
Yuccalechins A–C from the Yucca schidigera Roezl ex Ortgies Bark: Elucidation of the Relative and Absolute Configurations of Three New Spirobiflavonoids and Their Cholinesterase Inhibitory Activities

 ,
,  ,
,  ,
,  , ,
, , 


Abstract
:1. Introduction
2. Results
2.1. Identification of the Constituents Found in the Y. schidigera Ethyl Acetate Fraction
2.2. Structural Characterization of the New Phenolic Compounds
2.3. Anti-Cholinesterase Activities of trans-3,3′,5,5′-Tetrahydroxy-4′-methoxystilbene and Yuccalechins B and C
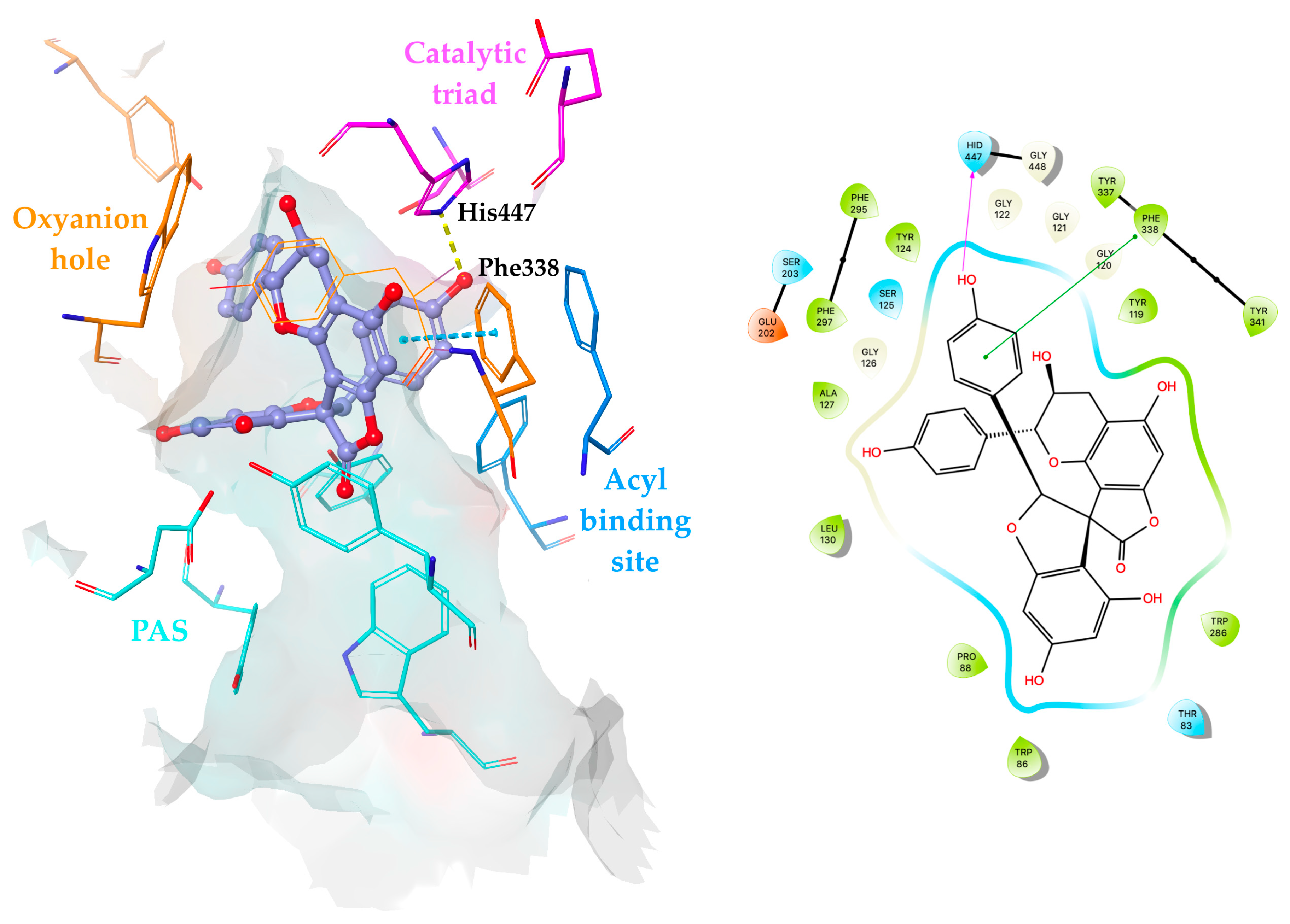
2.4. Molecular Docking Simulations of Yuccalechins B and C
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Plant Material
4.3. Extraction and Isolation
4.4. Semi-Preparative HPLC
4.5. High-Resolution LC-MS
4.6. NMR Spectroscopy
4.7. Optical Rotation [α]
4.8. Electronic Circular Dichroism (ECD) Spectroscopy
4.9. Enzyme Inhibition Assay
4.10. DP4+ Probability Calculation
4.11. ECD Spectra Calculation
4.12. Molecular Docking Studies
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ortgies, K.E. Gartenflora. Monatschrift für deutsche und schweizerische Garten-und Blumenkunde; Verlag von Ferdinand Enke: Erlangen, Germany, 1871; Available online: https://www.biodiversitylibrary.org/item/125737 (accessed on 7 October 2019).
- Cheeke, P.R. Saponins: Surprising Benefit of Desert Plants; Linus Pauling Institute Newsletter Oregon State University: Corvallis, OR, USA, 1998; pp. 4–5. [Google Scholar]
- Cheeke, P.R. Actual and Potential Applications of Yucca Schidigera and Quillaja Saponaria Saponins in Human and Animal Nutrition. In Saponins in Food, Feedstuffs and Medicinal Plants; Springer: Dordrecht, The Netherlands, 2000; pp. 241–254. [Google Scholar]
- Miyakoshi, M.; Tamura, Y.; Masuda, H.; Mizutani, K.; Tanaka, O.; Ikeda, T.; Ohtani, K.; Kasai, R.; Yamasaki, K. Antiyeast Steroidal Saponins from Yucca schidigera (Mohave Yucca), a New Anti-Food-Deteriorating Agent. J. Nat. Prod. 2000, 63, 332–338. [Google Scholar] [CrossRef]
- Oleszek, W.; Sitek, M.; Stochmal, A.; Piacente, S.; Pizza, C.; Cheeke, P. Steroidal saponins of Yucca schidigera Roezl. J. Agric. Food Chem. 2001, 49, 4392–4396. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, M.; Pecio, Ł.; Stochmal, A.; Oleszek, W. Qualitative and Quantitative Analysis of Steroidal Saponins in Crude Extract and Bark Powder of Yucca schidigera Roezl. J. Agric. Food Chem. 2011, 59, 8058–8064. [Google Scholar] [CrossRef] [PubMed]
- Piacente, S.; Montoro, P.; Oleszek, W.; Pizza, C. Yucca schidigera Bark: Phenolic Constituents and Antioxidant Activity. J. Nat. Prod. 2004, 67, 882–885. [Google Scholar] [CrossRef] [PubMed]
- Montoro, P.; Piacente, S.; Oleszek, W.; Pizza, C. Liquid chromatography/tandem mass spectrometry of unusual phenols from Yucca schidigera bark: Comparison with other analytical techniques. J. Mass Spectrom. 2004, 39, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Oleszek, W.; Sitek, M.; Stochmal, A.; Piacente, S.; Pizza, C.; Cheeke, P. Resveratrol and Other Phenolics from the Bark of Yucca schidigera Roezl. J. Agric. Food Chem. 2001, 49, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Piacente, S.; Bifulco, G.; Pizza, C.; Stochmal, A.; Oleszek, W. A novel phenolic spiro derivative, Yuccaone A, from Yucca schidigera bark. Tetrahedron Lett. 2002, 43, 9133–9136. [Google Scholar] [CrossRef]
- Fedorova, T.E.; Ivanova, S.Z.; Babkin, V.A. Spiroflavonoid compounds: Structure and distribution in nature review. Russ. J. Bioorgan. Chem. 2010, 36, 793–801. [Google Scholar] [CrossRef]
- Wenzig, E.M.; Oleszek, W.; Stochmal, A.; Kunert, O.; Bauer, R. Influence of Phenolic Constituents from Yucca schidigera Bark on Arachidonate Metabolism in Vitro. J. Agric. Food Chem. 2008, 56, 8885–8890. [Google Scholar] [CrossRef]
- Bassarello, C.; Bifulco, G.; Montoro, P.; Skhirtladze, A.; Benidze, M.; Kemertelidze, E.; Pizza, C.; Piacente, S. Yucca gloriosa: A Source of Phenolic Derivatives with Strong Antioxidant Activity. J. Agric. Food Chem. 2007, 55, 6636–6642. [Google Scholar] [CrossRef]
- Teponno, R.B.; Ponou, B.K.; Fiorini, D.; Barboni, L.; Tapondjou, L.A. Chemical Constituents from the Roots of Furcraea bedinghausii Koch. Int. Lett. Chem. Phys. Astron. 2013, 11, 9–19. [Google Scholar] [CrossRef]
- Nakashima, K.I.; Abe, N.; Oyama, M.; Inoue, M. Yuccalides A-C, three new phenolic compounds with spiro-structures from the roots of Yucca gloriosa. Fitoterapia 2016, 111, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Haslam, E.; Falshaw, C.P.; Begley, M.J. Procyanidins and polyphenols of Larix gmelini bark. Phytochemistry 1986, 25, 2629–2635. [Google Scholar] [CrossRef]
- Taniguchi, M.; Fujiwara, A.; Baba, K.; Wang, N.-H. Two biflavonoids from Daphne acutiloba. Phytochemistry 1998, 49, 863–867. [Google Scholar] [CrossRef]
- Yang, B.-H.; Zhang, W.-D.; Liu, R.-H.; Tan, C.-H.; Li, T.-Z.; Zhang, C.; Xu, X.-K.; Su, J. Spiro-biflavonoids from Larix olgensis Henry var. koreana Nakai. Helv. Chim. Acta 2005, 88, 2892–2896. [Google Scholar] [CrossRef]
- Li, Y.L.; Yang, X.W.; Li, S.M.; Tang, J.; Tian, J.M.; Peng, X.Y.; Huang, D.S.; Zhang, W.D. Two new spirobiflavonoids from Abies chensiensis with moderate NO production inhibitory activity. Planta Med. 2009, 75, 1534–1537. [Google Scholar] [CrossRef]
- Wada, S.; Hitomi, T.; Tanaka, R. Phenolic Compounds Isolated from the Bark of Abies sachalinensis. Helv. Chim. Acta 2009, 92, 1610–1620. [Google Scholar] [CrossRef]
- Buckingham, J.; Munasinghe, V.R.N. Dictionary of Flavonoids with CD-ROM; CRC Press: Boca Raton, FL, USA, 2015; ISBN 9781482282504. [Google Scholar]
- Dementia. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 7 October 2019).
