
Towards the Development of an In vivo Chemical Probe for Cyclin G Associated Kinase (GAK)

 ,
,
Abstract
:
1. Introduction
2. Results
2.1. Initial Investigation of P450 Metabolism of 1
2.2. Synthesis of Analogs of 1
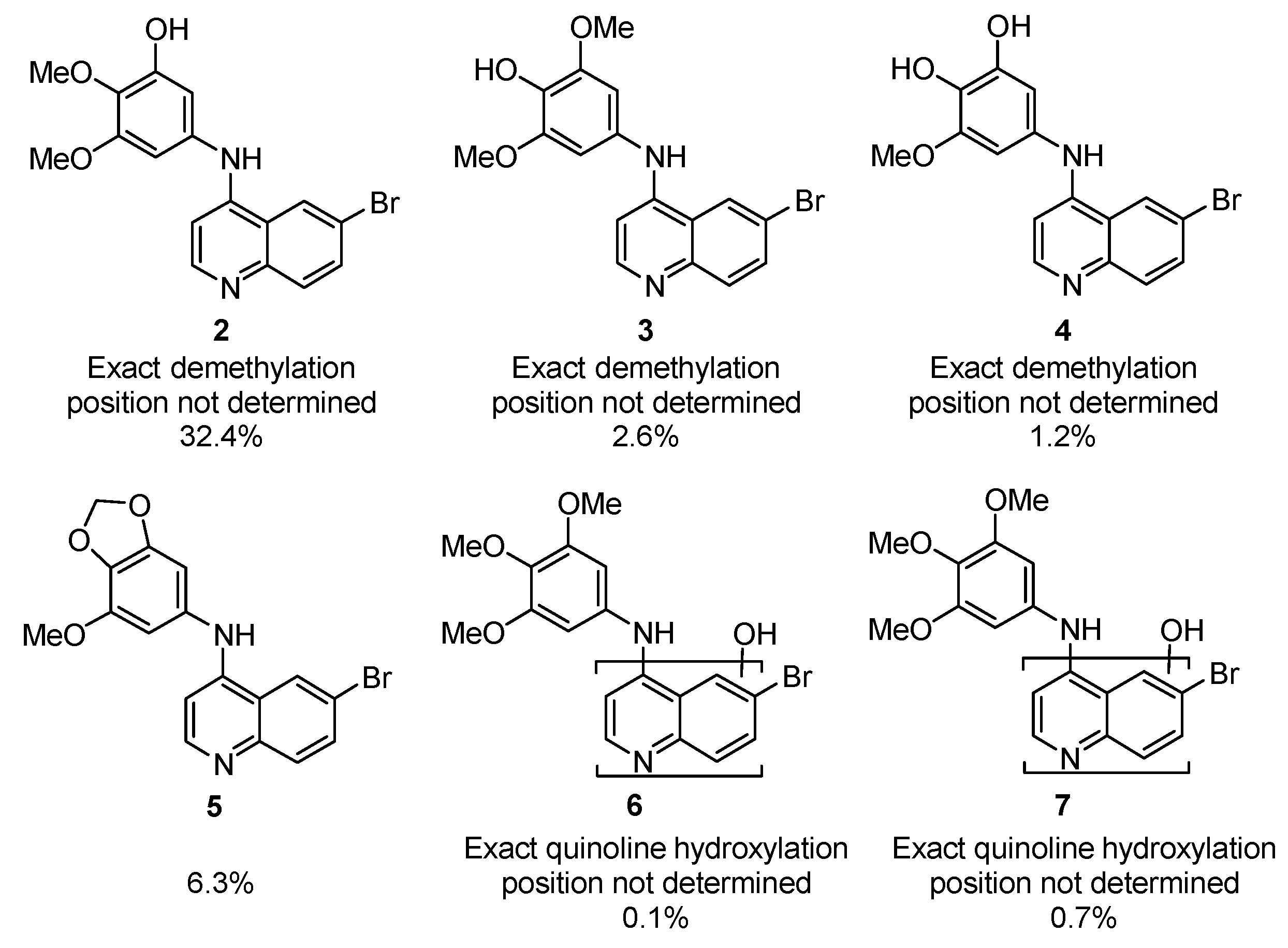
2.3. Metabolite Invesitigation Focused 4-Anilinoquinolines Analogs 8–13
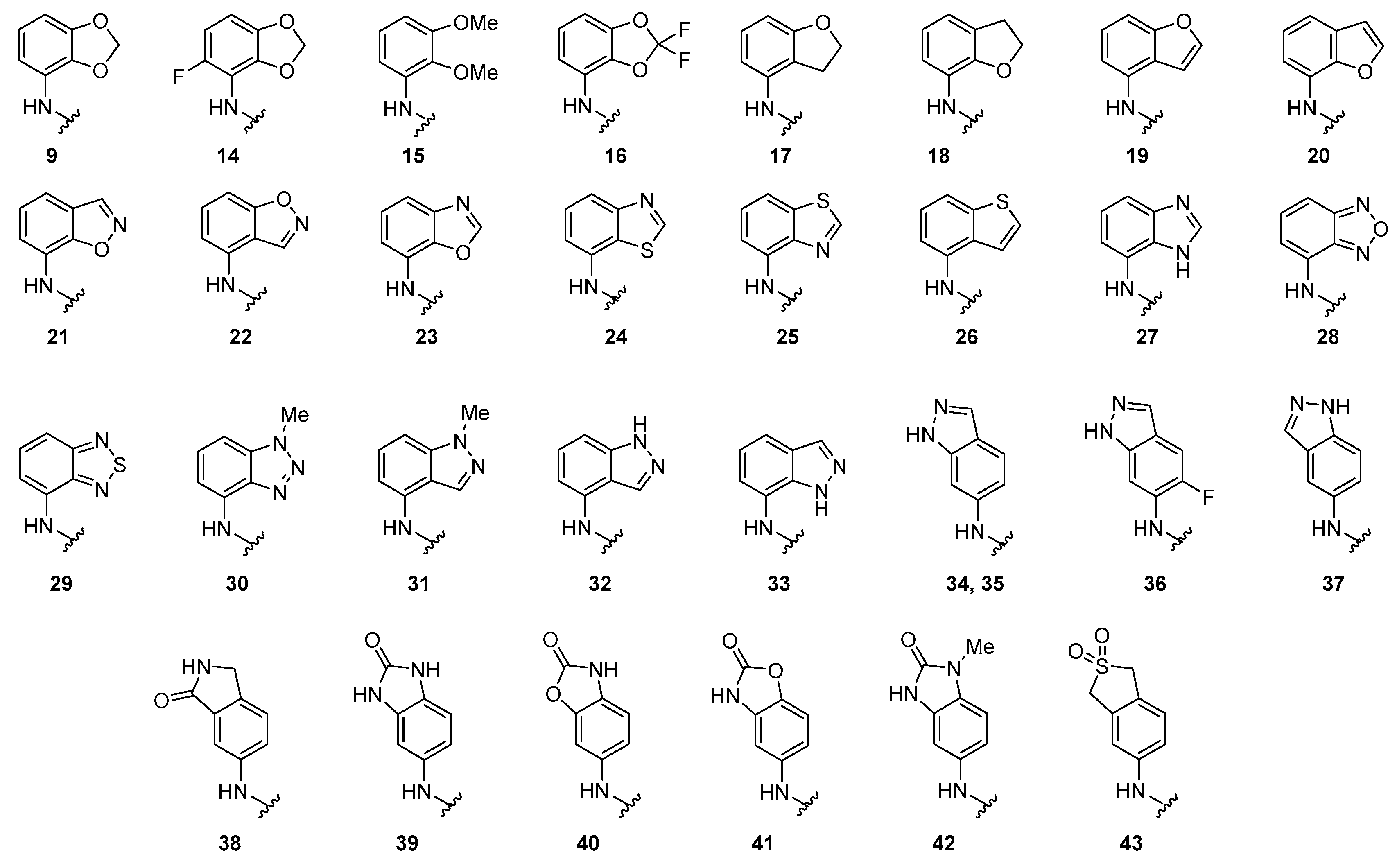
2.4. Design and Tesing of 4-Anilinoquinolines Analogs 14–43
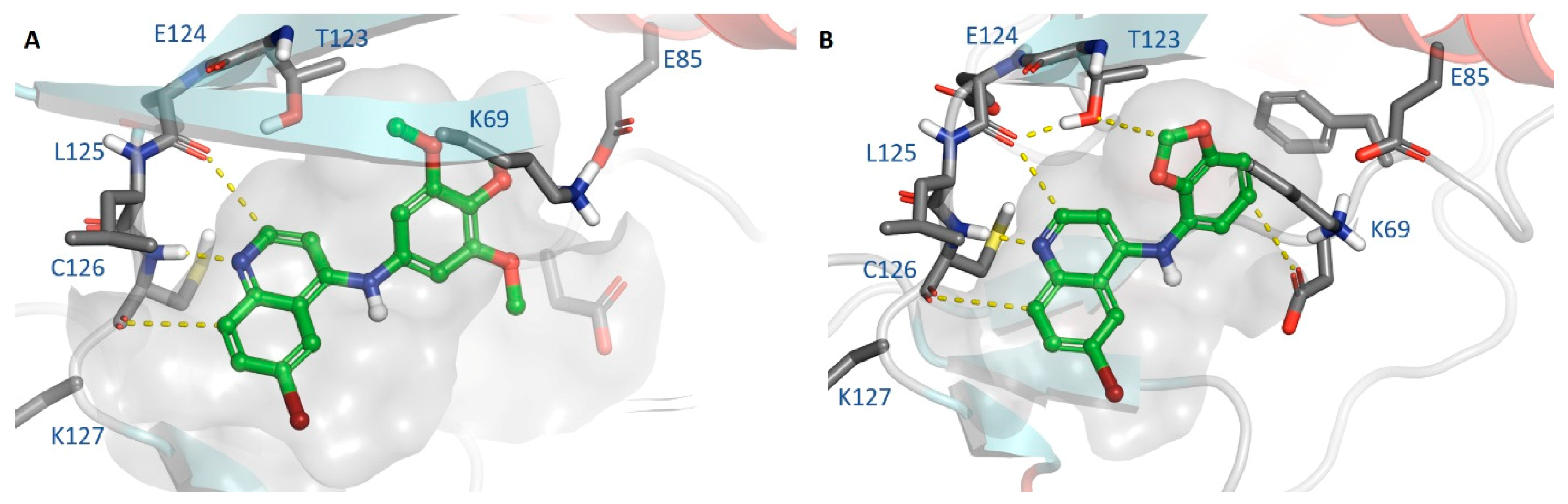
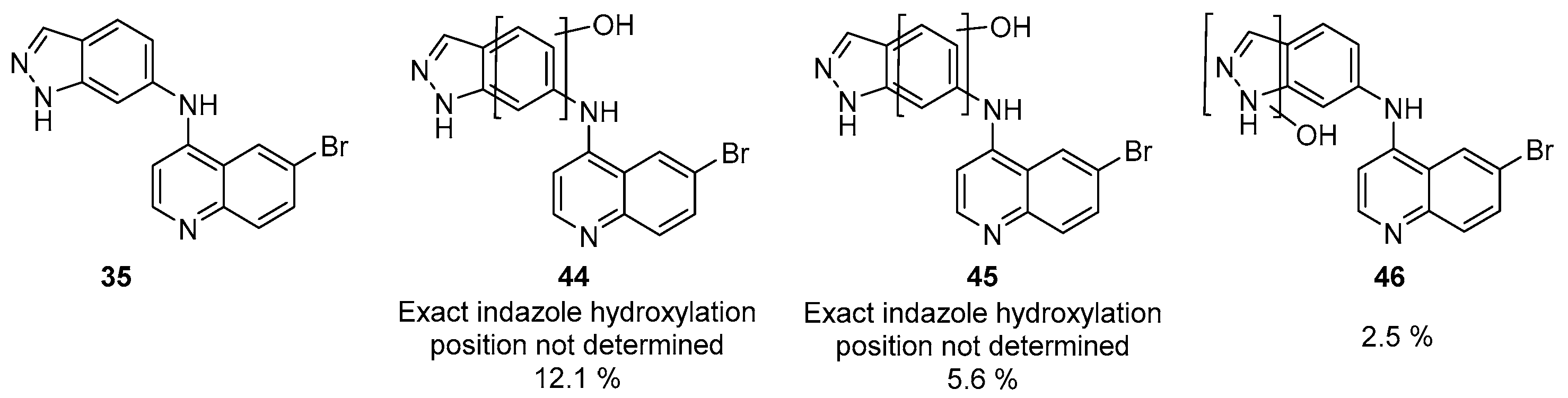
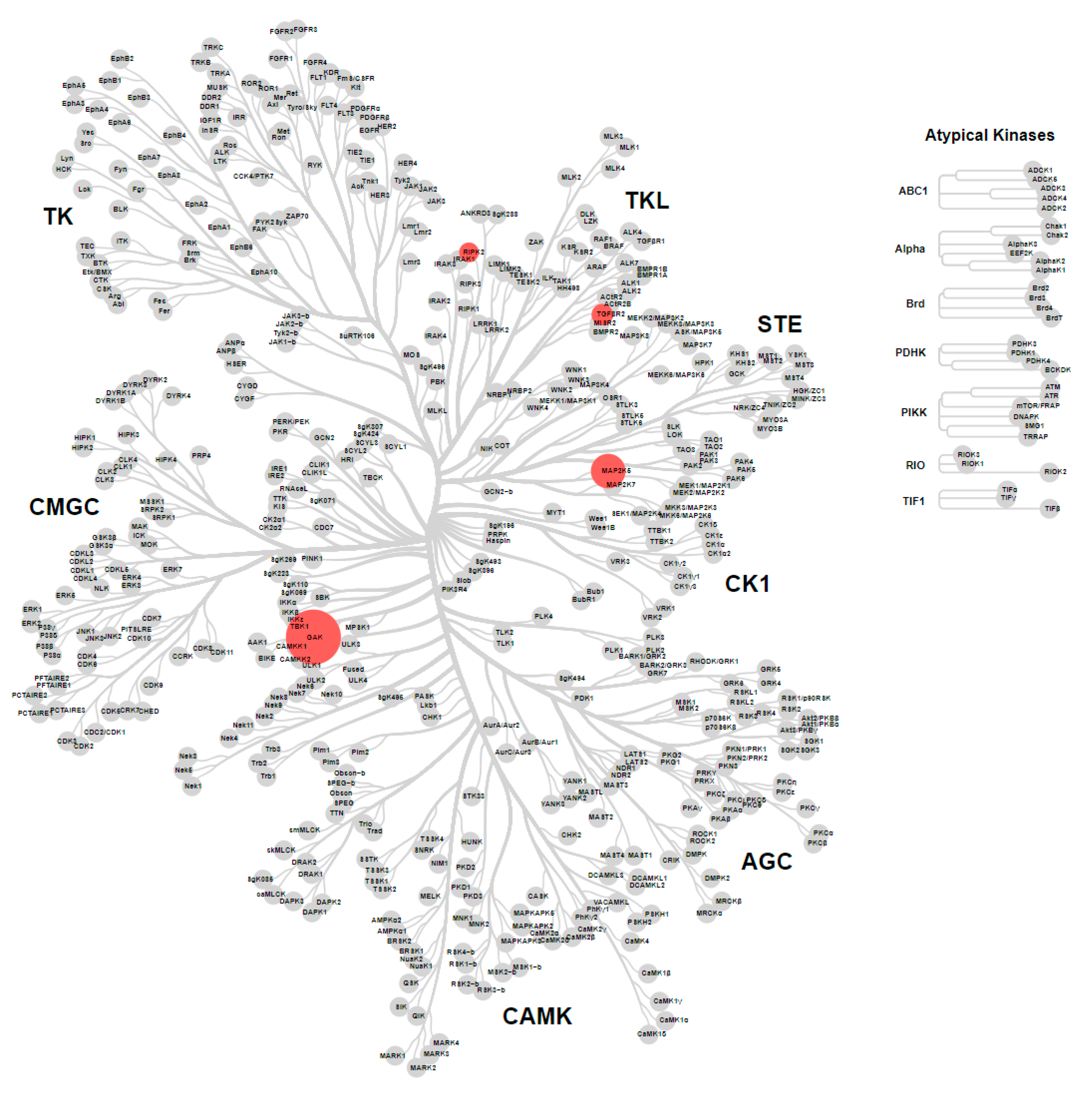
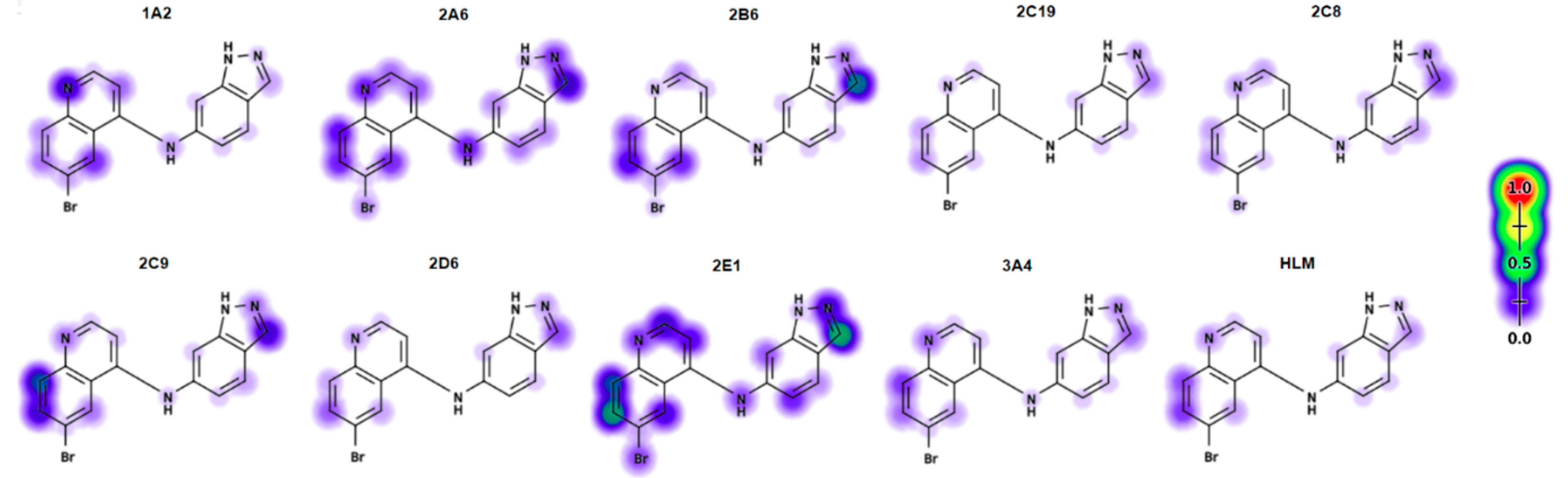
2.5. Metabolite ID Profiling and Kinome Scan of 35
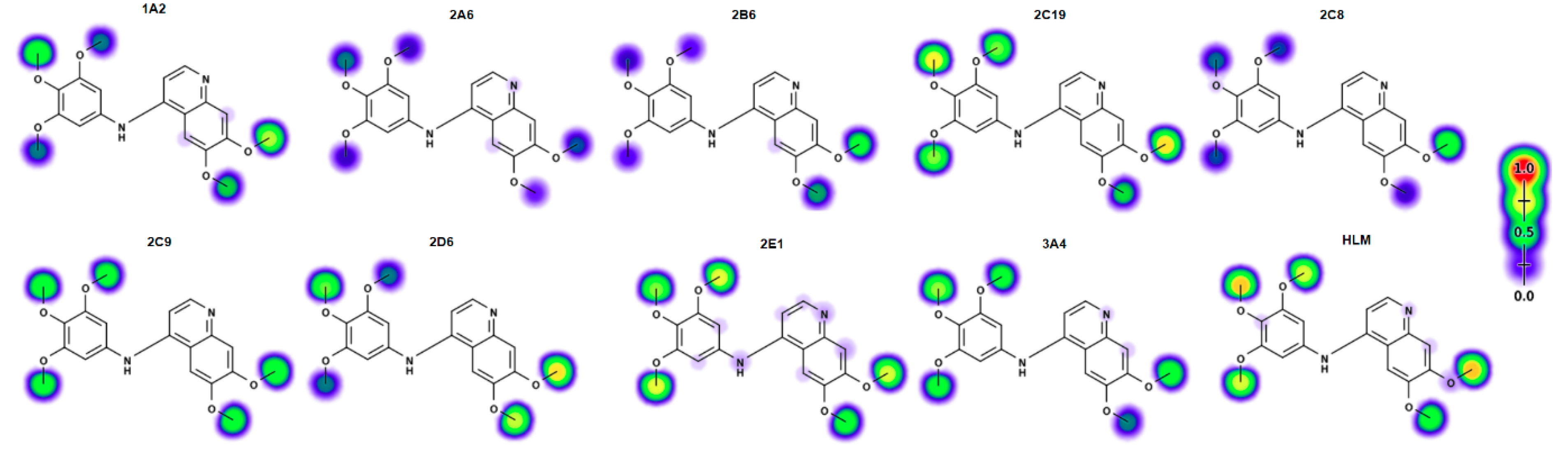
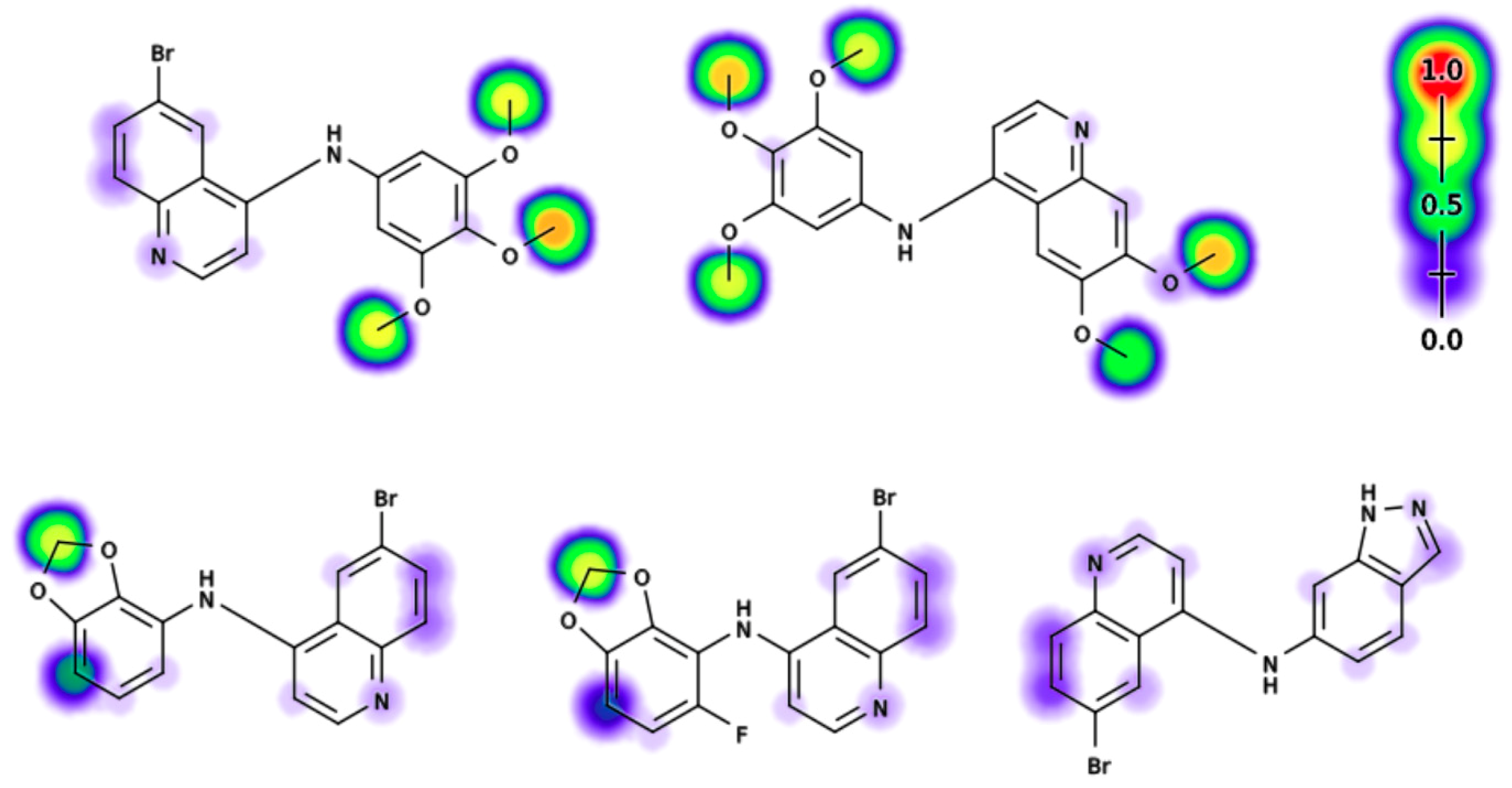
2.6. Electronic Fuikui Modelling Aniline of 4-Anilinoquinolines
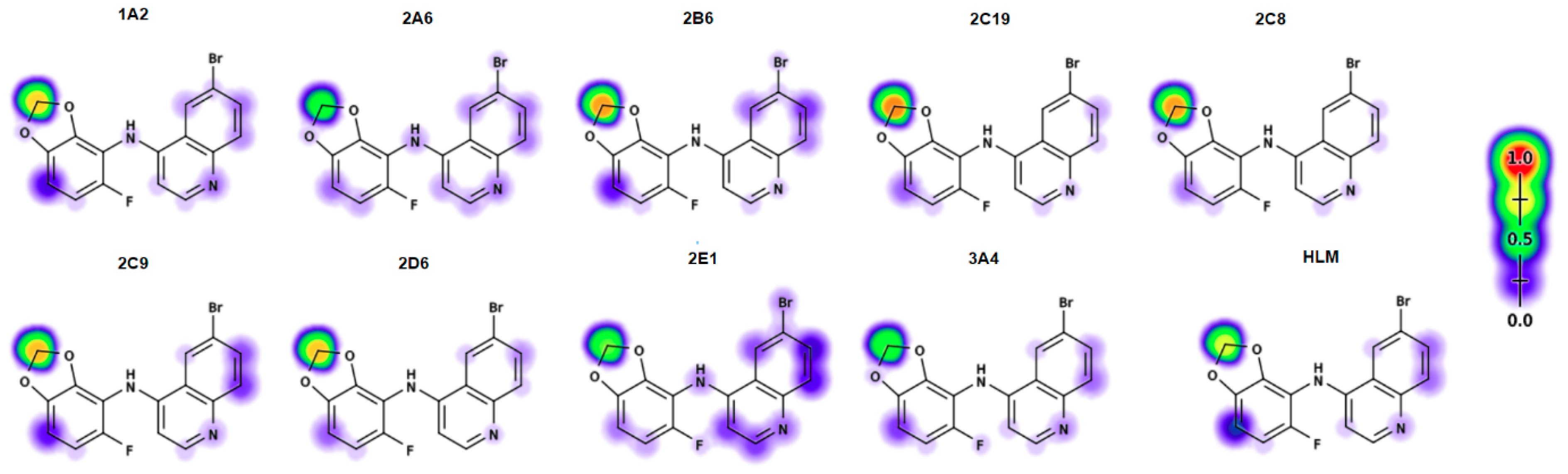
2.7. Modelling of Lead Compounds in XenoSite
2.8. 4-Anilinoquinolines Stablisation with ABT
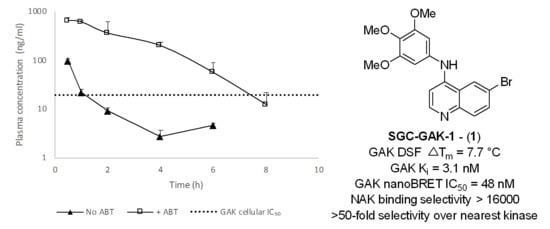
2.9. In Vivo Stabilisation of 1 and 8 with ABT
3. Discussion
4. Materials and Methods
4.1. Computational Methods
4.2. Biology Methods
4.2.1. NAK Family TR-FRET Assays
4.2.2. GAK NanoBRET Assay
4.2.3. Metabolite Identification
4.2.4. Pharmacokinetic Profiling
4.2.5. KinomeScan Assay
4.3. Chemistry Methods and Compound Characterization
4.3.1. General Information
4.3.2. General Procedure for the Synthesis of 4-anilinoquinolines (Scheme 1)
4.3.3. General Procedure for the Synthesis of 4-anilinoquinolines (Scheme 2)
4.3.4. Compound Characterization
4.3.5. Mass Spectrometry Method
4.4. Abbreviations Used
- cyclin G-associated kinase (GAK)
- mouse liver microsomes (MLM)
- nicotinamide adenine dinucleotide phosphate (NADPH)
- liquid chromatography mass spectra (LCMS)
- mitogen-activated protein kinase kinase 5 (MEK5/MAP2K5)
- transforming growth factor beta receptor 2 (TGFBR2)
