidized molecular species and damage it. This explains why, when wave R is not initiated in the CV scans of 3, 4 and 5, waves F1 and Ir become also chemically reversible (i.e. ipc/ipa ratios approach unity).
The one-electron iridium-based redox process for complexes 1–5 have Eo′ between 0.252–0.681 V versus FcH/FcH+ while for 6, Eo′ = 0.318 V (Table 3, Figure 3). The Ir redox process of 1, 2 and, to a lesser extent 5, are observed at potentials substantially larger than that of 6. ΔEo′ = Eo′1, 5 or 2 − Eoʹ6 = 364 (for 1), 416 (for 5) and 600 mV (for 2). The reason for this large shift to bigger potentials is the electron-withdrawing nature of the oxidized ferrocenium group(s) in the β-diketonato ligand. Eo′(IrIII/IV) of complex 2 is shifted to larger potentials than Eo′(IrIII/IV) of 1 as the Ir center of 2 is exposed to two oxidized ferrocenium moieties when it becomes redox active. The IrIII/IV couple of 5 is shifted the least because the ruthenocenyl group electronegativity is smaller than that of the CH3 group implying the ruthenocenyl group is more electron-donating than the CH3 group (χRc = 1.89; χCH3 = 2.34) [13,15]. Group electronegativity considerations, which also take into account charge changes on molecular fragments, therefore explain why the Eo′ value of complex 5 is shifted the least, that of complex 1 intermediately and that of complex 2 the most to more positive values when comparing IrIII/IVcouples of 1, 2 and 5 with that of 6. It also explains why Eo′(IrIII/IV) of complexes 3 (R = CH3, R′ = Rc, Eo′ = 0.283 V) and 4 (R = R′ = Rc, Eo′ = 0.252 V) are shifted in the negative (cathodic) direction to smaller Eo′ values than that of 6 having the CH3COCHCOCH3− ligand (Eo′ = 0.318 V) in the coordination sphere. The Ir center of complex 4 bearing two strongly electron-donating ruthenocenyl groups are oxidized the easiest in the present compound series 1–6.
In contrast to the ferrocenyl group’s reversible redox behavior of complexes 1, 2 and 5, irreversible electrochemistry was found for the ruthenocenyl centres of complexes 3–5. These Rc-groups displayed comparable peak anodic currents, ipa, at peak anodic potentials, Epa, in the potential range 0.771–1.179 V in their cyclic voltammograms (waves R1 and R2 in Figure 3 as well as Table 3), while ipc, which is associated with Epc, were of very weak intensity. The chemical irreversibility of Rc-based redox processes is evident from the ipc/ipa ratios of 3–5 ranging from 0.04 for R1a of complex 3 to 0.76 for R1 of complex 5. Complex 3 shows two ruthenocenyl oxidation waves labelled R1a and R1b (Figure 3). Half wave R1a is expected, but R1b not. Similarly, two ruthenocene oxidations are expected for complex 4, half waves R1 and R2a, but half wave R2b is not expected. Complex 5 shows no anomalies for the ruthenocenyl anodic process, only anodic half wave R1 is observed at scan rate 100 mV·s–1. However, at a slow scan rate (LSV at 2 mV·s–1 and OSW at 10 Hz, (Figure 3, left) a hint of an unexpected wave R1b is detected. A literature survey has shown that it is generally accepted that RuIV species arise in CV experiments involving ruthenocene derivatives in CH3CN/N(nBu)4PF6 solvent/supporting electrolyte systems [15,43,52]. We conclude that traces of a RuIV species such as [(C5H4R)RuIV(C5H5)]2+ are likely formed at wave R1b or R2b during the oxidation of the ruthenocenyl group, especially after Rc+ interacted with the ppy ligands of 3–5.
The iridium, ferrocenyl and ruthenocenyl waves all involve the same number of electrons (one) transferred, as is demonstrated for 5 in Figure 3, left, with the LSV scan. With the above background, the sequence of electrochemical events is first ferrocenyl oxidation at wave F1, then iridium oxidation at wave Ir and then ruthenocenyl oxidation at wave R1. An electrochemical scheme describing the redox processes of 5 can be written, see Scheme 2.

Scheme 2.
The sequence of electrochemical events associated with waves F1, Ir and R1 of [(ppy)2Ir(FcCOCHCORc)], 5. Eo′ values are valid for a scan rate of 0.1 V·s–1.
Electrochemical schemes for 1–4 and 6 may be found in the Supplementary Information, and the sequence of redox events may also be deduced from the Eo′ values in Table 3.
2.5. Quantification of the Relationship between ΣχR and Eo′
The range of IrIII/IVredox potentials from 0.252 to 0.681 V vs FcH/FcH+ (Table 3) was traced to changes in χR values (χR values may be found in Table 2) of β-diketonato side groups Fc, Fc+, CH3, Rc and Rc+. As an example of how this is done, utilizing the sum of the R-group group electronegativities, ΣχR, of the β-diketonato ligand of 5 when IrIII oxidation is initiated at wave Ir (Scheme 2 shows the reacting species for wave Ir as [(ppy)2Ir(Fc+COCHCORc)]+), ΣχR may be calculated as ΣχR = χFc+ + χRc = 2.82 +1.89 = 4.71. In a similar way, ΣχR may be calculated for all the β-diketonato ligands of 1–4 when IrIII oxidation is initiated in the CV experiments. This data is plotted in Figure 4 showing the Eo′ value of the IrIII/IV couple is directly proportional to ΣχR of the β-diketonato ligand.
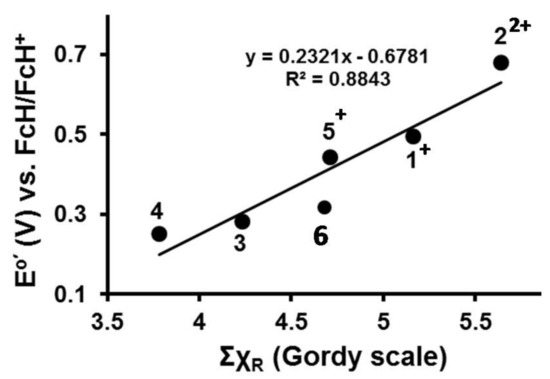
Figure 4.
Linear relationship between the sum of the group electronegativities, ΣχR, and the iridium-based redox potentials, E°′, for compounds 1–6.
A least square fitting of the data in Figure 4 allowed mathematical quantification as shown in Equation (1). The values in brackets are standard deviations.
Eo′(IrIII/IV) = 0.23(4) ΣχR – 0.68(20); R2 = 0.88
Equation (1) allows a rough estimation of the Eo′ value of the IrIII/IV couple in compounds of the type [(ppy)2Ir(RCOCHCOR′)] if the χR and χRʹ values of β-diketonato R and R′ side groups are known.
