
NAD Analogs in Aid of Chemical Biology and Medicinal Chemistry


Abstract
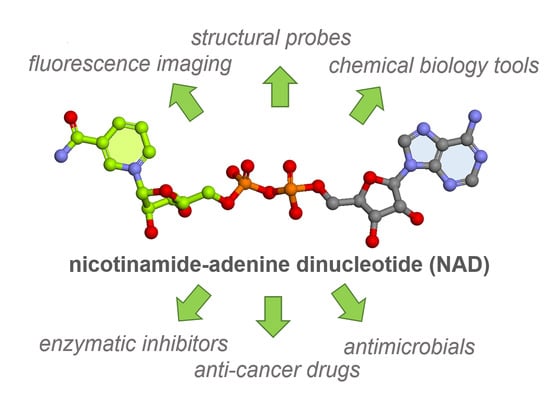
:
1. Introduction
- NAD analogs as fluorescent probes for real-time monitoring of miscellaneous NAD-processing enzymes
- NAD analogs for the detection and visualization of polyADPribose polymerase activity
- Potential therapeutic agents derived from NAD structure
- NAD analogs for the development of biorthogonal redox systems
- Methods and tools to advance our understanding of NAD-capped RNAs
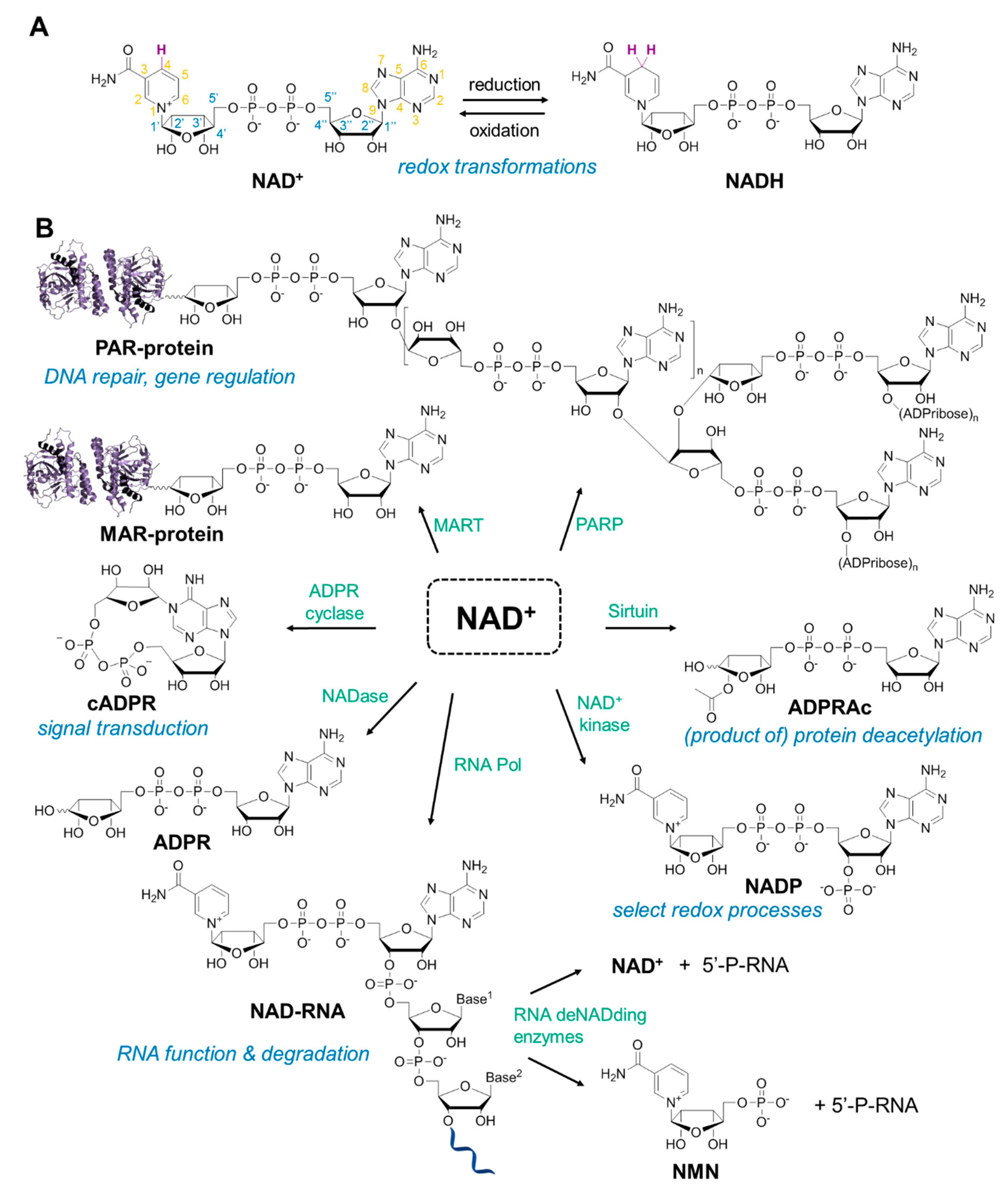
2. Fluorescent NAD Analogs for Real-Time Monitoring of NAD-Processing Enzymes
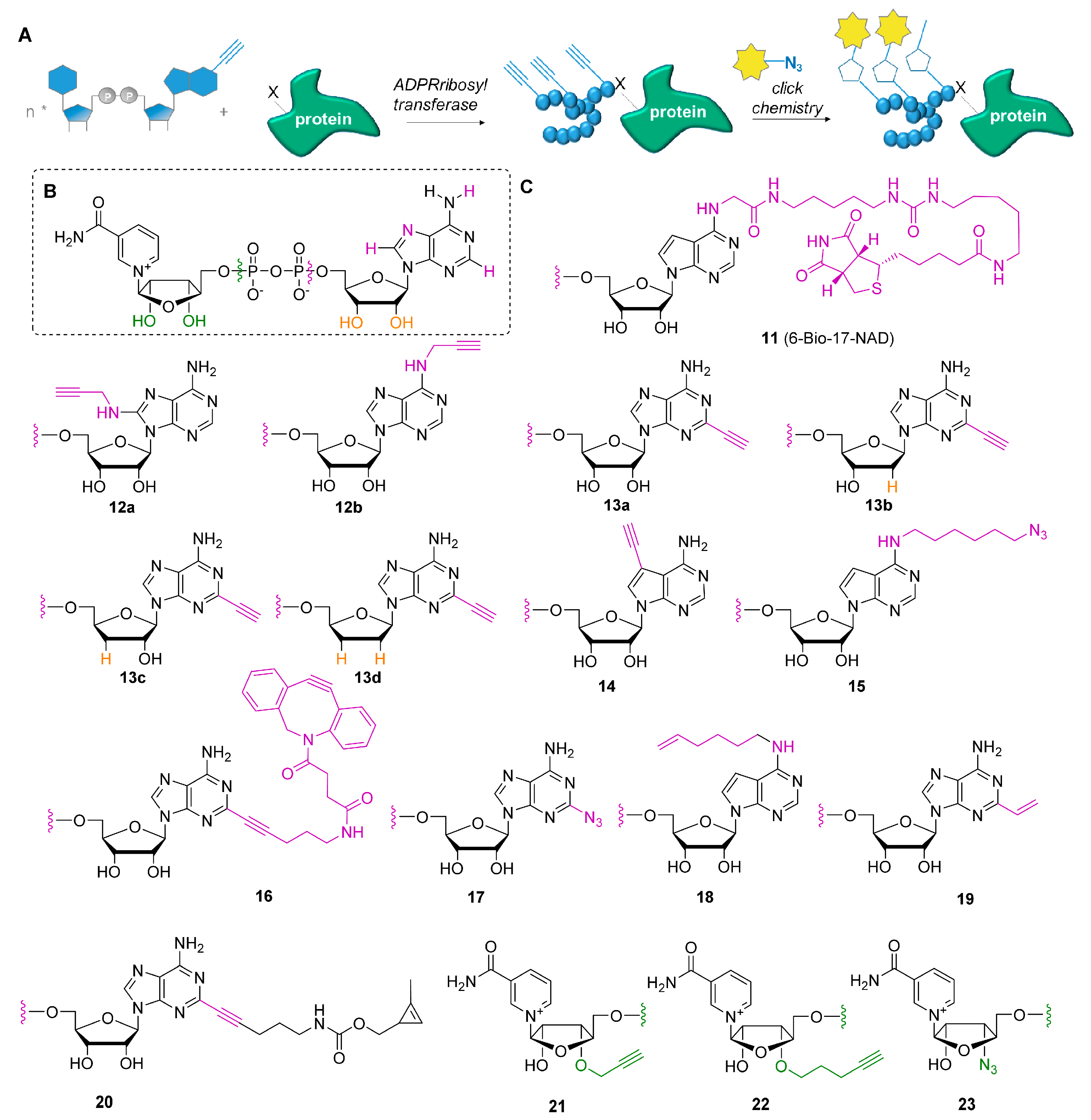
3. NAD Analogs to Study Protein ADP-Ribosylation
4. NAD-Derived Inhibitors of Biologically and Therapeutically Relevant Processes
4.1. NAD Analogs Targeting Enzymes that Attack the Glyosidic Bond
4.1.1. CD38 Ligands and Inhibitors
4.1.2. NAD Analogs Targeting Sirtuins and ADPribosyltransferases
4.2. NAD Analogs Targeting NAD-Dependent Metabolic Pathways
4.2.1. NAD Analogs Targeting Purine Biosynthesis Pathway
4.2.2. NADK Targeting Nucleotides and Pronucleotides
4.2.3. Miscellaneous and Unidentified Molecular Targets
5. NAD Analogs as Alternative Redox Co-Factors
6. NAD-Derived Chemical Tools to Study NAD-Capped RNAs
6.1. Chemoenzymatic Methods of NAD-RNA Detection
6.2. Synthesis of Molecular Tools to Study NAD-Capped RNAs
7. Conclusions and Perspectives for the Future
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAC | Azide alkyne cycloaddition |
| Ach | Acetylcholine |
| ACS | Acetyl-coA enzyme synthase |
| ADH | Alcohol dehydrogenase |
| ADPRC | ADP-ribosyl cyclase |