- receptor-interacting serine/threonine-protein kinase 2 (RIPK2)
- 1-aminobenzotriazole(ABT)
- milligrams per kilogram (MPK)
- N-methyl-2-pyrrolidoneolidone (NMP)
- Bis(dibenzylideneacetone)palladium(0) (Pd(dba)2)
- 2-Dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl (XPhos)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sorrell, F.J.; Szklarz, M.; Abdul Azeez, K.R.; Elkins, J.M.; Knapp, S. Family-wide structural analysis of human numb-associated protein kinases. Structure 2016, 24, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Kanaoka, Y.; Kimura, S.H.; Okazaki, I.; Ikeda, M.; Nojima, H. GAK: A cyclin G associated kinase contains a tensin/auxilin-like domain. FEBS Lett. 1997, 402, 73–80. [Google Scholar] [CrossRef]
- Sato, J.; Shimizu, H.; Kasama, T.; Yabuta, N.; Nojima, H. GAK, a regulator of clathrin-mediated membrane trafficking, localizes not only in the cytoplasm but also in the nucleus. Genes Cells 2009, 14, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Dzamko, N.; Zhou, J.; Huang, Y.; Halliday, G.M. Parkinson’s disease-implicated kinases in the brain; insights into disease pathogenesis. Front. Mol. Neurosci. 2014, 7, 57. [Google Scholar] [CrossRef] [PubMed]
- Chaikuad, A.; Keates, T.; Vincke, C.; Kaufholz, M.; Zenn, M.; Zimmermann, B.; Gutierrez, C.; Zhang, R.G.; Hatzos-Skintges, C.; Joachimiak, A.; et al. Structure of cyclin G-associated kinase (GAK) trapped in different conformations using nanobodies. Biochem. J. 2014, 459, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Susa, M.; Choy, E.; Liu, X.; Schwab, J.; Hornicek, F.J.; Mankin, H.; Duan, Z. Cyclin G-associated kinase is necessary for osteosarcoma cell proliferation and receptor trafficking. Mol. Cancer Ther. 2010, 9, 3342–3350. [Google Scholar] [CrossRef]