- Dolan, D.; Troncoso, J.; Resnick, S.M.; Crain, B.J.; Zonderman, A.B.; OșBrien, R.J. Age, Alzheimer’s disease and dementia in the Baltimore Longitudinal Study of Ageing. Brain 2010, 133, 2225–2231. [Google Scholar] [CrossRef]
- Orhan, G.; Orhan, I.; Subutay-Oztekin, N.; Ak, F.; Sener, B. Contemporary anticholinesterase pharmaceuticals of natural origin and their synthetic analogues for the treatment of Alzheimer’s disease. Recent Pat. CNS Drug Discov. 2009, 4, 43–51. [Google Scholar] [CrossRef]
- Tundis, R.; Loizzo, M.R.; Nabavi, S.M.; Orhan, I.E.; Skalicka-Woźniak, K.; D’Onofrio, G.; Aiello, F. Natural Compounds and Their Derivatives as Multifunctional Agents for the Treatment of Alzheimer Disease. In Discovery and Development of Neuroprotective Agents from Natural Products; Elsevier: Amsterdam, The Netherlands, 2018; pp. 63–102. ISBN 9780128097694. [Google Scholar]
- Abbas-Mohammadi, M.; Moridi, M.; Salehi, P.; Nejad, S.E.; Sonboli, A.; Kelso, C.; Skropeta, D. Acetylcholinesterase-inhibitory activity of Iranian plants: Combined HPLC/bioassay-guided fractionation, molecular networking and docking strategies for the dereplication of active compounds. J. Pharm. Biomed. Anal. 2018, 158, 471–479. [Google Scholar] [CrossRef]
- Brus, B.; Košak, U.; Turk, S.; Pišlar, A.; Coquelle, N.; Kos, J.; Stojan, J.; Colletier, J.-P.; Gobec, S. Discovery, Biological Evaluation, and Crystal Structure of a Novel Nanomolar Selective Butyrylcholinesterase Inhibitor. J. Med. Chem. 2014, 57, 8167–8179. [Google Scholar] [CrossRef] [PubMed]
- Musial, A.; Bajda, M.; Malawska, B. Recent Developments in Cholinesterases Inhibitors for Alzheimers Disease Treatment. Curr. Med. Chem. 2007, 14, 2654–2679. [Google Scholar] [CrossRef] [PubMed]
- Ferris, S.H. Switching previous therapies for Alzheimer’s disease to galantamine. Clin. Ther. 2001, 23, A3–A7. [Google Scholar] [CrossRef]
- Qian, Z.M.; Ke, Y. Huperzine A: Is it an Effective Disease-Modifying Drug for Alzheimer’s Disease? Front. Aging Neurosci. 2014, 6, 216. [Google Scholar] [CrossRef]
- Khaw, K.Y.; Choi, S.B.; Tan, S.C.; Wahab, H.A.; Chan, K.L.; Murugaiyah, V. Prenylated xanthones from mangosteen as promising cholinesterase inhibitors and their molecular docking studies. Phytomedicine 2014, 21, 1303–1309. [Google Scholar] [CrossRef]
- Orhan, I.E. Implications of Some Selected Flavonoids Towards Alzheimer’s Disease with the Emphasis on Cholinesterase Inhibition and their Bioproduction by Metabolic Engineering. Curr. Pharm. Biotechnol. 2014, 15, 352–361. [Google Scholar] [CrossRef]
- Chen, X.; Mukwaya, E.; Wong, M.-S.; Zhang, Y. A systematic review on biological activities of prenylated flavonoids. Pharm. Biol. 2014, 52, 655–660. [Google Scholar] [CrossRef]
- Al-Gubory, K.H.; Laher, I. (Eds.) Nutritional Antioxidant Therapies: Treatments and Perspectives; Springer International Publishing: Cham, Switzerland, 2017; ISBN 978-3-319-67623-4. [Google Scholar]
- Viña, J.; Lloret, A.; Ortí, R.; Alonso, D. Molecular bases of the treatment of Alzheimer’s disease with antioxidants: Prevention of oxidative stress. Mol. Asp. Med. 2004, 25, 117–123. [Google Scholar] [CrossRef]
- Zhu, X.; Raina, A.K.; Lee, H.; Casadesus, G.; Smith, M.A.; Perry, G. Oxidative stress signalling in Alzheimer’s disease. Brain Res. 2004, 1000, 32–39. [Google Scholar] [CrossRef]
- Orhan, I.; Kartal, M.; Naz, Q.; Ejaz, A.; Yilmaz, G.; Kan, Y.; Konuklugil, B.; Şener, B.; Iqbal Choudhary, M. Antioxidant and anticholinesterase evaluation of selected Turkish Salvia species. Food Chem. 2007, 103, 1247–1254. [Google Scholar] [CrossRef]
- Jang, M.H.; Piao, X.L.; Kim, J.M.; Kwon, S.W.; Park, J.H. Inhibition of cholinesterase and amyloid-β aggregation by resveratrol oligomers from Vitis amurensis. Phyther. Res. 2008, 22, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.-F.; Wang, X.-B.; Xie, S.-S.; Li, S.-Y.; Kong, L.-Y. Multitarget-directed resveratrol derivatives: Anti-cholinesterases, anti-β-amyloid aggregation and monoamine oxidase inhibition properties against Alzheimer’s disease. Medchemcomm 2014, 5, 609. [Google Scholar] [CrossRef]
- Gal, J. Pasteur and the art of chirality. Nat. Chem. 2017, 9, 604–605. [Google Scholar] [CrossRef] [PubMed]
- Mason, S. The origin of chirality in nature. Trends Pharmacol. Sci. 1986, 7, 20–23. [Google Scholar] [CrossRef]