| ALDH | Aldehyde dehydogenase |
| Aq | aqueous |
| ART | ADP-ribosyl transferase |
| ASST | Tiazofurin adenosine disulfide |
| BAD | Benzamide adenine dinucleotide |
| cADPR | Cyclic ADP-ribose |
| CDI | 1,1′-carbonyldiimidazole |
| CML | Chronic myleogenous leukemia |
| CRF | Chronic renal failure |
| CTA | Cholera toxin subunit A |
| CuAAC | Copper catalysed AAC |
| dpCoA | Dephospho coenzyme A |
| DHFR | dehydrofolate reductase |
| DIC | N,N-diidopropylcarbodiimide |
| DMF | Dimethylformamide |
| DMSO | Dimethylsulfoxide |
| DTA | di-5′-thioadenosine dinucleotide |
| FAD | Flavine adenine dinucleotide |
| FLIM-FRET | Fluorescence lifetime imaging microscopy–Förster resonance energy transfer |
| G6PDH | Glucose-6-phosphate dehydrogenase |
| GADPH | Glyceraldehyde phosphate dehydrogenase |
| GFP | Green fluorescent protein |
| IDH | Isocitrate dehydrogenase |
| IEDDA | Inverse electron demand Diels-Alder cycloaddition |
| IMP(DH) | Inosine-5′-monophosphate (dehydrogenase) |
| INH | Isoniazid |
| InhA | Enoyl-ACP reductase |
| IVT | in vitro transcription |
| LDH | Lactate dehydrogenase |
| MAD | Mycophenolic acid adenine dinucleotide |
| MART | MonoADP-ribosyl transferase |
| MeCN | Acetonitrile |
| MPA | Mycophenolic acid |
| Mtb | Mycobacterium |
| MTX | Methotrexate |
| NaAD | Nicotinic acid adenine dinucleotide |
| NAD | Nicotinamide adenine dinucleotide |
| NADase | NAD glycohydrolase |
| NADK | NAD kinase |
| NAD(P)S | Thionicotinamide adenine dinucleotide (phosphate) |
| Nam | Nicotinamide |
| NaMN | Nicotinic acid mononucleotide |
| NaMNAT | NaMN adenylyl transferase |
| NCIN | Non-canonical initiating nucleotides |