2.6. DFT view of Iridium Oxidation
Oxidation of a molecule involves in most cases the removal of an electron from the highest occupied molecular orbital (HOMO) of the molecule [53]. The character of the HOMO thus gives the locus of the oxidation. The ease of oxidation is inversely proportional to the energy of the HOMO, since the larger the energy of the HOMO, the easier it will be to remove the electron at a lower redox potential. The HOMOs of molecules 3, 4, and 6 are iridium dx2-y2 based (see Figure 5) and the energy of the HOMOs, EHOMO, decreases as the Eo′ values of ferrocenyl group oxidation (peak F1) increase (Data in Table S9 of the Supplementary Information).
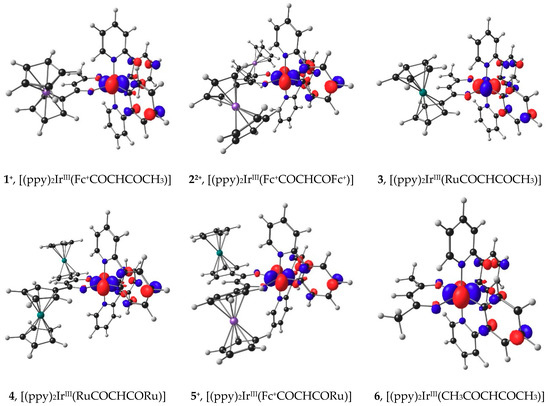
Figure 5.
B3LYP/6-311G(d,p)/def2-TZVPP(SDD) calculated HOMOs of molecules involved in the IrIII oxidation. Calculations was done using DCM as solvent. A contour of 0.06 eÅ-3 was used for the MO plots. Gas phase calculated HOMOs look similar and are provided in the Supporting Information Figure S2.
For complexes 1, 5 (both have only one ferrocenyl group in its structure) and 2 (this complex has two ferrocenyl groups in its structure) ferrocenyl oxidation occurs before the iridium center is oxidized. Thus, the HOMOs of the cations 1+ and 5+ will be associated with IrIII oxidation, while the HOMO of the di-cation 22+ is associated with iridium, see Figure 5.
The lowest unoccupied molecular orbitals (LUMO’s) of 1+, 5+ and 22+, the orbitals where the electron(s) were removed during the initial ferrocenyl group oxidation, are located on iron, since ferrocenyl oxidation of 1, 5 and 2 occur before iridium oxidation, as illustrated in Figure 6. The Eo′ values of the IrIII/IV couple of 1–6 relate to gas phase HOMO energies of 1+, 22+, 3, 4, 5+ and 6 by Equation (2). Figure 7, black data points, shows an Eo′ – EHOMO plot of available gas phase data points. When taking the experimental solvent used for electrochemical analysis, namely dichloromethane, into account in the DFT calculations, a relationship to the same level of accuracy is obtained see equation 3, though the slope of the Eo′ – EHOMO plot is much steeper, as previously found when relating redox potentials with frontier orbital energies [54] (Figure 7, blue data points).
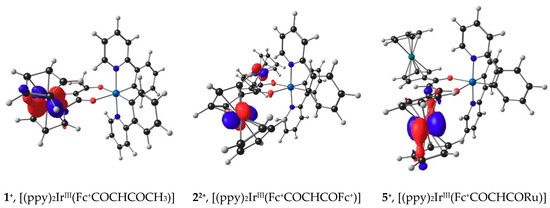
Figure 6.
B3LYP/6-311G(d,p)/def2-TZVPP(SDD) calculated LUMOs of molecules after ferrocenyl oxidation. Calculations was done using DCM as solvent. A contour of 0.06 eÅ–3 was used for the MO plots. Gas phase calculated LUMOs look similar and are provided in the Supporting Information Figure S3.
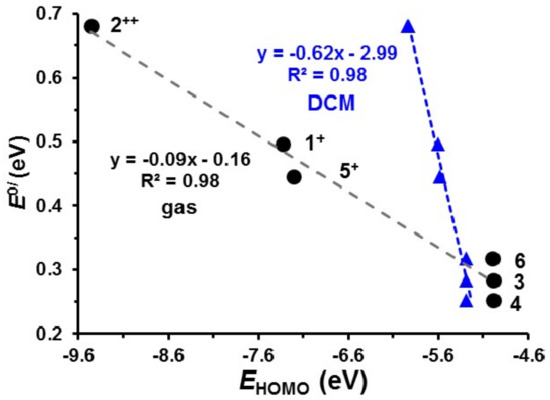
Figure 7.
Relationship between the redox potential of the IrIII/IV couple and the HOMO energy (EHOMO) of the species involved. The black data points were obtained by DFT calculations in gas phase, while the blue data points were obtained using DCM as solvent in the DFT calculations. Data in Table S9 of the Supporting Information.
Gas Phase: Eo′ (IrIII/IV) = −0.087(7) EHOMO − 0.16(5); R2 = 0.98
In DCM: Eoʹ (IrIII/IV) = −0.65(5) EHOMO − 3.0(3); R2 = 0.98
In addition to Equation (1), equations (2) or (3) thus provide methods to estimate the Eo′ value of the IrIII/IV couple in compounds of the type [(ppy)2Ir(RCOCHCOR′)] by optimizing the most stable structure of the species involved in the iridium oxidation, using DFT calculations. EHOMO of the optimized DFT calculated structure may then be obtained from the output DFT file.
2.7. Phosphorescence and Spectroelectrochemistry
Complex 6 showed similar solid-state phosphorescence (irradiation with a UV-lamp at wavelengths 365 and 254 nm with a sample on a TLC plate) and CH2Cl2 solution photoluminescence as previously reported in literature [55] with an ambient temperature emission λmax = 518 nm and lifetime of 1.1 μs, Figure 8. For 6 the lowest excited state is a triplet and as a result emission to the singlet ground state is forbidden without spin-orbit coupling (SOC). The emitting state is assigned as a 3MLCT (metal-to-ligand charge transfer) state and involves mainly occupied Ir 5d and unoccupied ppy π* orbitals [55]. The majority of complexes of the type [(C^N)2Ir(LX)] (where C^N = cyclometallating ligand such as ppy, and LX = monoanionic ligand such as acac) reported in literature showed efficient phosphorescence [10,11,12]. Upon performing bulk electrolysis (0.8 V vs. Ag wire) the IrIII core is oxidized to IrIV. Complex 6+ lost all phosphorescence, see Figure 8, left. This shows an IrIII core is required for photoluminescence; IrIV quenches it. Figure 8, right, shows how the UV-Vis spectrum of 6 changed after bulk electrolysis converted it to 6+ having an IrIV core. The UV-vis spectra of 1, 1+ and 12+ are also shown.