- Maier, M.E. Structural revisions of natural products by total synthesis. Nat. Prod. Rep. 2009, 26, 1105–1124. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Shah, A.A.; Korman, H.; Khan, T.; Shi, L.; Worawalai, W.; Theodorakis, E.A. Total Synthesis and Structural Revision of Antibiotic CJ-16,264. Angew. Chemie Int. Ed. 2015, 54, 9203–9208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y. Structural revision of glabramycins B and C, antibiotics from the fungus Neosartorya glabra by DFT calculations of NMR chemical shifts and coupling constants. RSC Adv. 2015, 5, 36858–36864. [Google Scholar] [CrossRef]
- Burns, D.C.; Mazzola, E.P.; Reynolds, W.F. The role of computer-assisted structure elucidation (CASE) programs in the structure elucidation of complex natural products. Nat. Prod. Rep. 2019, 36, 919–933. [Google Scholar] [CrossRef]
- Smith, S.G.; Goodman, J.M. Assigning Stereochemistry to Single Diastereoisomers by GIAO NMR Calculation: The DP4 Probability. J. Am. Chem. Soc. 2010, 132, 12946–12959. [Google Scholar] [CrossRef]
- Bagno, A.; Rastrelli, F.; Saielli, G. Toward the Complete Prediction of the 1H and 133C NMR Spectra of Complex Organic Molecules by DFT Methods: Application to Natural Substances. Chem. A Eur. J. 2006, 12, 5514–5525. [Google Scholar] [CrossRef]
- Bagno, A. Complete Prediction of the 1H NMR Spectrum of Organic Molecules by DFT Calculations of Chemical Shifts and Spin-Spin Coupling Constants. Chemistry 2001, 7, 1652–1661. [Google Scholar]
- Barone, G.; Duca, D.; Silvestri, A.; Gomez-Paloma, L.; Riccio, R.; Bifulco, G. Determination of the Relative Stereochemistry of Flexible Organic Compounds by Ab Initio Methods: Conformational Analysis and Boltzmann-Averaged GIAO 13C NMR Chemical Shifts. Chem. A Eur. J. 2002, 8, 3240. [Google Scholar] [CrossRef]
- Barone, G.; Gomez-Paloma, L.; Duca, D.; Silvestri, A.; Riccio, R.; Bifulco, G. Structure Validation of Natural Products by Quantum-Mechanical GIAO Calculations of 13C NMR Chemical Shifts. Chem. A Eur. J. 2002, 8, 3233. [Google Scholar] [CrossRef]
- Grimblat, N.; Zanardi, M.M.; Sarotti, A.M. Beyond DP4: An Improved Probability for the Stereochemical Assignment of Isomeric Compounds using Quantum Chemical Calculations of NMR Shifts. J. Org. Chem. 2015, 80, 12526–12534. [Google Scholar] [CrossRef] [PubMed]
- Nugroho, A.E.; Morita, H. Circular dichroism calculation for natural products. J. Nat. Med. 2014, 68, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bringmann, G.; Bruhn, T.; Maksimenka, K.; Hemberger, Y. The Assignment of Absolute Stereostructures through Quantum Chemical Circular Dichroism Calculations. Eur. J. Org. Chem. 2009, 2009, 2717–2727. [Google Scholar] [CrossRef]
- Crawford, T.D.; Tam, M.C.; Abrams, M.L. The Current State of Ab Initio Calculations of Optical Rotation and Electronic Circular Dichroism Spectra. J. Phys. Chem. A 2007, 111, 12057–12068. [Google Scholar] [CrossRef]
- Bassarello, C.; Bifulco, G.; Montoro, P.; Skhirtladze, A.; Kemertelidze, E.; Pizza, C.; Piacente, S. Gloriosaols A and B, two novel phenolics from Yucca gloriosa: Structural characterization and configurational assignment by a combined NMR-quantum mechanical strategy. Tetrahedron 2007, 63, 148–154. [Google Scholar] [CrossRef]
- Sun, J.; Liang, F.; Bin, Y.; Li, P.; Duan, C. Screening Non-colored Phenolics in Red Wines using Liquid Chromatography/Ultraviolet and Mass Spectrometry/Mass Spectrometry Libraries. Molecules 2007, 12, 679–693. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Parkin, K.L. Isolation and identification of potential cancer chemopreventive agents from methanolic extracts of green onion (Allium cepa). Phytochemistry 2007, 68, 1059–1067. [Google Scholar] [CrossRef]
- Simmler, C.; Antheaume, C.; André, P.; Bonté, F.; Lobstein, A. Glucosyloxybenzyl Eucomate Derivatives from Vanda teres Stimulate HaCaT Cytochrome c Oxidase. J. Nat. Prod. 2011, 74, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Cerezo, A.B.; Espartero, J.L.; Winterhalter, P.; Garcia-Parrilla, M.C.; Troncoso, A.M. (+)-Dihydrorobinetin: A Marker of Vinegar Aging in Acacia (Robinia pseudoacacia) Wood. J. Agric. Food Chem. 2009, 57, 9551–9554. [Google Scholar] [CrossRef] [PubMed]
- Mabry, T.J.; Markham, K.R.; Thomas, M.B. The Systematic Identification of Flavonoids; Springer: Berlin/Heidelberg, Germany, 1970; ISBN 978-3-642-88460-3. [Google Scholar]
- Rogerio-Candelera, M.A. Science, Technology and Cultural Heritage; CRC Press: London, UK, 2014; ISBN 9781315712420. [Google Scholar]