- Fabian, M.A.; Biggs, W.H.; Treiber, D.K.; Atteridge, C.E.; Azimioara, M.D.; Benedetti, M.G.; Carter, T.A.; Ciceri, P.; Edeen, P.T.; Floyd, M.; et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23, 329–336. [Google Scholar] [CrossRef]
- Asquith, C.R.M.; Berger, B.T.; Wan, J.; Bennett, J.M.; Capuzzi, S.J.; Crona, D.J.; Drewry, D.H.; East, M.P.; Elkins, J.M.; Fedorov, O.; et al. SGC-GAK-1: A chemical probe for Cyclin G Associated Kinase (GAK). J. Med. Chem. 2019, 62, 2830–2836. [Google Scholar] [CrossRef]
- Asquith, C.R.M.; Naegeli, K.M.; East, M.P.; Laitinen, T.; Havener, T.M.; Wells, C.I.; Johnson, G.L.; Drewry, D.H.; Zuercher, W.J.; Morris, D.C. Design of a Cyclin G Associated Kinase (GAK)/Epidermal Growth Factor Receptor (EGFR) inhibitor set to interrogate the relationship of EGFR and GAK in Chordoma. J. Med. Chem. 2019, 62, 4772–4778. [Google Scholar] [CrossRef]
- Sakurai, M.A.; Ozaki, Y.; Okuzaki, D.; Naito, Y.; Sasakura, T.; Okamoto, A.; Tabara, H.; Inoue, T.; Hagiyama, M.; Ito, A.; et al. Gefitinib and luteolin cause growth arrest of human prostate cancer PC-3 cells via inhibition of cyclin G-associated kinase and induction of miR-630. PLoS ONE 2014, 9, e100124. [Google Scholar] [CrossRef]
- Asquith, C.R.M.; Tizzard, G. 6-Bromo-N-(2-methyl-2H-benzo[d][1,2,3]triazol-5-yl)quinolin-4-amine. Molbank 2019, 4, M1087. [Google Scholar] [CrossRef]
- Asquith, C.R.M.; Laitinen, T.; Bennett, J.M.; Godoi, P.H.; East, M.P.; Tizzard, G.J.; Graves, L.M.; Johnson, G.L.; Dornsife, R.E.; Wells, C.I.; et al. Identification and optimization of 4-anilinoquinolines as inhibitors of cyclin G associated kinase. Chem. Med. Chem. 2018, 13, 48–66. [Google Scholar] [CrossRef] [PubMed]
- Asquith, C.R.M.; Treiber, D.K.; Zuercher, W.J. Utilizing comprehensive and mini-kinome panels to optimize the selectivity of quinoline inhibitors for cyclin G Associated Kinase (GAK). Bioorg. Med. Chem. Lett. 2019, 29, 1727–1731. [Google Scholar] [CrossRef] [PubMed]