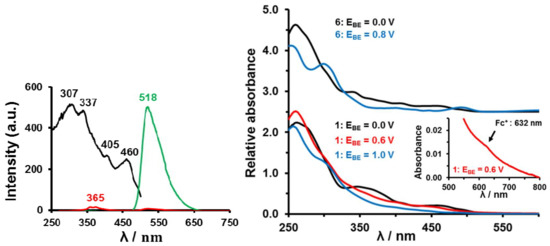
Figure 8.
Left: Excitation spectrum (black) for observing emission at 518 nm and photoluminescence spectra (green and red) at 20 oC of a CH2Cl2 solution containing 0.050 mmol dm-3 [(ppy)2Ir(acac)], 6, and 0.05 mol dm-3 [N(nBu)4][PF6], before (green) and after (red) bulk electrolysis at 0.8 V vs. Ag wire. After bulk electrolysis, IrIII was converted to IrIV, but the same excitation caused hardly any emission (red spectrum). Right: UV–Vis spectra of 0.05 mmol·dm−3 CH2Cl2 solutions of [(ppy)2Ir(acac)], 6, (top) and [(ppy)2Ir(FcCOCHCOCH3)], 1, (bottom) in CH2Cl2 in the presence of 0.05 mol·dm−3 [N(nBu)4][PF6] at 20 oC after bulk electrolysis (BE) at the indicated potentials (EBE vs. Ag/Ag+). Black spectra are for 1 and 6 in the ground state. The red spectrum was obtained after Fc oxidation in 1 to generate 1+; blue spectra are the result of IrIII oxidation generating 12+ or 6+. The red insert highlights an electronic band associated with the Fc+ moiety of 1+.
The photophysical properties of complexes 1–5 are also quenched. Unlike the pale yellow [(ppy)2Ir(acac)] complex 6, solid complexes 1–5 displayed hardly any phosphorescence when irradiated with a UV-lamp at wavelengths 365 and 254 nm. Neither did they display any phosphorescence in solution or after bulk electrolysis at 0.6 V vs. Ag wire (to oxidise Fc to Fc+ generating for example 1+) and at 1.0 V (to oxidize IrIII to IrIV generating 12+). Complexes 2–5 behaved in the same way showing that photoluminescence is quenched in 1–5 by the ferrocenyl, ferrocenium, ruthenocenyl and IrIV molecular fragments. Our results therefor mirror those of other researchers that indicated ruthenocenyl and ferrocenyl groups are photoluminescence quenchers [56].
3. Materials and Methods
3.1. General Information
Solid reagents ferrocene, ruthenocene (Sigma-Aldrich, Johannesburg, South Africa) and di-μ-chlorotetrakis[2-(2-pyridinyl-κN)phenyl-κC]diiridium(III) (STREM, Newburyport, MA, United States of America) were used without any further purification. Liquid reagents (Sigma-Aldrich and Merck, Johannesburg, South Africa) were used without any further purification unless specified otherwise. Dichloromethane was dried by distillation from calcium hydride and, to remove photochemically generated HCl, passed through basic alumina prior to use. For electrochemical experiments, spectrochemical grade dichloromethane (Aldrich) was used and stored under an argon atmosphere. Distilled water was used throughout. Metallocene-containing β-diketones FcCOCH2COR (7, R = CH3; 8, R = Fc) [13] and RcCOCH2COR (9, R = CH3; 10, R = Rc; 11, R = Fc) [15] were synthesized utilizing published procedures with care being taken to separate it from FcCOCH=C(CH3)Fc, the aldol self-condensation product of acetyl ferrocene [57]. The complexes [(ppy)Ir(CH3COCHCOCH3)] (6, [32]) as well as tetrabutylammonium tetrakispentafluorophenyl-borate [58], and [N(nBu)4][B(C6F5)4] were prepared as described before. Column chromatography was performed on Kieselgel 60 (Merck, grain size 0.040−0.063 nm) using hexane/diethyl ether (1:1) as mobile phase, unless otherwise specified.
Infrared spectra were recorded on a Tensor 27 spectrophotometer (Bruker, Johannesburg, South Africa) equipped with a Bruker Platinum ATR accessory (diamond crystal), running OPUS software (Version 6.5) (Bruker, Johannesburg, South Africa). UV–Vis spectra were recorded on a Cary 5000 Probe UV-Vis-NIR spectrophotometer (Varian, Johannesburg, South Africa). Luminescence measurements were carried out on a Varian Cary Eclipse Fluorescence spectrometer. 1H-NMR spectra were recorded on a 300 MHz FOURIER NMR spectrometer or a 600 MHz AVANCE II NMR spectrometer (Bruker, Johannesburg, South Africa) operating at 25 °C with chemical shifts presented as δ values referenced to SiMe4 at 0.00 ppm utilizing CDCl3 as solvent. Melting points were determined using analytically pure samples on a BX51 microscope (Olympus, Johannesburg, South Africa) using a TMS 600 hot stage (LINKAM Johannesburg, South Africa). Elemental analyses were performed by Canadian Microanalytical Service (Delta, BC, Canada).
3.2. Synthesis
The [(ppy)2Ir(RCOCHCOR′)] complexes 1–5 were prepared via modification of a published method [32] for synthesis of 6 to be compatible with complexes having metallocene-containing ligands. General procedure: A mixture of [(ppy)2(μ-Cl)Ir]2 (0.150 g, 0.14 mmol), the relevant β-diketone (6–10) (2.2 eq., 0.31 mmol) and sodium carbonate (2.2 eq., 0.31 mmol) were refluxed in 2-ethoxyethanol (7 mL) under inert atmosphere for 16 hours. The reaction mixture was allowed to cool to room temperature and the coloured precipitate was filtered off and washed with water and 10 mL of dry ethanol. The precipitate was then purified via chromatography on silica with a CH2Cl2/n-hexane/acetone (60:35:5) mobile phase. After removal of solvents, the product was dried under reduced pressure for 16 hours. Small crystallographic quality needle-like crystals were obtained for these complexes from slow evaporation of a CH2Cl2/n-heptane solution.