- Zhang, J.; Xu, W.; Wang, P.; Huang, J.; Bai, J.; Huang, Z.; Liu, X.; Qiu, X. Chemical Analysis and Multi-Component Determination in Chinese Medicine Preparation Bupi Yishen Formula Using Ultra-High Performance Liquid Chromatography With Linear Ion Trap-Orbitrap Mass Spectrometry and Triple-Quadrupole Tandem Mass Spectrometry. Front. Pharmacol. 2018, 9, 568. [Google Scholar] [CrossRef] [Green Version]
- Mari, A.; Montoro, P.; Pizza, C.; Piacente, S. Liquid chromatography tandem mass spectrometry determination of chemical markers and principal component analysis of Vitex agnus-castus L. fruits (Verbenaceae) and derived food supplements. J. Pharm. Biomed. Anal. 2012, 70, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Cuyckens, F.; Claeys, M. Mass spectrometry in the structural analysis of flavonoids. J. Mass Spectrom. 2004, 39, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Montoro, P.; Skhirtladze, A.; Bassarello, C.; Perrone, A.; Kemertelidze, E.; Pizza, C.; Piacente, S. Determination of phenolic compounds in Yucca gloriosa bark and root by LC–MS/MS. J. Pharm. Biomed. Anal. 2008, 47, 854–859. [Google Scholar] [CrossRef] [PubMed]
- Min, K.; Hwang, B.; Lim, H.-S.; Kang, B.-S.; Oh, G.-J.; Lee, J.; Kang, S.-H.; Lee, K.; Ro, J.; Kim, Y. (-)-Epiafzelechin: Cyclooxygenase-1 Inhibitor and Anti-Inflammatory Agent from Aerial Parts of Celastrus orbiculatus. Planta Med. 1999, 65, 460–462. [Google Scholar] [CrossRef]
- Shen, Z.; Falshaw, C.P.; Haslam, E.; Begley, M.J. A Novel Spiro-Biflavonoid from Larix gmelini. J. Chem. Soc. Chem. Commun. 1985, 1135. [Google Scholar] [CrossRef]
- Hemingway, R.W.; Tobiason, F.L.; Wayne McGraw, G.; Steynberg, J.P. Conformation and Complexation of Tannins: NMR Spectra and Molecular Search Modeling of Flavan-3-ols. Magn. Reson. Chem. 1996, 34, 424–433. [Google Scholar] [CrossRef]
- Kríž, Z.; Koča, J.; Imberty, A.; Charlot, A.; Auzély-Velty, R. Investigation of the complexation of (+)-catechin by β-cyclodextrin by a combination of NMR, microcalorimetry and molecular modeling techniques. Org. Biomol. Chem. 2003, 1, 2590–2595. [Google Scholar] [CrossRef]
- Clark-Lewis, J.; Jackman, L.; Spotswood, T. Nuclear magnetic resonance spectra, stereochemistry, and conformation of flavan derivatives. Aust. J. Chem. 1964, 17, 632. [Google Scholar] [CrossRef]
- Usman, A.; Thoss, V.; Nur-e-Alam, M. Isolation of (-)-Epicatechin from Trichilia emetica Whole Seeds. Am. J. Org. Chem. 2016, 21, 77–82. [Google Scholar]
- Perlin, A.S.; Casu, B. Carbon-13 and proton magnetic resonance spectra of D-glucose-13C. Tetrahedron Lett. 1969, 10, 2921–2924. [Google Scholar] [CrossRef]
- Juaristi, E.; Cuevas, G. Manifestations of Stereoelectronic Interactions in 1JC-H One-Bond Coupling Constants. Acc. Chem. Res. 2007, 40, 961–970. [Google Scholar] [CrossRef]
- Alabugin, I.V.; Manoharan, M.; Zeidan, T.A. Homoanomeric Effects in Six-Membered Heterocycles. J. Am. Chem. Soc. 2003, 125, 14014–14031. [Google Scholar] [CrossRef]
- Alabugin, I.V. Stereoelectronic Interactions in Cyclohexane, 1, 3-Dioxane, 1, 3-Oxathiane, and 1, 3-Dithiane: W-Effect, σC-X↔ σ* CH Interactions, Anomeric Effect What Is Really Important? J. Org. Chem. 2000, 65, 3910–3919. [Google Scholar] [CrossRef]
- Crews, P.; Rodriguez, J.; Jaspars, M. Organic Structure Analysis, 2nd ed.; Oxford University Press: New York, NY, USA, 2010; ISBN 9780195336047. [Google Scholar]
- Castañar, L.; Sistaré, E.; Virgili, A.; Williamson, R.T.; Parella, T. Suppression of phase and amplitude J(HH) modulations in HSQC experiments. Magn. Reson. Chem. 2015, 53, 115–119. [Google Scholar] [CrossRef]
- Koźmiński, W.; Nanz, D. Sensitivity Improvement and New Acquisition Scheme of Heteronuclear Active-Coupling-Pattern-Tilting Spectroscopy. J. Magn. Reson. 2000, 142, 294–299. [Google Scholar] [CrossRef]
- Alabugin, I.V. Probing Stereoelectronic Effects with Spectroscopic Methods. In Stereoelectronic Effects: A Bridge Between Structure and Reactivity; Hoboken, N.J., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2016; pp. 1–7. ISBN 9781118906378. [Google Scholar]