- Asquith, C.R.M.; Fleck, N.; Torrice, C.D.; Crona, D.J.; Grundner, C.; Zuercher, W.J. Anti-tubercular activity of novel 4-anilinoquinolines and 4-anilinoquinazolines Bioorg. Med. Chem. Lett. 2019, 18, 2695–2699. [Google Scholar] [CrossRef]
- Vasta, J.D.; Corona, C.R.; Wilkinson, J.; Zimprich, C.A.; Hartnett, J.R.; Ingold, M.R.; Zimmerman, K.; Machleidt, T.; Kirkland, T.A.; Huwiler, K.G.; et al. Quantitative, wide-spectrum kinase profiling in live cells for assessing the effect of cellular ATP on target engagement. Cell Chem. Biol. 2018, 25, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Kellenberger, E.; Rodrigo, J.; Muller, P.; Rognan, D. Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins 2004, 57, 225–242. [Google Scholar] [CrossRef]
- Ohbayashi, N.; Murayama, K.; Kato-Murayama, M.; Kukimoto-Niino, M.; Uejima, T.; Matsuda, T.; Ohsawa, N.; Yokoyoma, S.; Nojima, H.; Shirouzu, M. Structural basis for the inhibition of cyclin G-associated kinase by gefitinib. ChemistryOpen 2018, 7, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Kovackova, S.; Chang, L.; Bekerman, E.; Neveu, G.; Barouch-Bentov, R.; Chaikuad, A.; Heroven, C.; Sala, M.; De Jonghe, S.; Knapp, S.; et al. Selective inhibitors of cyclin G Associated Kinase (GAK) as anti-hepatitis C agents. J. Med. Chem. 2015, 58, 3393–3410. [Google Scholar] [CrossRef]
- Mineva, T.; Russo, N. Atomic Fukui indices and orbital hardnesses of adenine, thymine, uracil, guanine and cytosine from density functional computations. J. Mol. Struct. Theochem. 2010, 943, 71–76. [Google Scholar] [CrossRef]
- Matlock, M.K.; Hughes, T.B.; Swamidass, S.J. XenoSite server: A web-available site of metabolism prediction tool. Bioinformatics 2015, 31, 1136–1137. [Google Scholar] [CrossRef]