3.3. Characterization Data
bis(2-Phenylpyridinato-C2,N)(1-ferrocenylbutane-1,3-dione-κ2-O,O′)iridium(III) (1)
Rf: 0.83. Yield: 105 mg (0.14 mmol, 48% based on [(ppy)2(μ-Cl)Ir]2. 1H-NMR (300 MHz, CDCl3): δ, ppm 8.69 (d, 1H, J 5.8 Hz, ArH), 8.56 (d, 1H, J 5.8 Hz, ArH), 7.88 (d, 2H, J 7.7 Hz, ArH), 7.72 (m, 2H, ArH), 7.60 (d, 2H, J 7.8 Hz, ArH), 7.19 (m, 1H, ArH), 7.09 (m, 1H, ArH), 6.85 (m, 2H), 6.73 (m, 2H, ArH), 6.40 (dd, 1H, J 7.6, 1.2 Hz, ArH), 6.31 (dd, 1H, J 7.6, 1.1 Hz, ArH), 5.52 (s, 1H), 4.62 (m, 1H), 4.53 (m, 1H), 4.21 (m, 2H), 3.77 (s, 5H, FeC5H5), 1.89 (s, 3H). Calculated for C36H29FeIrN2O2: C, 56.18; H, 3.80; N, 3.64%. Found: C, 55.82; H, 3.78; N, 3.32%. M.p. >200 oC (dec). IR: ν(CO) 1560 (s), 1546 (s), 1510 (vs) cm−1.
bis(2-Phenylpyridinato-C2,N)(1,3-diferrocenylpropane-1,3-dionato-κ2-O,O′)-iridium(III) (2)
Rf: 0.76. Yield: 90 mg (0.10 mmol, 34% based on [(ppy)2(μ-Cl)Ir]2. 1H-NMR (300 MHz, CDCl3): δ, ppm 8.56 (dd, 2H, J 5.8, 0.9 Hz, ArH), 7.90 (d, 2H, J 8.0 Hz, ArH), 7.68 (m, 4H, ArH), 7.12 (m, 2H, ArH), 6.90 (td, 2H, J 7.4, 1.3 Hz, ArH), 6.77 (td, 2H, J 7.5, 1.3 Hz, ArH), 6.44 (dd, 2H, J 7.6, 1.0 Hz, ArH), 5.86 (s, 1H), 4.71 (m, 2H), 4.61 (m, 2H), 4.24 (t, 4H, J 1.9 Hz), 3.78 (s, 10H, 2 × FeC5H5). Calculated for C45H37Fe2IrN2O2: C, 57.39; H, 3.96; N, 2.97%. Found: C, 57.08; H, 3.71; N, 3.14%. M.p. > 250 oC (dec). IR: ν(CO) 1542 (vs), 1510 (s) cm−1.
bis(2-Phenylpyridinato-C2,N)(1-ruthenocenylbutane-1,3-dione-κ2-O,O′)iridium(III) (3)
Rf: 0.78. Yield: 126 mg (0.16 mmol, 55% based on [(ppy)2(μ-Cl)Ir]2. 1H-NMR (300 MHz, CDCl3): δ, ppm 8.60 (dd, 1H, J 5.9, 0.8 Hz, ArH), 8.50 (dd, 1H, J 5.9, 0.9 Hz, ArH), 7.88 (d, 2H, J 8.1 Hz, ArH), 7.74 (td, 2H, J 7.2, 1.6 Hz, ArH), 7.57 (dd, 2H, J 7.6, 3.3 Hz, ArH), 7.16 (m, 2H), 6.83 (m, 2H, ArH), 6.71 (m, 2H, ArH), 6.34 (dd, 1H, J 7.5, 1.1 Hz, ArH), 6.27 (dd, 1H, J 7.6, 1.0 Hz, ArH), 5.44 (s, 1H), 4.95 (m, 2H), 4.54 (t, 2H, J 1.8 Hz), 4.28 (s, 5H, RuC5H5), 1.84 (s, 3H). Calculated for C36H29IrN2O2Ru: C, 53.06; H, 3.59; N, 3.44%. Found: C, 53.16; H, 3.75; N, 3.14%. M.p. >200 oC (dec). IR: ν(CO) 1561 (s), 1510 (vs) cm−1.
bis(2-Phenylpyridinato-C2,N)(1,3-diruthenocenylpropane-1,3-dionato-κ2-O,O′)iridium(III) (4)
Rf: 0.89. Yield: 210 mg (0.20 mmol, 71% based on [(ppy)2(μ-Cl)Ir]2. 1H-NMR (300 MHz, CDCl3): δ, ppm 8.56 (dd, 2H, J 5.7, 0.8 Hz, ArH), 7.88 (d, 2H, J 7.9 Hz, ArH), 7.73 (td, 2H, J 7.6, 1.6 Hz, ArH), 7.59 (dd, 2H, J 7.7, 1.3 Hz, ArH), 7.15 (td, 2H, J 6.6, 1.4 Hz, ArH), 6.85 (td, 2H, J 7.4, 1.2 Hz, ArH), 6.72 (td, 2H, J 7.5, 1.3 Hz, ArH), 6.33 (dd, 2H, J 7.6, 0.9 Hz, ArH), 5.71 (s, 1H), 4.99 (m, 4H), 4.56 (m, 4H), 4.28 (s, 10H, 2 × RuC5H5). Calculated for C45H37IrN2O2Ru2: C, 52.36; H, 3.61; N, 2.71%. Found: C, 52.71; H, 3.27; N, 2.68%. M.p. > 250 oC (dec.). IR: ν(CO) 1546 (vs), 1514 (s) cm−1.
bis(2-Phenylpyridinato-C2,N)(1-ferrocenyl-3-ruthenocenyl-1,3-propanedionato-κ2-O,O′)iridium(III) (5)
Rf: 0.90. Yield: 84 mg (0.09 mmol, 32% based on [(ppy)2(μ-Cl)Ir]2. 1H-NMR (300 MHz, CDCl3): δ, ppm 8.63 (m, 2H, ArH), 7.89 (d, 2H, J 8.3 Hz, ArH), 7.71 (m, 2H, ArH), 7.62 (m, 2H, ArH), 7.14 (m, 2H, ArH), 6.87 (m, 2H, ArH), 6.74 (m, 2H, ArH), 6.38 (td, 2H, J 7.2, 0.9 Hz, ArH), 5.79 (s, 1H), 5.04 (t, 2H, J 1.7 Hz), 4.66 (m, 1H), 4.57 (m, 3H), 4.29 (s, 5H, RuC5H5), 4.22 (t, 2H, J 1.9 Hz), 3.77 (s, 5H, FeC5H5). Calculated for C45H37FeIrN2O2Ru: C, 54.76; H, 3.78; N, 2.84%. Found: C, 54.69; H, 3.42; N, 2.69%. M.p. > 250 oC (dec.). IR: ν(CO) 1545 (vs), 1514 (s) cm−1.