- Hameed, R.; van Mourik, T.; Khan, A. 13C-1H coupling constants as a conformational tool for structural assignment of quinic and octulosonic acid. J. Mol. Model. 2018, 24, 324. [Google Scholar] [CrossRef] [Green Version]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Orhan, I.E.; Jedrejek, D.; Senol, F.S.; Salmas, R.E.; Durdagi, S.; Kowalska, I.; Pecio, L.; Oleszek, W. Molecular modeling and in vitro approaches towards cholinesterase inhibitory effect of some natural xanthohumol, naringenin, and acyl phloroglucinol derivatives. Phytomedicine 2018, 42, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Ślusarczyk, S.; Senol Deniz, F.S.; Woźniak, D.; Pecio, Ł.; Pérez-Sánchez, H.; Cerón-Carrasco, J.P.; Stochmal, A.; den-Haan Alonso, H.; Matkowski, A.; Orhan, I.E. Selective in vitro and in silico cholinesterase inhibitory activity of isoflavones and stilbenes from Belamcandae chinensis rhizoma. Phytochem. Lett. 2019, 30, 261–272. [Google Scholar] [CrossRef]
- Rosenberry, T.; Brazzolotto, X.; Macdonald, I.; Wandhammer, M.; Trovaslet-Leroy, M.; Darvesh, S.; Nachon, F. Comparison of the Binding of Reversible Inhibitors to Human Butyrylcholinesterase and Acetylcholinesterase: A Crystallographic, Kinetic and Calorimetric Study. Molecules 2017, 22, 2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, H.; Bladt, S. Plant Drug Analysis: A Thin Layer Chromatography Atlas, 2nd ed.; Elgazzar, A.H., Ed.; Springer: Berlin/Heidelberg, Gemany; New York, NY, USA, 2001; ISBN 3-540-58676-8. [Google Scholar]
- Tyburn, J.-M.; Coutant, J. TopSpin ERETIC 2; Bruker Corporation: Billerica, MA, USA, 2015. [Google Scholar]
- Burlec, A.F.; Pecio, Ł.; Mircea, C.; Cioancă, O.; Corciovă, A.; Nicolescu, A.; Oleszek, W.; Hăncianu, M. Chemical Profile and Antioxidant Activity of Zinnia elegans Jacq. Fractions. Molecules 2019, 24, 2934. [Google Scholar] [CrossRef] [Green Version]
- Kozachok, S.; Pecio, Ł.; Orhan, I.E.; Deniz, F.S.S.; Marchyshyn, S.; Oleszek, W. Reinvestigation of Herniaria glabra L. saponins and their biological activity. Phytochemistry 2020, 169, 112162. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Rev. A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of Human Acetylcholinesterase in Complex with Pharmacologically Important Ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef]
- Luo, H.-J.; Wang, J.-Z.; Huang, N.-Y.; Deng, W.-Q.; Zou, K. Induced-fit docking and virtual screening for 8-hydroxy-3-methoxy-5H-pyrido [2,1-c] pyrazin-5-one derivatives as inducible nitric oxide synthase inhibitors. J. Chem. Pharm. Res. 2014, 6, 1187–1194. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Compound Name | RT (min) | λmax (nm) | Neutral Formula | Error * (ppm) | Mσ ** | Observed [M − H]− | Major Fragments (%) | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1. | Mixture of polar compounds | 0.66 | - | - | - | - | - | - | - |
| 2. | p-Hydroxybenzoic acid | 2.20 | 255 | C7H6O3 | 1.5 | 1.4 | 137.0242 | 137.0243 (100), 93.0342 (69) | [56] |
| 3. | Hydroxymethoxyacetophenone | 2.73 | - | C9H10O3 | 2.2 | 6.2 | 165.0554 | 165.0553 (100), 123.0450 (25), 152.0112 (6) | [57] |
| 4. | Hydroxybenzylmalic acid | 2.73 | - | C11H12O6 | 2.2 | 1.9 | 239.0556 | 179.0347 (100), 239.0557 (77), 177.0553 (55), 149.0604 (32), 133.0656 (11), 195.0659 (11) | [58] |
| 5. | Caffeic acid | 2.86 | 215, 323 | C9H8O4 | 2.3 | 0.7 | 179.0346 | 135.0449 (100), 179.0347 (65) | [56] |
| 6. | Unknown | 3.02 | - | C23H18O10 | 0.9 | 20.4 | 453.0823 | 310.0484 (100), 325.0716 (49), 376.0585 (48), 256.0372 (39) | |
| 7. | (Epi)afzelechin | 3.43 | - | C15H14O5 | 1.1 | 1.4 | 273.0766 | 273.0766 (100), 205.0867 (39), 255.0660 (29), 137.0247 (29), 229.0868 (28) | [11] |
| 8. | Unknown | 4.00 | - | C22H16O8 | -0.1 | 17.4 | 407.0773 | 345.0768 (100), 163.0037 (92), 269.0455 (80), 293.0819 (68), 279.0661 (67) | - |
| 9. | Unknown | 4.42 | - | C29H22O9 | 0.7 | 9.6 | 513.1187 | 293.0817 (100), 379.0823 (70), 275.0713 (65), 335.0924 (61), 361.0721 (59) | - |
| 10. | Unknown | 5.39 | - | C20H24O7 | 0.2 | 23.0 | 375.1448 | 297.1133 (100), 282.0900 (28), 151.0401 (22), 315.1256 (9), 136.0186 (6) | - |
| 11. | Unknown | 5.77 | 323 | C14H12O4 | -0.3 | 1.4 | 243.0663 | 243.0661 (100), 201.0554 (7), 225.0558 (3) | - |
| 12. | Dihydrorobinetin | 5.77 | 263 | C15H12O7 | -0.3 | 0.9 | 303.0511 | 183.0299 (100), 139.0402 (67), 165.0195 (20), 137.0246 (9), 97.0294 (4), 95.0500 (3) | [59] |
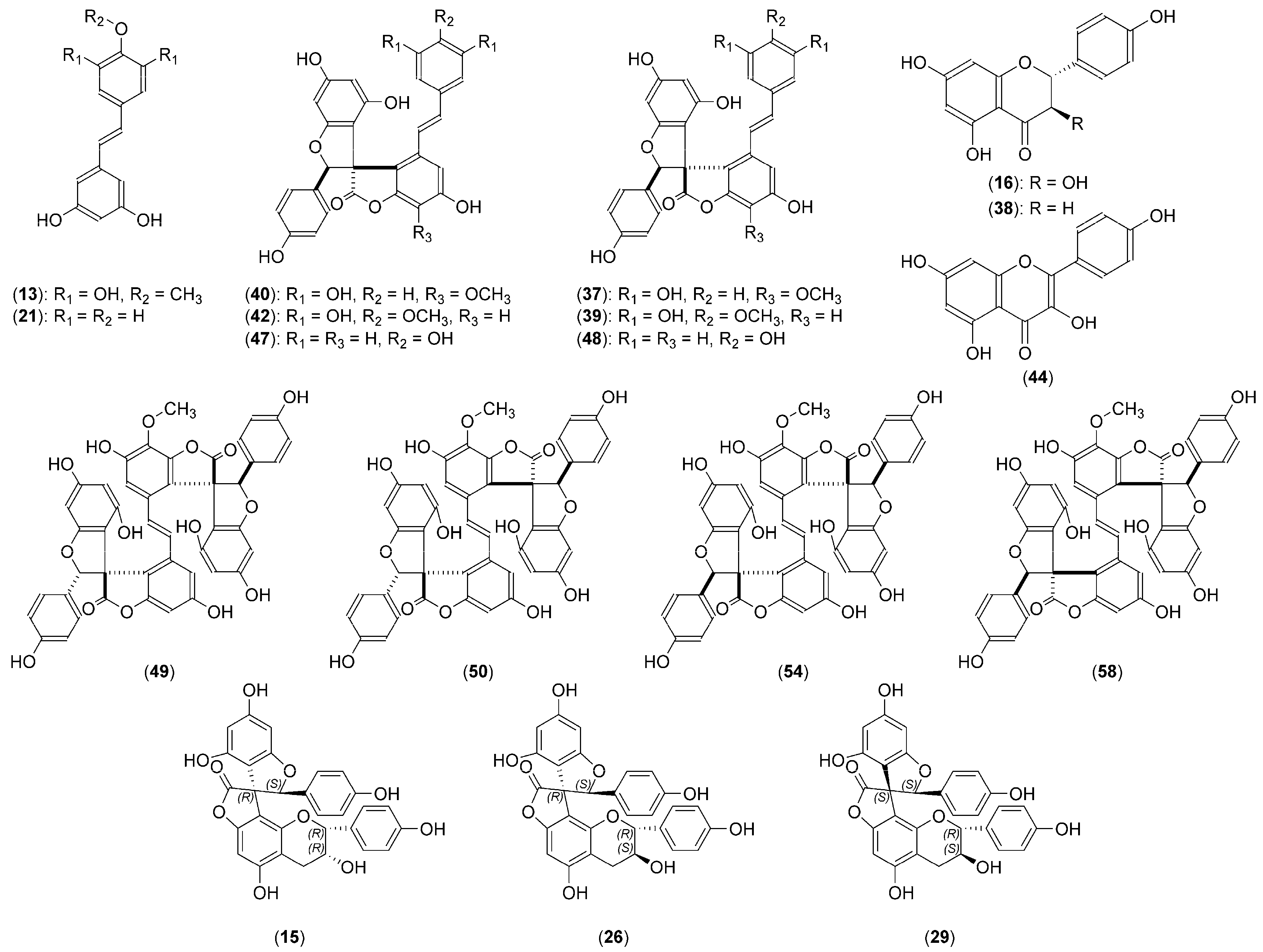
| 13. | Trans-3,3′,5,5′-tetrahydroxy-4′-methoxystilbene | 6.15 | 227, 315 | C15H14O5 | 1.0 | 2.6 | 273.0766 | 258.0526 (100), 240.0434 (9), 273.0766 (5), 196.0528 (4), 188.0477 (2), 172.0532 (2) | [8,9] |
| 14. | Unknown | 6.43 | - | C30H24O11 | 2.4 | 18.5 | 559.1232 | 373.0715 (100), 433.0922 (42), 418.0688 (32), 401.0657 (22), 125.0245 (16) | - |
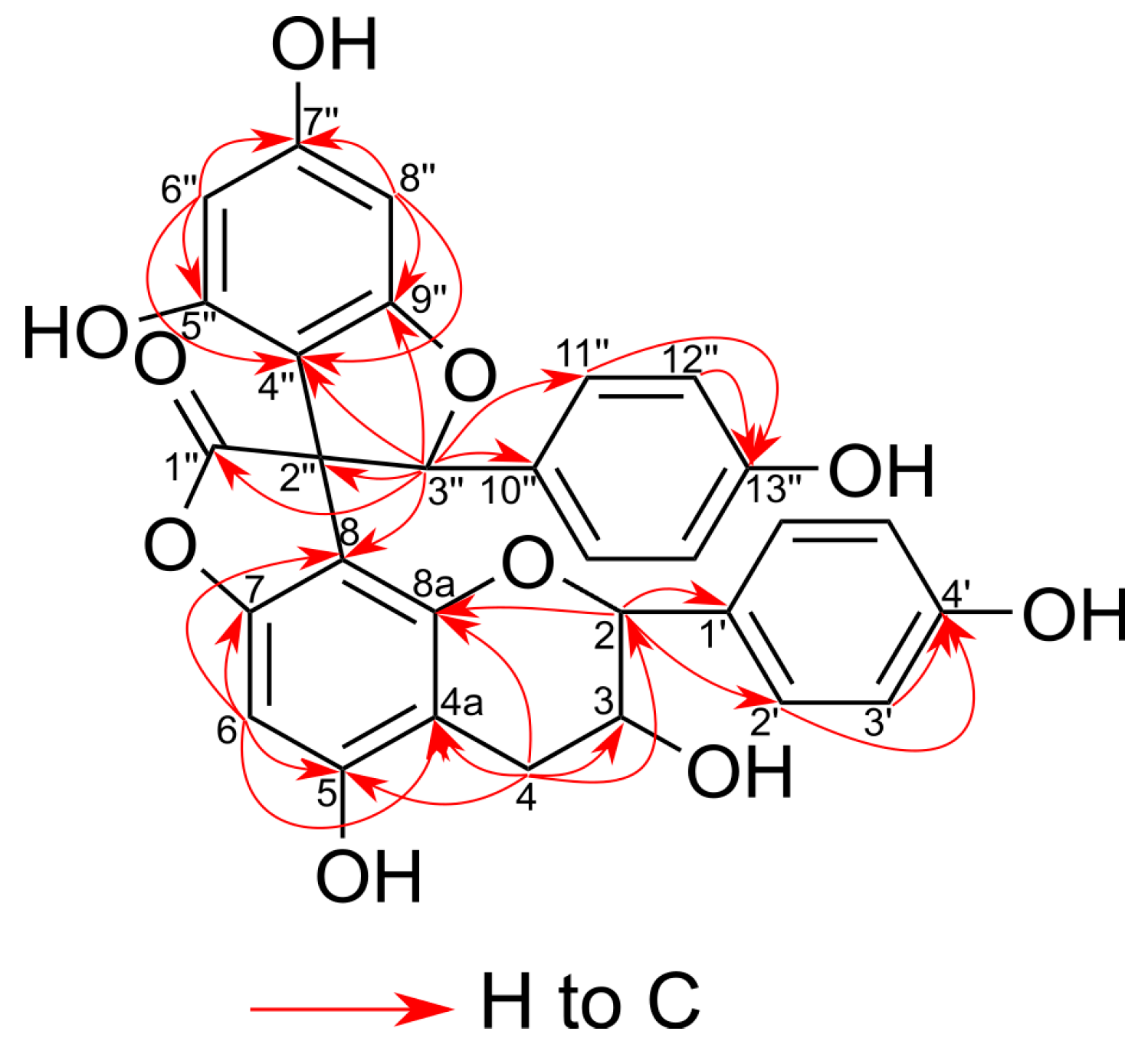