- Zaretzki, J.; Rydberg, P.; Bergeron, C.; Bennett, K.P.; Olsen, L.; Breneman, C.M. RS-Predictor models augmented with SMARTCyp reactivities: Robust metabolic regioselectivity predictions for nine CYP isozymes. J. Chem. Inf. Model. 2012, 52, 1637–1659. [Google Scholar] [CrossRef] [PubMed]
- Zaretzki, J.; Matlock, M.; Swamidass, S.J. XenoSite: Accurately predicting CYP-mediated sites of metabolism with neural networks. J. Chem. Inf. Model. 2013, 53, 3373–3383. [Google Scholar] [CrossRef] [PubMed]
- De Montellano, P.R.O. 1-Aminobenzotriazole: A mechanism-based cytochrome P450 inhibitor and probe of cytochrome P450 biology. Med. Chem. 2018, 8, 038. [Google Scholar] [CrossRef] [PubMed]
- Linder, C.D.; Renaud, N.A.; Hutzler, J.M. Is 1-aminobenzotriazole an appropriate in vitro tool as a nonspecific cytochrome P450 inactivator? Drug Metab. Dispos. 2009, 37, 10–13. [Google Scholar] [CrossRef]
- Stringer, R.A.; Ferreira, S.; Rose, J.; Ronseaux, S. Application of osmotic pumps for sustained release of 1-aminobenzotriazole and inhibition of cytochrome P450 enzymes in mice: Model comparison with the hepatic P450 reductase null mouse. Drug Metab. Dispos. 2016, 44, 1213–1216. [Google Scholar] [CrossRef]
- Frye, S.V. The art of the chemical probe. Nat. Chem. Biol. 2010, 6, 159–161. [Google Scholar] [CrossRef]
- Arrowsmith, C.H.; Audia, J.E.; Austin, C.; Baell, J.; Bennett, J.; Blagg, J.; Bountra, C.; Brennan, P.E.; Brown, P.J.; Bunnage, M.E.; et al. The promise and peril of chemical probes. Nat. Chem. Biol. 2015, 11, 536–541. [Google Scholar] [CrossRef] [Green Version]
- Di, L.; Kerns, E.H.; Ma, X.J.; Huang, Y.; Carter, G.T. Applications of high throughput microsomal stability assay in drug discovery. Comb. Chem. High Throughput Screen. 2008, 11, 469–476. [Google Scholar] [CrossRef]
- Interactive Substructure Search of the Drugbank Database. Available online: http://www.cheminfo.org/flavor/cheminformatics/Database/Drugbank_structure_search/index.html (accessed on 11 October 2019).