3.4. Crystal Structure Determination of 3
Single crystal diffraction studies on complex 3 were done using Quazar multi-layer optics monochromated Mo Kα radiation (k = 0.71073 Å) on a Bruker D8 Venture kappa geometry diffractometer (Bruker, Johannesburg, South Africa) with duo Iμs sources, a Photon 100 CMOS detector and APEX II control software [59]. X-ray diffraction measurements were performed at 150(2) K. Data reduction was performed using SAINT+ [59] and the intensities were corrected for absorption using SADABS [59]. The structures were solved by direct methods using SHELXT [60], using the SHELXL-2014/7 [61] program. The non-hydrogen atoms were refined anisotropically. All H atoms were placed in geometrically idealised positions and constrained to ride on their parent atoms. For tables containing the data collection and refinement parameters, bond lengths and bond angles, see Tables S1–S8 in the Supplementary Information as well as CCDC 1916701.
3.5. Electrochemistry
Cyclic voltammograms (CV’s), square wave voltammograms (SW’s) and linear sweep voltamograms (LSV’s) were recorded using a computer-controlled BAS model 100 B potentiostat (Analytical Science Technology, Cape Town, South Africa). All experiments were performed in a dry cell under an argon atmosphere. Platinum wires were used for the pseudo internal reference electrode as well as the auxiliary electrode, while a glassy carbon working electrode (surface diameter 3.00 mm) was utilized. Between each set of scans the working electrode was polished on a Buhler polishing mat with 1 micron and then with ¼ micron diamond paste. All electrode potentials are reported using the potential of the ferrocene/ferrocenium redox couple FcH+/FcH (FcH = FeII(η5–C5H5)2, Eo′ = 0.00 V) as reference. Decamethylferrocene, Fc* [62], was used as internal standard to prevent signal overlap with ferrocenyls or iridium of complexes 1–5 (Eo′ = −0.608 V versus free ferrocene with ΔE = 0.077 V and ipc/ipa = 1 under the conditions employed). Analyte concentrations were ca. 0.5 mM in CH2Cl2 and 0.1 M tetrabutylammonium tetrakispenta- fluorophenylborate, [N(nBu)4][B(C6F5)4] was used as supporting electrolyte. Data was exported to a spread sheet program for adjustment and diagram preparation.
Bulk electrolysis (BE) experiments were performed using a computer-controlled BAS model 100 B potentiostat ((Analytical Science Technology, Cape Town, South Africa). BE experiments were conducted in a three-compartment cell with a Pt-mesh working electrode, Pt-wire auxiliary electrode and Ag-wire reference electrode. Solutions containing 0.05 mmol·dm−3 analyte and 0.05 mmol·dm−3 [N(nBu)4][PF6] as electrolyte were subjected to a set potential until the current ratio reached 1.0 % of its original value.
3.6. DFT Calculations
All calculations were carried out using DFT in the gas or solvent phase using the Gaussian 16 program [63]. The B3LYP (Becke 1993 and Lee-Yang-Parr) hybrid functional [64,65] (UB3LYP for open shell species) with the triple-ζ basis set 6-311G(d,p) basis set was used for the lighter atoms (C, H, N, O, Fe), whereas the Stuttgart/Dresden (SDD) pseudopotential was used to describe the Ru and Ir electronic cores, while the metal valence electrons were described using the def2-TZVPP basis set [66]. For solvent calculations, the polarizable continuum model (PCM) [67] as implemented in Gaussian 16 was used with dichloromethane as solvent.
4. Conclusions
Ferrocene- and ruthenocene-containing iridium(III) heteroleptic complexes of the type [(ppy)2Ir(RCOCHCOR′)] may easily be synthesized in moderate (30–70%) yields. These complexes are stable up to 200 oC and may be stored for long times in air. A single crystal structure determination of 3 showed that ppy coordination is such that the pyridine fragments are trans relative to each other giving rise to a N-Ir-N angle approaching 180o. UV/vis spectroscopy shows complexes to absorb strongly in the UV region. Two weaker absorption bands are found in the visible light region between ca. 350 and 530 nm. The sequence of redox events is in the order Fc oxidation, then IrIII oxidation and finally ruthenocene oxidation, all in one-electron transfer steps. The ferrocenyl, IrIII and ruthenocenyl centres are all electrochemically reversibly oxidized, but generation of the ruthenocenium cation, Rc+, leads to compound degradation and loss of chemical reversibility of the Fc+/Fc and IrIII/IV redox couples. The precise potential at which IrIII are oxidized is dependent on the sum of Gordy scale group electronegativities, ΣχR, of R groups of the β-diketonato ligand. DFT calculations showed that the energy of the HOMOs of the species involved in the IrIII/IV oxidation, are inverse proportional to the redox potential of the IrIII/IV couple. This relationship provides a linear correlation with 98 % accurate prediction of the redox potential of the IrIII/IV couple of complexes of the type [(ppy)2Ir(RCOCHCORʹ)]. While [(ppy)2Ir(H3CCOCHCOCH3)] (6) exhibits phosphorescence with emission λmax = 518 nm at 25 oC, the ferrocenyl and ruthenocenyl molecular fragments of 1–5 quench all phosphorescence. Electrochemical generation of the oxidized species 6+ by generation of an IrIV moiety also cause phosphorescence quenching. Spectroelectrochemical results showed that IrIV generation led to very weak absorption patterns above 430 nm. These complexes may have potential as new antineoplastic drugs and they may also act as precursors towards iridium oxide and ruthenium oxide nano-electrocatalysts in electrochemical water splitting reactions.
Supplementary Materials
1H-NMR Spectra of 1–5; IR Spectra 1–5; Electrochemical Schemes for 1–6; Crystallographic C-H···O interactions within 3; Tables providing crystallographic information; DFT figures, data and optimized coordinates in the gas phase as well as in DCM.
Author Contributions
Investigation, B.E.B., J.C. and F.P.M.; Supervision, J.W.H.N. and J.C.S.; Writing—original draft, B.E.B., J.C., F.P.M., J.W.H.N. and J.C.S.; Writing—review & editing, J.C.S.
Funding
The authors acknowledge the South African National Research Foundation (J.C.S., Grant number 96123, J.C., Grant numbers 113327 and 96111 and F.P.M., 117995) the Central Research Fund of the University of the Free State, Bloemfontein, South Africa (J.C.S., J.C. and B.E.B.) for financial support. Funding is also acknowledged from Synfuels China Technology Co. Ltd., Beijing-Huairou, P.R. China (J.W.N.) and Syngaschem BV, The Netherlands (J.C.S. and B.E.B.).
Conflicts of Interest
The authors declare no conflicts of interest.
Data Availability
CCDC 1916701 contains the supplementary crystallographic data for COMPLEX 3 of this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: deposit@ccdc.cam.ac.uk).