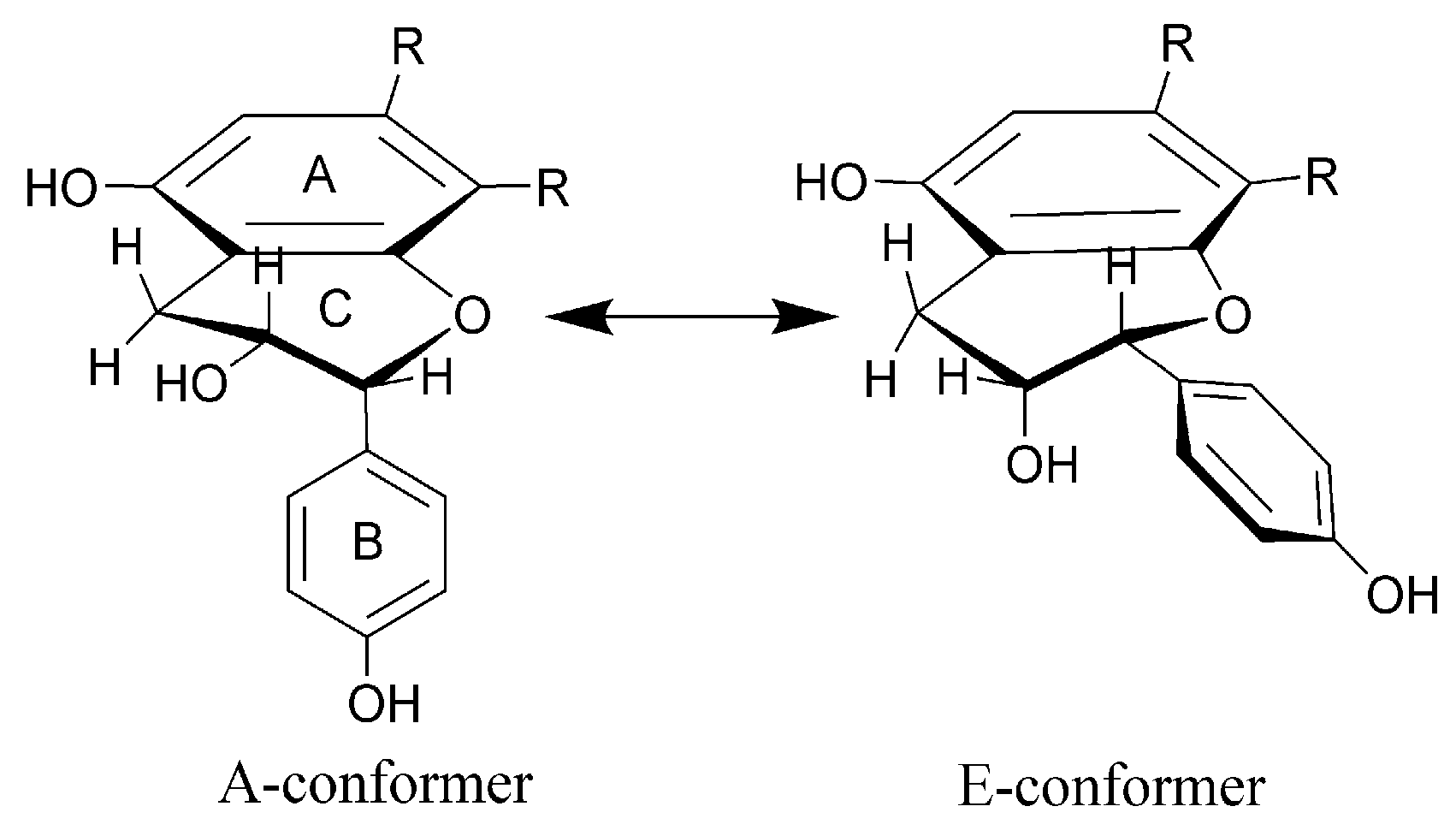
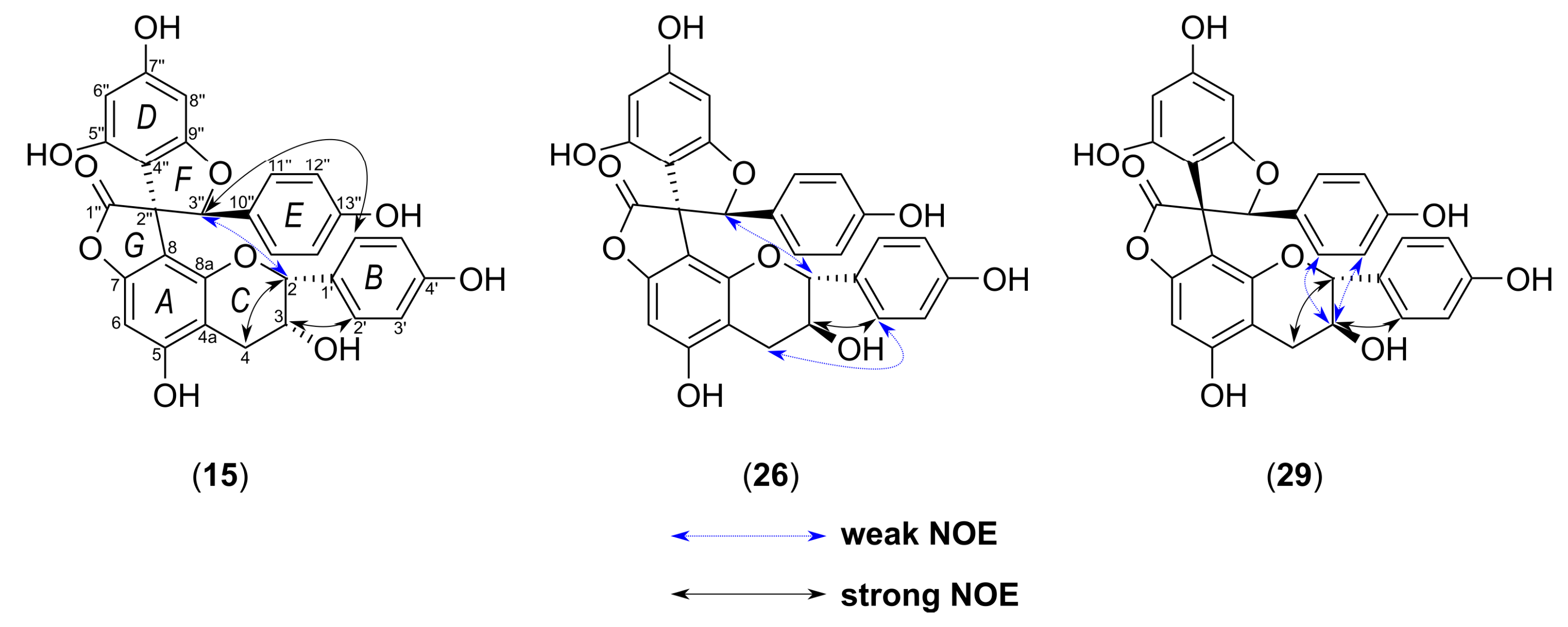
| 15. | Yuccalechin A | 6.60 | 215 | C30H22O10 | 1.8 | 6.0 | 541.1131 | 308.0346 (100), 281.0468 (88), 267.0313 (60), 269.0453 (41), 415.0843 (30) | - |
| 16. | Aromadendrin (dihydrokaempferol) | 6.70 | 295 | C15H12O6 | 1.3 | 4.8 | 287.0557 | 259.0608 (100), 125.0243 (68), 287.0243 (39), 243.0658 (25), 201.0554 (14) | [60] |
| 17. | Unknown | 6.81 | - | C29H24O10 | 1.9 | 25.9 | 531.1286 | 257.0459 (100), 393.0605 (65), 531.1293 (64), 378.0752 (45), 269.0489 (40) | - |
| 18. | Glyceryl azelate | 6.81 | - | C12H22O6 | 1.3 | 11.9 | 261.1340 | 187.0974 (100), 125.0971 (31), 261.1342 (15), 169.0869 (13) | [61] |
| 19. | Azelaic acid | 6.94 | - | C9H16O4 | 1.8 | 0.4 | 187.0972 | 187.0973 (100), 125.0970 (73), 169.0868 (20), 97.0655 (3) | [62] |
| 20. | Myricetin | 7.10 | 370 | C15H10O8 | 0.8 | 1.4 | 317.0300 | 317.0301 (100), 178.9983 (59), 151.0035 (42), 137.0244 (19) | [60] |
| 21. | Trans-resveratrol | 7.42 | 215, 307 | C14H12O3 | 3.3 | 4.5 | 227.0706 | 227.0707 (100), 185.0603 (8), 183.0809 (2) | [9,63] |
| 22. | Oxododecanedioic acid | 7.60 | - | C12H20O5 | 2.9 | 10.7 | 243.1231 | 225.1124 (100), 243.1232 (45), 181.1230 (24), 207.1021 (20), 199.1335 (17) | - |
| 23. | Tetrahydroxyflavone | 7.60 | - | C15H10O6 | 2.2 | 14.5 | 285.0398 | 285.0400 (100), 151.0033 (91), 257.0450 (55), 107.0137 (15), 213.0545 (11) | [64] |
| 24. | Unknown | 7.77 | - | C28H22O8 | 2.5 | 5.6 | 485.1230 | 257.0451 (100), 485.1227 (61), 391.0819 (35), 347.0921 (34), 227.0706 (24) | - |
| 25. | Unknown | 8.21 | - | C20H26O6 | 3.0 | 4.8 | 361.1646 | 361.1645 (100), 346.1417 (51), 165.0548 (27), 179.0701 (8), 122.0374 (7) | - |
| 26. | Yuccalechin B | 8.39 | 210 | C30H22O10 | 3.1 | 16.5 | 541.1123 | 308.0334 (100), 281.0459 (99), 267.0331 (71), 415.0829 (32), 361.0719 (31) | - |
| 27. | Unknown | 8.51 | 319 | C29H24O19 | 3.6 | 1.9 | 515.1329 | 362.0784 (100), 515.1332 (70), 241.0500 (59), 253.0507 (42), 291.0664 (19) | - |
| 28. | Unknown | 8.64 | 319 | C15H12O6 | 2.8 | 13.3 | 287.0553 | 287.0553 (100), 219.0656 (25), 199.0423 (16), 227.0354 (11), 185.0238 (8) | - |
| 29. | Yuccalechin C | 8.93 | 211 | C30H22O10 | 3.5 | 4.7 | 541.1121 | 308.0337 (100), 281.0459 (93), 267.0306 (43), 269.0449 (39), 415.0828 (30) | - |
| 30. | Larixinol isomer IV | 9.07 | - | C30H22O10 | 3.0 | 15.3 | 541.1124 | 267.0300 (100), 269.0450 (71), 281.0463 (62), 308.0339 (48), 445.1277 (46) | - |
| 31. | Quercetin | 9.14 | 219, 367 | C15H10O7 | 2.7 | 3.0 | 301.0346 | 301.0344 (100), 151.0031 (77), 178.9979 (51), 121.0292 (10), 273.0401 (10) | [64] |
| 32. | Unknown | 9.30 | - | C30H22O10 | 3.6 | 6.7 | 541.1121 | 269.0444 (100), 390.0732 (39), 362.0798 (15), 308.0335 (15), 281.0459 (13) | - |
| 33. | Unknown | 9.30 | - | C16H12O8 | 2.7 | 14.8 | 331.0451 | 316.0214 (100), 331.0448 (9) | - |
| 34. | Unknown | 9.48 | - | C15H12O5 | 3.3 | 2.1 | 271.0603 | 253.0496 (100), 227.0704 (52), 271.0602 (26), 185.0598 (4), 225.0551 (3) | - |
| 35. | Unknown | 9.62 | 315 | C24H20O8 | 3.9 | 5.3 | 435.1068 | 420.0834 (100), 435.1069 (96), 281.0462 (62), 393.0964 (50), 240.0420 (31) | - |
| 36. | Larixinol isomer V | 9.62 | 315 | C30H22O10 | 3.4 | 4.1 | 541.1122 | 308.0329 (100), 281.0460 (97), 267.0328 (60), 415.0826 (34), 497.1219 (23) | [20] |
| 37. | Yuccaol E | 9.96 | 207, 319 | C30H22O10 | 3.7 | 8.9 | 541.1120 | 498.0936 (100), 513.1171 (52), 267.0310 (27), 429.0974 (11), 375.0511 (11) | [7,63] |
| 38. | Naringenin | 10.27 | 211, 291 | C15H12O5 | 3.5 | 1.6 | 271.0612 | 271.0603 (100), 151.0031 (83), 119.0499 (23), 177.0188 (11), 107.0133 (6) | [64] |
| 39. | Yuccaol C | 10.33 | 211, 319 | C30H22O10 | 3.2 | 4.5 | 541.1123 | 267.0303 (100), 513.1171 (65), 498.0934 (63), 239.0348 (36), 429.0981 (23) | [9,63,65] |
| 40. | Yuccalide A | 10.57 | 211, 323 | C30H22O10 | 4.9 | 12.0 | 541.1114 | 498.0934 (100), 267.0304 (76), 513.1168 (72), 239.0348 (26), 429.0979 (18) | [15] |
| 41. | Unknown | 10.57 | 211, 323 | C29H20O9 | 3.6 | 8.9 | 511.1016 | 267.0302 (100), 483.1069 (36), 239.0345 (33), 385.0713 (26), 399.0857 (22) | - |
| 42. | Yuccaol D | 10.64 | 211, 323 | C30H22O10 | 3.6 | 7.4 | 541.1121 | 498.0934 (100), 267.0303 (89), 513.1168 (77), 239.0348 (30), 429.0979 (21) | [7,63,65] |
| 43. | Unknown | 10.76 | 215 | C30H22O9 | 4.0 | 5.3 | 525.1170 | 399.0868 (100), 267.0300 (84), 269.0443 (66), 361.0695 (42), 333.0766 (38) | - |