- Strelevitz, T.J.; Foti, R.S.; Fisher, M.B. In Vivo use of the P450 inactivator 1-aminobenzotriazole in the rat: Varied dosing route to elucidate gut and liver contributions to first-pass and systemic clearance. J. Pharm. Sci. 2006, 95, 1334–1341. [Google Scholar] [CrossRef]
- Larson, K.B.; Wang, K.; Delille, C.; Otofokun, I.; Acosta, E.P. Pharmacokinetic enhancers in HIV therapeutics. Clin. Pharm. 2014, 53, 865–872. [Google Scholar] [CrossRef]
- Zhao, Y.; Lee, M.H.; Paderu, P.; Lee, A.; Jimenez-Ortigosa, C.; Park, S.; Mansbach, R.S.; Shaw, K.J.; Perlin, D.S. Significantly improved pharmacokinetics enhances. Antimicrob. Agents Chemother. 2018, 62, e00425-18. [Google Scholar] [CrossRef] [PubMed]
- Stringer, R.A.; Weber, E.; Tigani, B.; Lavan, P.; Medhurst, S.; Sohal, B. 1-Aminobenzotriazole modulates oral drug pharmacokinetics through cytochrome P450 inhibition and delay of gastric emptying in rats. Drug Metab. Dispos. 2014, 42, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Balani, S.K.; Li, P.; Nguyen, J.; Cardoza, K.; Zeng, H.; Mu, D.X.; Wu, J.T.; Gan, L.S.; Lee, F.W. Effective dosing regimen of 1-aminobenzotriazole for inhibition of antipyrine clearance in guinea pigs and mice using serial sampling. Drug Metab. Dispos. 2004, 32, 1092–1095. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Y.; Gao, Y.; Xiang, Y.; Zhang, W.; Lu, C.; Zhuang, X. Atipamezole is a promising non-discriminative inhibitor against pan-CYP450 including diclofenac 4′-hydroxylation: A comparison with ABT for drug ADME optimization and mechanism study. Eur. J. Pharm. Sci. 2019, 130, 156–165. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1, 8–43 are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cmpd | R | X | T1/2 (min) a | T1/2 Ratio b | Primary Metabolism Site | Primary Metabolite Mass |