| NCD | Nicotinamide cytidine dinucleotide |
| NdAD | Nicotinamide-2′-deoxy adenosine dinucleotide |
| NGD | Nicotinamide guanosine dinucleotide |
| NMN | Nicotinamide mononucleotide |
| NMNAT | NMN adenylyl transferase |
| NMO | 4-methylmorpholine N-oxide |
| NRK | Nicotinamide riboside kinase |
| NUD | Nicotinamide uridine dinucleotide |
| PAR | Poly(ADP-ribose) |
| PARG | Poly(ADP-ribose) glycohydrolase |
| PARP/ARTD | Poly(ADP-ribose) polymerase |
| Pdh | Phosphite dehydrogenase |
| PPi | Pyrophosphate |
| ROS | Reactive oxygen species |
| Rt | Room temperature |
| SIRT | Sirtuin |
| SPAAC | Strain promoted AAC |
| TFA | Trifluoroacetic acid |
| TIPS-Cl | 2,4,6-triisopropylbenzenesulfonyl chloride |
| thA | Thieno[4,3-d]pyrimidine riboside |
| TMR | Tetramethylrhodamine |
| TN | Thionicotinamide |
| TPPTS | 3,3′,3″-Phosphanetriyltris(benzenesulfonic acid) trisodium salt |
| TSST | Ditiazofurin disulfide |
| TXPTS | Tris(2,4-dimethyl-5-sulfophenyl)phosphine trisodium salt |
| tzA | Isothiazolo[4,3-d]pyrimidine riboside |
References
- Agledal, L.; Niere, M.; Ziegler, M. The phosphate makes a difference: Cellular functions of NADP. Redox Rep. 2010, 15, 2–10. [Google Scholar] [CrossRef]
- Di Stefano, M.; Conforti, L. Diversification of NAD biological role: The importance of location. FEBS J. 2013, 280, 4711–4728. [Google Scholar] [CrossRef]
- Katsyuba, E.; Auwerx, J. Modulating NAD+ metabolism, from bench to bedside. EMBO J. 2017, 36, 2670–2683. [Google Scholar] [CrossRef]
- Stromland, O.; Niere, M.; Nikiforov, A.A.; VanLinden, M.R.; Heiland, I.; Ziegler, M. Keeping the balance in NAD metabolism. Biochem. Soc. Trans. 2019, 47, 119–130. [Google Scholar] [CrossRef]