| 44. | Kaempferol | 11.02 | 265, 363 | C15H10O6 | 3.3 | 8.2 | 285.0395 | 285.0394 (100) | [64] |
| 45. | Unknown | 11.16 | - | C30H22O9 | 3.3 | 4.3 | 525.1174 | 399.0865 (100), 267.0310 (72), 361.0707 (60), 307.0607 (38), 293.0445 (36) | - |
| 46. | Gloriosaol isomer I | 11.24 | 320 | C45H30O15 | 3.6 | 22.9 | 809.1483 | 267.0296 (100), 239.0343 (55), 211.0395 (11), 513.1166 (3), 541.1121 (3) | - |
| 47. | Yuccaol A | 11.58 | 207, 327 | C29H20O8 | 3.1 | 7.2 | 495.1070 | 267.0310 (100), 467.1120 (67), 239.0347 (60), 399.1224 (21), 357.1122 (16) | [9,63,65] |
| 48. | Yuccaol B | 11.83 | 207, 327 | C29H20O8 | 2.6 | 9.2 | 495.1073 | 267.0312 (100), 239.0349 (50), 467.1122 (44), 399.1229 (16), 357.1124 (12) | [9,63,65] |
| 49. | Gloriosaol E | 11.98 | 207, 323 | C45H30O15 | 3.8 | 12.0 | 809.1481 | 267.0294 (100), 239.0343 (44), 211.0395 (8), 541.1118 (3), 513.1171 (3) | [13,65] |
| 50. | Gloriosaol D | 12.05 | 207, 323 | C45H30O15 | 3.6 | 7.3 | 809.1483 | 267.0294 (100), 239.0343 (45), 211.0395 (9), 541.1117 (3), 513.1171 (3) | [13,65] |
| 51. | Unknown | 12.33 | - | C30H22O9 | 3.1 | 5.3 | 525.1175 | 267.0314 (100), 497.1226 (94), 239.0359 (41), 413.1018 (29), 482.0994 (25) | - |
| 52. | Gloriosaol isomer II | 12.33 | - | C45H30O15 | 3.3 | 14.3 | 809.1485 | 267.0297 (100), 239.0345 (43), 211.0396 (9), 541.1125 (4), 513.1166 (3) | - |
| 53. | Unknown | 12.53 | 283 | C16H16O3 | 2.7 | 15.0 | 255.1020 | 255.1020 (100) | - |
| 54. | Gloriosaol A | 12.53 | 207, 319 | C45H30O15 | 3.3 | 3.9 | 809.1485 | 267.0295 (100), 239.0343 (49), 211.0395 (10), 541.1117 (4), 513.1172 (3) | [55,65] |
| 55. | Trihydroxyoctadecadienoic acid | 12.71 | - | C18H32O5 | 2.7 | 5.1 | 327.2168 | 327.2168 (100), 211.1334 (39), 229.1439 (32), 171.1021 (19), 239.1284 (8) | - |
| 56. | Unknown | 12.79 | 215 | C30H22O9 | 3.3 | 7.3 | 525.1174 | 267.0312 (100), 399.0868 (95), 361.0711 (70), 307.0603 (40), 349.0712 (39) | - |
| 57. | Unknown | 12.97 | - | C17H16O6 | 2.2 | 3.9 | 315.0867 | 315.0867 (100), 178.9981 (23), 297.0761 (20), 194.0216 (15), 152.0111 (11) | - |
| 58. | Gloriosaol C | 13.14 | 207, 327 | C45H30O15 | 3.0 | 2.2 | 809.1488 | 267.0295 (100), 239.0344 (42), 211.0396 (8), 541.1121 (4), 513.1173 (3) | [13] |
| 59. | Unknown | 13.28 | - | C18H20O6 | 1.6 | 16.8 | 331.1182 | 285.1127 (100) | - |
| 60. | Gloriosaol isomer III | 13.63 | - | C45H30O15 | 3.2 | 38.4 | 809.1486 | 267.0301 (100), 239.0349 (36), 508.0790 (12), 365.0655 (11), 463.0835 (10) | - |
| 61. | Trihydroxyoctadecenoic acid | 13.82 | - | C18H34O5 | 2.1 | 7.1 | 329.2327 | 329.2326 (100), 211.1334 (35), 229.1440 (26), 171.1022 (13), 139.1126 (3) | - |
| 62. | Unknown | 14.14 | - | C45H30O14 | 2.3 | 28.1 | 793.1545 | 269.0452 (100), 267.0297 (99), 399.0872 (68), 125.0239 (37), 639.1304 (19) | - |
| 63. | Unknown | 14.14 | - | C30H18O11 | 2.0 | 21.9 | 553.0765 | 375.0508 (100), 267.0304 (74), 525.0813 (69), 509.0871 (60), 457.0916 (59) | - |
| 64. | Unknown | 14.80 | - | C17H16O4 | 1.4 | 3.5 | 283.0972 | 283.0972 (100), 162.0320 (16), 134.0372 (2), 268.0743 (2) | - |
| 65. | Unknown | 14.89 | - | C15H12O4 | 1.4 | 1.6 | 255.0659 | 255.0659 (100) | - |
| 66. | Unknown | 16.68 | - | C17H16O5 | 2.0 | 2.4 | 299.0919 | 299.0919 (100), 178.0267 (18), 150.0320 (2) | - |
| 67. | Unknown | 18.82 | - | C34H52O11 | 3.2 | 47.2 | 635.3416 | 101.0241 (100), 589.3345 (11) | - |
| 68. | Unknown | 18.93 | - | C18H28O4 | 2.0 | 24.8 | 307.1909 | 223.1334 (100), 137.0970 (63), 265.1806 (30), 307.1913 (20), 185.1183 (17) | - |
| 69. | Unknown | 19.59 | - | C34H54O11 | 4.4 | 10.3 | 637.3565 | 161.0450 (100), 113.0244 (68), 591.3534 (16) | - |
| 70. | Unknown | 23.78 | - | C34H56O10 | 3.2 | 46.4 | 623.3781 | 577.3724 (100), 159.0298 (63) | - |
| 71. | Unknown | 26.68 | - | C28H42O6 | 3.3 | 7.0 | 473.2893 | 175.0395 (100), 473.2893 (23), 297.2427 (13), 160.0168 (11), 193.0499 (10) | - |
| Position | Type | 15 | 26 | 29 | |||
|---|---|---|---|---|---|---|---|
| δH (J, Hz) | δC (ppm) | δH (J, Hz) | δC (ppm) | δH (J, Hz) | δC (ppm) | ||
| 2 | CH | 4.91 br s | 79.4 | 4.99 d (5.8) | 81.6 | 4.19 d (9.5) | 82.7 |
| 3 | CH | 4.46 ddd (4.4, 2.9, 1.6) | 65.6 | 4.05 dt (5.8, 5.5) | 68.1 | 3.85 ddd (9.8, 9.5, 5.8) | 67.7 |
| 4α | CH2 | 2.90 dd (17.0, 2.9) | 29.6 | 2.68 d (5.5) | 26.8 | 2.39 dd (16.3, 9.8) | 30.5 |
| 4β | 2.96 dd (17.0, 4.4) | 2.93 dd (16.3, 5.8) | |||||
| 5 | C | 158.4 | 158.0 | 157.6 | |||
| 6 | CH | 6.16 s | 91.6 | 6.14 s | 91.5 | 6.05 s | 91.1 |
| 7 | C | 153.8 | 154.1 | 152.8 | |||