|---|---|---|---|---|---|---|
| 8 | 3,4,5-(OMe)3 | 6,7-(OMe)2 | 12.2 | 1.6 | Aniline | Parent − CH2 |
| 9 | 2,3-O-CH2-O- | 6-Br | 8.9 | 1.9 | Aniline | Parent + O |
| 10 | 2,5-F | 6-Br | 1.7 | 0.17 | Aniline | Parent + O |
| 11 | 2,4-F | 6-Br | 3.2 | 0.47 | Aniline | Parent − F + OH |
| 12 | 2,3,4,5,6-F5 | 6-CF3 | 45.6 | 7.0 | nt | nt |
| 13 | 3,4,5-(OMe)3 | 6-CF3 | 6.4 | 0.78 | Aniline | Parent + O |
| Cmpd | R | X | Biochemical a GAK Ki (nM) | Live Cell b GAK IC50 (nM) c |
|---|---|---|---|---|
| 8 | 3,4,5-(OMe)3 | 6,7-(OMe)2 | 0.54 d | 25 d |
| 9 | 2,3-O-CH2-O- | 6-Br | 1.9 | 22 |
| 10 | 2,5-F | 6-Br | 15 | 870 |
| 11 | 2,4-F | 6-Br | 35 | 2700 |
| 12 | 2,3,4,5,6-F5 | 6-CF3 | 880 | nt |
| 13 | 3,4,5-(OMe)3 | 6-CF3 | 3.9 d | 180 d |
| Cmpd a | X | Biochemical b GAK Ki (nM) | Live Cell c GAK IC50 (nM) d | T1/2 (Min) d | T1/2 Ratio e |
|---|---|---|---|---|---|
| 9 | Br | 1.9 | 22 | 8.9 | 1.9 |
| 14 | Br | 1.4 | 120 | 1.8 | 0.27 |
| 15 | Br | 740 | >5000 | 1.0 | 0.12 |
| 16 | Br | 460 | >5000 | 2.0 | 0.24 |
| 17 | Br | 5.2 | >5000 | 1.7 | 0.16 |
| 18 | Br | 29 | 2500 | 0.82 | 0.10 |
| 19 | Br | 1.9 | 180 | 1.7 | 0.16 |
| 20 | Br | 4.3 | 570 | 0.9 | 0.08 |
| 21 | Br | 37 | 1400 | 5.2 | 0.64 |
| 22 | Br | 19 | >5000 | 1.4 | 0.17 |
| 23 | Br | 19 | 340 | 1.7 | 0.21 |
| 24 | Br | 11 | 220 | 5.3 | 0.68 |
| 25 | Br | 26 | >5000 | 2.6 | 0.32 |
| 26 | Br | 23 | 3300 | 4.1 | 0.50 |
| 27 | Br | 4.1 | >5000 | 8.1 | 1.0 |
| 28 | Br | 41 | 2500 | 1.2 | 0.15 |
| 29 | Br | 5.3 | 400 | 1.6 | 0.20 |
| 30 | Br | 2.8 | >5000 | 0.73 | 0.09 |
| 31 | Br | 3.6 | >5000 | 0.80 | 0.10 |
| 32 | Br | 14 | 760 | 2.5 | 0.32 |
| 33 | CF3 | 720 | >5000 | 5.9 | 0.90 |
| 34 | CF3 | 34 | 2600 | 46.4 | 9.8 |
| 35 | Br | 20 | 1400 | 45.6 | 9.8 |
| 36 | Br | nt | >5000 | 14.8 | 2.2 |
| 37 | Br | 47 | >5000 | 7.5 | 0.92 |