- Davar, D.; Beumer, J.H.; Hamieh, L.; Tawbi, H. Role of PARP inhibitors in cancer biology and therapy. Curr. Med. Chem. 2012, 19, 3907–3921. [Google Scholar] [CrossRef]
- Pant, S.; Maitra, A.; Yap, T.A. PARP inhibition—Opportunities in pancreatic cancer. Nat. Rev. Clin. Oncol. 2019, 16, 595–596. [Google Scholar] [CrossRef]
- David Adams, J.; Klaidman, L.K. Sirtuins, Nicotinamide and Aging: A Critical Review. Lett. Drug Des. Discov. 2007, 4, 44–48. [Google Scholar] [CrossRef]
- Abdelraheim, S.R.; Spiller, D.G.; McLennan, A.G. Mammalian NADH diphosphatases of the Nudix family: Cloning and characterization of the human peroxisomal NUDT12 protein. Biochem. J. 2003, 374, 329–335. [Google Scholar] [CrossRef]
- Cahova, H.; Winz, M.L.; Hofer, K.; Nubel, G.; Jaschke, A. NAD captureSeq indicates NAD as a bacterial cap for a subset of regulatory RNAs. Nature 2015, 519, 374–377. [Google Scholar] [CrossRef]
- Kiledjian, M. Eukaryotic RNA 5′-End NAD+ Capping and DeNADding. Trends Cell Biol. 2018, 28, 454–464. [Google Scholar] [CrossRef]
- Jaschke, A.; Hofer, K.; Nubel, G.; Frindert, J. Cap-like structures in bacterial RNA and epitranscriptomic modification. Curr. Opin. Microbiol. 2016, 30, 44–49. [Google Scholar] [CrossRef]
- Walters, R.W.; Matheny, T.; Mizoue, L.S.; Rao, B.S.; Muhlrad, D.; Parker, R. Identification of NAD+ capped mRNAs in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2017, 114, 480–485. [Google Scholar] [CrossRef]
- Julius, C.; Yuzenkova, Y. Bacterial RNA polymerase caps RNA with various cofactors and cell wall precursors. Nucleic Acids Res. 2017, 45, 8282–8290. [Google Scholar] [CrossRef]