| 8 | C | 106.7 | 105.8 | 106.1 | |||
| 4a | C | 104.8 | 104.9 | 105.6 | |||
| 8a | C | 153.1 | 152.1 | 153.2 | |||
| 1′ | C | 130.6 | 131.2 | 130.1 | |||
| 2′ | CH | 7.15 d (8.5) | 128.7 | 6.98 d (8.5) | 128.2 | 7.20 d (8.5) | 130.4 |
| 3′ | CH | 6.72 d (8.5) | 115.8 | 6.66 d (8.5) | 115.9 | 6.83 d (8.5) | 115.7 |
| 4′ | C | 157.7 | 157.8 | 158.3 | |||
| 5′ | CH | 6.72 d (8.5) | 115.8 | 6.66 d (8.5) | 115.9 | 6.83 d (8.5) | 115.7 |
| 6′ | CH | 7.15 d (8.5) | 128.7 | 6.98 d (8.5) | 128.2 | 7.20 d (8.5) | 130.4 |
| 1″ | C | 177.1 | 177.1 | 180.6 | |||
| 2″ | C | 61.5 | 61.5 | 61.9 | |||
| 3″ | CH | 6.25 s | 91.2 | 6.13 s | 90.6 | 5.72 s | 94.5 |
| 4″ | C | 105.7 | 105.8 | 106.1 | |||
| 5″ | C | 156.1 | 156.2 | 155.1 | |||
| 6″ | CH | 5.81 d (2.0) | 96.7 | 5.91 d (2.0) | 96.7 | 5.78 d (2.0) | 96.5 |
| 7″ | C | 161.3 | 161.4 | 161.0 | |||
| 8″ | CH | 5.98 d (2.0) | 90.6 | 5.94 d (2.0) | 90.8 | 5.66 d (2.0) | 90.7 |
| 9″ | C | 164.8 | 164.6 | 163.8 | |||
| 10″ | C | 128.3 | 128.1 | 128.1 | |||
| 11″ | CH | 7.06 d (8.5) | 128.5 | 7.03 d (8.5) | 128.4 | 7.07 d (8.5) | 128.4 |
| 12″ | CH | 6.68 d (8.5) | 115.8 | 6.68 d (8.5) | 115.8 | 6.60 d (8.5) | 115.5 |
| 13″ | C | 158.6 | 158.6 | 158.3 | |||
| 14″ | CH | 6.68 d (8.5) | 115.8 | 6.68 d (8.5) | 115.8 | 6.60 d (8.5) | 115.5 |
| 15″ | CH | 7.06 d (8.5) | 128.5 | 7.03 d (8.5) | 128.4 | 7.07 d (8.5) | 128.4 |
| J-Type | (+)-Catechin | (−)-Epicatechin | 15 | 26 | 29 |
|---|---|---|---|---|---|
| 1JC2-H2 | 146.1 | 142.9 | 144.0 | 148.9 | 144.7 |
| 1JC3-H3 | 145.3 | 146.2 | 146.2 | 146.0 | 144.8 |
| 2JC3-H2 | −4.0 | +1.0 | +1.4 | −4.7 | −2.5 |
| 2JC2-H3 | −1.6 | +1.9 | +1.5 | 0.0 | −3.3 |
| 2JC4-H3 | −1.0 | +2.0 | −2.2 | −1.0 | −0.9 |
| 3JC4-H2 | +3.2 | +1.8 | +2.1 | +3.5 | +3.0 |
| Parameters | 15 | 26 | 29 | |||
|---|---|---|---|---|---|---|
| 2″R,3″S,2S,3R | 2″R,3″S,2R,3R | 2″R,3″S,2R,3S | 2″R,3″S,2S,3S | 2″R,3″R,2S,3R | 2″R,3″R,2R,3S | |
| 1H-MAE | 3.5 | 3.40 | 3.8 | 3.53 | 0.38 | 0.4 |
| 13C-MAE | 5.6 | 5.40 | 5.5 | 5.18 | 5.23 | 4.95 |
| Total MAE | 4.85 | 4.44 | 4.92 | 4.60 | 3.54 | 3.37 |
| 1H-CMAE | 4.16 | 3.69 | 3.29 | 3.9 | 0.83 | 0.7 |
| 13C-CMAE | 10.92 | 10.74 | 9.53 | 10.45 | 10.65 | 9.85 |
| Total CMAE | 8.57 | 8.03 | 8.40 | 8.17 | 7.23 | 6.67 |
| r H | 0.9703 | 0.9724 | 0.9772 | 0.9826 | 0.98460 | 0.93860 |
| r C | 0.9966 | 0.9977 | 0.9983 | 0.9986 | 0.99900 | 0.99770 |
| Total r | 0.98995 | 0.99203 | 0.99377 | 0.99506 | 0.99608 | 0.98812 |
| Compound | Inhibition (% ± S.D.a) at 1000 µMb | |
|---|---|---|
| AChE | BChE | |
| 13 | 19.33 ± 3.03 | 25.21 ± 2.58 |
| 26 | 80.53 ± 1.22 (IC50 = 294.18 ± 5.26 µM) | 48.46 ± 2.29 |
| 29 | 52.55 ± 2.60 (IC50 = 655.18 ± 6.35 µM) | 33.41 ± 1.37 |
| Referencec | 97.92 ± 0.01 (IC50 = 2.29 ± 0.33 µM) | 91.52 ± 1.63 (IC50 = 124.03 ± 4.05 µM) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pecio, Ł.; Alilou, M.; Kozachok, S.; Erdogan Orhan, I.; Eren, G.; Senol Deniz, F.S.; Stuppner, H.; Oleszek, W. Yuccalechins A–C from the Yucca schidigera Roezl ex Ortgies Bark: Elucidation of the Relative and Absolute Configurations of Three New Spirobiflavonoids and Their Cholinesterase Inhibitory Activities. Molecules 2019, 24, 4162. https://doi.org/10.3390/molecules24224162
Pecio Ł, Alilou M, Kozachok S, Erdogan Orhan I, Eren G, Senol Deniz FS, Stuppner H, Oleszek W. Yuccalechins A–C from the Yucca schidigera Roezl ex Ortgies Bark: Elucidation of the Relative and Absolute Configurations of Three New Spirobiflavonoids and Their Cholinesterase Inhibitory Activities. Molecules. 2019; 24(22):4162. https://doi.org/10.3390/molecules24224162
Chicago/Turabian StylePecio, Łukasz, Mostafa Alilou, Solomiia Kozachok, Ilkay Erdogan Orhan, Gokcen Eren, Fatma Sezer Senol Deniz, Hermann Stuppner, and Wiesław Oleszek. 2019. "Yuccalechins A–C from the Yucca schidigera Roezl ex Ortgies Bark: Elucidation of the Relative and Absolute Configurations of Three New Spirobiflavonoids and Their Cholinesterase Inhibitory Activities" Molecules 24, no. 22: 4162. https://doi.org/10.3390/molecules24224162
APA StylePecio, Ł., Alilou, M., Kozachok, S., Erdogan Orhan, I., Eren, G., Senol Deniz, F. S., Stuppner, H., & Oleszek, W. (2019). Yuccalechins A–C from the Yucca schidigera Roezl ex Ortgies Bark: Elucidation of the Relative and Absolute Configurations of Three New Spirobiflavonoids and Their Cholinesterase Inhibitory Activities. Molecules, 24(22), 4162. https://doi.org/10.3390/molecules24224162