References
- Jones, J.H. The Cativa™ Process for the Manufacture of Acetic Acid. Platin. Met. Rev. 2000, 44, 94–105. [Google Scholar]
- Sunley, G.J.; Watson, D.J. High productivity methanol carbonylation catalysis using iridium—The Cativa™ process for the manufacture of acetic acid. Catal. Today 2000, 58, 293–307. [Google Scholar] [CrossRef]
- Hetterscheid, D.G.H.; Van der Ham, C.J.M.; Diaz-Morales, O.; Verhoefen, M.W.G.M.; Longo, A.; Banerjee, D.; Niemantsverdriet, J.W.; Reek, J.N.H.; Feiters, M.C. Early stages of catalyst aging in the iridium mediated water oxidation reaction. Phys. Chem. Chem. Phys. 2016, 18, 10931–10940. [Google Scholar] [CrossRef]
- Sapountzi, F.M.; Gracia, J.M.; Westrate, C.J.; Fredriksson, H.O.A.; Niemantsverdriet, J.W. Electrocatalysts for the generation of hydrogen, oxygen and synthesis gas. Prog. Energy Combust. Sci. 2017, 58, 1–35. [Google Scholar] [CrossRef]
- Bennett, M.A.; Mitchell, T.R.B. γ-Carbon-bonded 2,4-pentanedionato complexes of trivalent iridium. Inorg. Chem. 1976, 15, 2936–2938. [Google Scholar] [CrossRef]
- Collins, J.E.; Castellani, M.; Rheingold, A.L.; Miller, E.J.; Geiger, W.E.; Rieger, A.L.; Rieger, P.H. Synthesis, characterization, and molecular structure of bis (tetraphenylcyclopentdienyl) rhodium (II). Organometallics 1995, 14, 1232–1238. [Google Scholar] [CrossRef]
- Han, F.-Q.; Han, C.-M.; Xu, H. Recent progress in functionalized electrophosphorescent iridium (III) complexes. Chin. Chem. Lett. 2016, 27, 1193–1200. [Google Scholar] [CrossRef]
- Kwong, R.C.; Sibley, S.; Dubovoy, T.; Balbo, M.A.; Forrest, S.R.; Thompson, M.E. Efficient, saturated red organic light emitting devices based on phosphorescent platinum (II) porphyrins. Chem. Mater. 1999, 11, 3709–3713. [Google Scholar] [CrossRef]
- Tung, Y.-L.; Wu, P.-C.; Liu, C.-S.; Chi, Y.; Yu, J.-K.; Hu, J.-K.; Chou, P.-T.; Peng, S.-M.; Lee, G.-H.; Tao, Y.; et al. Efficient red phosphorescent osmium (II) complexes for OLED applications. Organometallics 2004, 23, 3745–3748. [Google Scholar] [CrossRef]
- Ragni, R.; Maiorano, V.; Pugliese, M.; Maggiore, A.; Orselli, E.; Babudri, F.; Gigli, G.; De Cola, L.; Farinola, G.M. A highly fluorinated iridium complex as a blue-green emitting component for white electroluminescence. Synth. Met. 2017, 227, 148–155. [Google Scholar] [CrossRef]
- Holder, E.; Langeveld, B.M.W.; Schubert, U.S. New trends in the use of transition metal-ligand complexes for applications in electroluminescent devices. Adv. Mater. 2005, 17, 1109–1121. [Google Scholar] [CrossRef]
- Baldo, M.A.; O’Brien, D.F.; You, Y.; Shoustikov, A.; Sibley, S.; Thompson, M.E.; Forrest, S.R. Highly efficient phosphorescent emission from organic electroluminescent devices. Nature 1998, 395, 151–154. [Google Scholar] [CrossRef]
- Du Plessis, W.C.; Erasmus, J.J.C.; Lamprecht, G.J.; Conradie, J.; Cameron, T.S.; Aquino, M.A.S.; Swarts, J.C. Cyclic voltammetry of ferrocene-containing β-diketones as a tool to obtain group electronegativities. The structure of 3-ferrocenoyl-1,1,1-trifluoro-2-hydroxyprop-2-ene. Can. J. Chem. 1999, 77, 378–386. [Google Scholar] [CrossRef]
- Swarts, J.C.; Vosloo, T.G.; Cronje, S.J.; Du Plessis, W.C.; Van Rensburg, C.E.J.; Kreft, E.; Van Lier, J.E. Cytotoxicity of a series of ferrocene-containing beta-diketones. Anticancer Res. 2008, 28, 2781–2784. [Google Scholar] [PubMed]
- Kemp, K.C.; Fourie, E.; Conradie, J.; Swarts, J.C. Ruthenocene-containing β-diketones: Synthesis, pKa′ values, keto-enol isomerization kinetics, and electrochemical aspects. Organometallics 2008, 27, 353–362. [Google Scholar] [CrossRef]
- Kemp, K.C.; Nell, M.J.; Van Rensburg, C.E.J.; Swarts, J.C. Cytotoxicity of ruthenocene-containing β-diketones. Anticancer Res. 2012, 32, 2915–2918. [Google Scholar]
- Bryant, J.R.; Taves, J.E.; Mayer, J.M. Oxidations of hydrocarbons by manganese (III) tris (hexafluoroacetylacetonate). Inorg. Chem. 2002, 41, 2769–2776. [Google Scholar] [CrossRef]
- Lamprecht, G.J.; Swarts, J.C.; Conradie, J.; Leipoldt, J.G. Structure of carbonyl[(ferrocenecarbonyl)trifluoroacetonato-κO,κO]triphenylphosphinerhodium (I). Acta Cryst. 1993, C49, 82–84. [Google Scholar] [CrossRef]
- Erasmus, E.; Conradie, J.; Muller, A.; Swarts, J.C. Synthesis, crystal structure and electrochemistry of tetrahedral mono-β-diketonato titanocenyl complexes. Inorg. Chim. Acta 2007, 360, 2277–2283. [Google Scholar] [CrossRef]
- Imai, H.; Yoshikatsu, Y. Syntheses of Acetoacetylferrocene and Its Metal Chelates. Nippon Kagaku Zasshi 1970, 91, 452–457. [Google Scholar] [CrossRef]
- Atim, S.; Yang, L.; Nesterov, V.; Wang, X.; Richmond, M.G. Synthesis of the labile rhenium (I) complexes fac-Re(CO)3(L)[k2-O,O-FcC(O)CHC(O)Me] (where Fc = ferrocenyl; L = THF, H2O, alkyne) and alkyne addition to the diketonate ligand. J. Organomet. Chem. 2018, 874, 87–100. [Google Scholar] [CrossRef]
- Obuah, C.; Ainooson, M.K.; Darkwa, J. Effects of electrochemical properties of ferrocenylpyrazolylnickel (II) and palladium (II) compounds on their catalytic activities in ethylene oligomerisation reactions. RSC Adv. 2018, 8, 5362–5371. [Google Scholar] [CrossRef]