| 38 | Br | 62 | >5000 | 8.1 | 1.0 |
| 39 | Br | 360 | >5000 | >200 | >25 |
| 40 | Br | 680 | >5000 | 55.9 | 6.9 |
| 41 | Br | 110 | >5000 | 27.8 | 3.4 |
| 42 | Br | 1600 | >5000 | 33.6 | 4.2 |
| 43 | Br | 1700 | >5000 | 15.6 | 1.9 |
| Cmpd | T1/2 (min) a | T1/2 Ratio | Clint (mL/min/kg) | T1/2 (min) a | T1/2 Ratio b | Clint (mL/min/kg) |
|---|---|---|---|---|---|---|
| (−) ABT | (+) ABT | |||||
| 1 | 5.7 | 0.58 | 990 | 130 | 13.2 | 40 |
| 8 | 18 | 1.3 | 320 | >200 | 14.7 | <28 |
| 9 | 0.74 | 0.05 | 7610 | 1.8 | 0.13 | 3140 |
| 14 | 0.74 | 0.05 | 7550 | 2.5 | 0.18 | 2290 |
| 35 | 46 | 9.8 | 120 | 170 | 17.5 | 35 |
| Experiment | Tmax (h) | Cmax (ng/mL) | AUCinf (h·ng/mL) |
|---|---|---|---|
| 10 mpk 1 | 0.5 | 97 | 98 |
| 2 h pretreat 50 mpk ABT 10 mpk 1 | 0.5 | 650 | 1900 |
| 10 mpk 8 | 1.0 | 19 | 71 |
| 2 h pretreat 50 mpk ABT 10 mpk 8 | 2.0 | 107 | 569 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asquith, C.R.M.; Bennett, J.M.; Su, L.; Laitinen, T.; Elkins, J.M.; Pickett, J.E.; Wells, C.I.; Li, Z.; Willson, T.M.; Zuercher, W.J. Towards the Development of an In vivo Chemical Probe for Cyclin G Associated Kinase (GAK). Molecules 2019, 24, 4016. https://doi.org/10.3390/molecules24224016
Asquith CRM, Bennett JM, Su L, Laitinen T, Elkins JM, Pickett JE, Wells CI, Li Z, Willson TM, Zuercher WJ. Towards the Development of an In vivo Chemical Probe for Cyclin G Associated Kinase (GAK). Molecules. 2019; 24(22):4016. https://doi.org/10.3390/molecules24224016
Chicago/Turabian StyleAsquith, Christopher R. M., James M. Bennett, Lianyong Su, Tuomo Laitinen, Jonathan M. Elkins, Julie E. Pickett, Carrow I. Wells, Zengbiao Li, Timothy M. Willson, and William J. Zuercher. 2019. "Towards the Development of an In vivo Chemical Probe for Cyclin G Associated Kinase (GAK)" Molecules 24, no. 22: 4016. https://doi.org/10.3390/molecules24224016