- Koroteev, P.S.; Dobrokhotova, Z.V.; Ilyukhin, A.B.; Efimov, N.N.; Rouzières, M.; Kiskin, M.A.; Clérac, R.; Novotortsev, V.M. Synthesis, structure, and physical properties of new rare earth ferrocenoylacetonates. Dalton Trans. 2016, 45, 6405–6417. [Google Scholar] [CrossRef] [PubMed]
- Koroteev, P.S.; Dobrokhotova, Z.V.; Ilyukhin, A.B.; Efimov, N.N.; Novotortsev, V.M. Synthesis, structure, and magnetic properties of lanthanide ferrocenoylacetonates with nitrate and 2,2′-bipyridine ligands. J. Coord. Chem. 2016, 69, 2723–2735. [Google Scholar] [CrossRef]
- Buitendach, B.E.; Gągor, A.; Swarts, J.C. Electrochemical Evidence of Intramolecular Electronic Communication in Zr and Hf Phthalocyanines Bearing Ferrocene-Containing β-Diketonato Axial Ligands: Structure of [PcHf(FcCOCHCOC6H5)2]. Inorg. Chem. 2013, 52, 10245–10257. [Google Scholar] [CrossRef]
- Gericke, H.J.; Muller, A.J.; Swarts, J.C. Electrochemical illumination of intramolecular communication in ferrocene-containing tris-β-diketonato aluminum (III) complexes; cytotoxicity of Al(FcCOCHCOCF3)3. Inorg. Chem. 2012, 51, 1552–1561. [Google Scholar] [CrossRef]
- Buitendach, B.E.; Erasmus, E.; Landman, M.; Niemantsverdriet, J.W.; Swarts, J.C. Consequences of electron-density manipulations on the X-ray photoelectron spectroscopic properties of ferrocenyl-β-diketonato complexes of manganese (III). Structure of [Mn(FcCOCHCOCH3)3]. Inorg. Chem. 2016, 55, 1992–2000. [Google Scholar] [CrossRef]
- Joubert, C.C.; Van As, L.; Jakob, A.; Speck, M.J.; Lang, H.; Swarts, J.C. Intramolecular electronic communication in ferrocene-based β-diketonato copper (II) complexes as observed by an electrochemical study. Polyhedron 2013, 55, 80–86. [Google Scholar] [CrossRef]
- Conradie, J.; Swarts, J.C. Oxidative addition of CH3I and CO migratory insertion in a series of ferrocene-containing carbonyl phosphine betadiketono rhodium (I) complexes. Organometallics 2009, 28, 1018–1026. [Google Scholar] [CrossRef]
- Vosloo, T.G.; du Plessis, W.C.; Swarts, J.C. Kinetics of substitution of ferrocenyl-containing β-diketonatoligands by phenanthroline fromβ-diketonato-1,5-cyclooctadienerhodium (I) complexes. Inorg. Chim. Acta 2002, 331, 188–193. [Google Scholar] [CrossRef]
- Conradie, J. A DFT study of the reactivity of β-diketonato-1,5-cyclo-octadieneiridium(I) complexes. Polyhedron 2013, 51, 164–167. [Google Scholar] [CrossRef]
- Lamansky, S.; Djurovich, P.; Murphy, D.; Abdel-Razzaq, F.; Kwong, R.; Tsyba, I.; Bortz, M.; Mui, B.; Thompson, M.E. Synthesis and Characterization of Phosphorescent Cyclometalated Iridium Complexes. Inorg. Chem. 2001, 40, 1704–1711. [Google Scholar] [CrossRef] [PubMed]
- Chepelin, O.; Ujma, J.; Wu, X.; Slawin, A.M.Z.; Pitak, M.B.; Coles, S.J.; Michel, J.; Jones, A.C.; Barran, P.E.; Lusby, P.J. Luminescent, Enantiopure, Phenylatopyridine Iridium-Based Coordination Capsules. J. Am. Chem. Soc. 2012, 134, 19334–19337. [Google Scholar] [CrossRef] [PubMed]
- Takayasu, S.; Suzuki, T.; Shinozaki, K. Intermolecular Interactions and Aggregation of fac-Tris(2-phenylpyridinato-C2,N)iridium (III) in Nonpolar Solvents. J. Phys. Chem. B 2013, 117, 9449–9456. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.H.; Davies, J.E.; Galloy, J.J.; Johnson, O.; Kennard, O.; Macrae, C.F.; Mitchell, E.M.; Mitchell, G.F.; Smith, J.M.; Watson, D.G. The development of versions 3 and 4 of the Cambridge Structural Database System. J. Chem. Inf. Comput. Sci. 1991, 31, 187–204. [Google Scholar] [CrossRef]
- Du Plessis, W.C.; Davis, W.L.; Cronje, S.J.; Swarts, J.C. Structural, thermodynamic and kinetic consequences of a spectroscopic study of the equilibrium between isomeric forms of ferrocene-containing β-diketones. Inorg. Chim. Acta 2001, 314, 97–104. [Google Scholar] [CrossRef]
- Von Chrzanowski, L.S.; Lutz, M.; Spek, A.L. γ-Tris(2,4-pentanedionato-κ2O,O′) aluminium (III) at 110 K. Acta Cryst. 2006, E62, 3318–3320. [Google Scholar] [CrossRef]
- Dulatas, L.T.; Brown, S.N.; Ojomo, E.; Noll, B.C.; Cavo, M.J.; Holt, P.B.; Wopperer, M.M. Intermetallic Communication in Titanium (IV) Ferrocenyldiketonates. Inorg. Chem. 2009, 48, 10789–10799. [Google Scholar] [CrossRef]
- Woisetschlager, O.E.; Geisbauer, A.; Polborn, K.; Beck, W.Z. Kohlenwasserstoffverbrückte Metallkomplexe. XLIX Koordinationschemie von bis (ferrocenyl) substituierten 1,3-Diketonaten mit Ruthenium, Rhodium, Iridium und Palladium. Anorg. Allg. Chem. 2000, 626, 766–774. [Google Scholar] [CrossRef]
- Bell, W.; Crayston, J.A.; Glidewell, C.; Mazid, M.A.; Hursthause, M.B. The constitution of a ferrocenyl diketone: Solution and solid state NMR spectroscopy. Crystal structure of 1-ferrocenyl-3-hydroxybut-2-en-1-one. J. Organomet. Chem. 1992, 434, 115–121. [Google Scholar] [CrossRef]
- Woisetschläger, O.E.; Geisbauer, A.; Polborn, K.; Sünkel, K.; Beck, W.Z. Spacer-verbrückte Bis-, Tris-und Tetrakis (ferrocenyl)-1,3-Diketone. Anorg. Allg. Chem. 1999, 625, 2164–2168. [Google Scholar] [CrossRef]
- Chang, C.-J.; Yang, C.-H.; Chen, K.; Chi, Y.; Shu, C.-F.; Ho, M.-L.; Yeh, Y.-S.; Chou, P.-T. Color tuning associated with heteroleptic cyclometalated Ir (III) complexes: Influence of the ancillary ligand. Dalton Trans. 2007, 36, 1881–1890. [Google Scholar] [CrossRef]
- Swarts, J.C.; Nafady, A.; Roudebush, J.H.; Trupia, S.; Geiger, W.E. One-Electron Oxidation of Ruthenocene: Reactions of the Ruthenocenium Ion in Gentle Electrolyte Media. Inorg. Chem. 2009, 48, 2156–2165. [Google Scholar] [CrossRef] [PubMed]
- Barrière, F.; Geiger, W.E. Use of Weakly Coordinating Anions to Develop an Integrated Approach to the Tuning of ΔE1/2 Values by Medium Effects. J. Am. Chem. Soc. 2006, 128, 3980–3989. [Google Scholar] [CrossRef] [PubMed]
- Cook, M.J.; Chambrier, I.; White, G.F.; Fourie, E.; Swarts, J.C. Electrochemical and EPR studies of two substituted bis-cadmium tris-phthalocyanine complexes: Elucidation of unexpectedly different free-radical character. Dalton Trans. 2009, 7, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Kissinger, P.T.; Heineman, W.R. Cyclic Voltammetry. J. Chem. Educ. 1983, 60, 702–706. [Google Scholar] [CrossRef]
- Fourie, E.; Swarts, J.C.; Chambrier, I.; Cook, M.J. Electrochemical and spectroscopic detection of self-association of octa-alkyl phthalocyaninato cadmium compounds into dimeric species. Dalton Trans. 2009, 7, 1145–1154. [Google Scholar] [CrossRef]
- Gericke, H.J.; Barnard, N.I.; Erasmus, E.; Swarts, J.C.; Cook, M.J.; Aquino, M.A.S. Solvent and Electrolyte Effects in Enhancing the Identification of Intramolecular Electronic Communication in a Multi Redox-Active Diruthenium Tetraferrocenoate Complex, a Triple-Sandwiched Dicadmium Phthalocyanine and a Ruthenocene-Containing β-Diketone. Inorg. Chim. Acta 2010, 363, 2222–2232. [Google Scholar] [CrossRef]
- Cohen, S.G. Gordy Scale Group Electronegativities, χR, Are Empirical Numbers that Express the Combined Tendency of a Group of Atoms, Like R = CF3 or ferrocenyl (Fc), to Attract Electrons (Including Those in a Covalent Bond) as a Function of the Number of Valence Electrons, n, and the Covalent Radius, r (Å), of Groups as Discussed in Reference 62 and also by Wells, P.R. In Progress in Physical Organic Chemistry; John Wiley & Sons, Inc.: New York, NY, USA, 1968; Volume 6, pp. 111–145. [Google Scholar]
- Kagarise, R.E. Relationship Between the Electronegativities of Adjacent Substituents and the Stretching Frequency of the Carbonyl Group. J. Am. Chem. Soc. 1955, 77, 1377–1379. [Google Scholar] [CrossRef]
- Pavlishchuk, V.V.; Addison, A.W. Conversion constants for redox potentials measured versus different reference electrodes in acetonitrile solutions at 25 °C. Inorg. Chim. Acta 2000, 298, 97–102. [Google Scholar] [CrossRef]
- Hill, M.G.; Lamanna, W.M.; Mann, K.R. Tetrabutylammonium tetrakis [3,5-bis(trifluoromethyl)phenyl]borate as a noncoordinating electrolyte: Reversible 1e- oxidations of ruthenocene, osmocene, and Rh2 (TM4)42+ (TM4 = 2,5-diisocyano-2,5-dimethylhexane). Inorg. Chem. 1991, 30, 4687–4690. [Google Scholar] [CrossRef]
- Conradie, J. A Frontier orbital energy approach to redox potentials. J. Phys. Conf. Ser. 2015, 633, 12045–12050. [Google Scholar] [CrossRef]
- Ferreira, H.; Conradie, M.M.; Conradie, J. Electrochemical properties of a series of Co (II) complexes, containing substituted phenanthrolines. Electrochim. Acta 2018, 292, 489–501. [Google Scholar] [CrossRef]
- Lamansky, S.; Djurovich, P.; Murphy, D.; Abdel-Razzaq, F.; Lee, H.-E.; Adachi, C.; Burrows, P.E.; Forrest, S.R.; Thompson, M.E. Highly phosphorescent bis-cyclometalated iridium complexes: Synthesis, photophysical characterization, and use in organic light emitting diodes. J. Am. Chem. Soc. 2001, 123, 4304–4312. [Google Scholar] [CrossRef] [PubMed]
- Traverso, O.; Rossi, R.; Magon, L.; Cinquantini, A.; Kemp, T.J. The quenching of excited uranyl ion by d 6 metallocenes. J. Chem. Soc. Dalton Trans. 1978, 6, 569–572. [Google Scholar] [CrossRef]
- Erasmus, J.J.C.; Lamprecht, G.J.; Swarts, J.C.; Roodt, A.; Oskarsson, Å. (E)-1,3-Diferrocenyl-2-buten-1-one-Water (4/1). Acta Cryst. 1996, C52, 3000–3002. [Google Scholar] [CrossRef]
- LeSuer, R.J.; Buttolph, C.; Geiger, W.E. Comparison of the Conductivity Properties of the Tetrabutylammonium Salt of Tetrakis (pentafluorophenyl) borate Anion with Those of Traditional Supporting Electrolyte Anions in Nonaqueous Solvents. Anal. Chem. 2004, 76, 6395–6401. [Google Scholar] [CrossRef]
- Bruker AXS Inc. APEXII and SADABS; Bruker AXS Inc.: Madison, WI, USA, 2012. [Google Scholar]
- Sheldrick, G.M. Shelxt—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Ruiz Aranzaes, J.; Daniel, M.-C.; Astruc, D. Metallocenes as references for the determination of redox potentials by cyclic voltammetry: Permethylated iron and cobalt sandwich complexes, inhibition by polyamine dendrimers and the role of hydroxyl-containing ferrocenes. Can. J. Chem. 2006, 84, 288–299. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).