- Jiao, X.; Doamekpor, S.K.; Bird, J.G.; Nickels, B.E.; Tong, L.; Hart, R.P.; Kiledjian, M. 5′ End Nicotinamide Adenine Dinucleotide Cap in Human Cells Promotes RNA Decay through DXO-Mediated deNADding. Cell 2017, 168, 1015–1027. [Google Scholar] [CrossRef]
- Wang, Y.; Li, S.; Zhao, Y.; You, C.; Le, B.; Gong, Z.; Mo, B.; Xia, Y.; Chen, X. NAD+-capped RNAs are widespread in the Arabidopsis transcriptome and can probably be translated. Proc. Natl. Acad. Sci. USA 2019, 116, 12094–12102. [Google Scholar] [CrossRef]
- Frindert, J.; Zhang, Y.; Nubel, G.; Kahloon, M.; Kolmar, L.; Hotz-Wagenblatt, A.; Burhenne, J.; Haefeli, W.E.; Jaschke, A. Identification, Biosynthesis, and Decapping of NAD-Capped RNAs in B. subtilis. Cell Rep. 2018, 24, 1890–1901. [Google Scholar] [CrossRef]
- Chen, Y.G.; Kowtoniuk, W.E.; Agarwal, I.; Shen, Y.; Liu, D.R. LC/MS analysis of cellular RNA reveals NAD-linked RNA. Nat. Chem. Biol. 2009, 5, 879–881. [Google Scholar] [CrossRef]
- Bird, J.G.; Zhang, Y.; Tian, Y.; Panova, N.; Barvik, I.; Greene, L.; Liu, M.; Buckley, B.; Krasny, L.; Lee, J.K.; et al. The mechanism of RNA 5′ capping with NAD+, NADH and desphospho-CoA. Nature 2016, 535, 444–447. [Google Scholar] [CrossRef]
- Julius, C.; Riaz-Bradley, A.; Yuzenkova, Y. RNA capping by mitochondrial and multi-subunit RNA polymerases. Transcription 2018, 9, 292–297. [Google Scholar] [CrossRef]
- Barvik, I.; Rejman, D.; Panova, N.; Sanderova, H.; Krasny, L. Non-canonical transcription initiation: The expanding universe of transcription initiating substrates. FEMS Microbiol. Rev. 2017, 41, 131–138. [Google Scholar] [CrossRef]
- Bird, J.G.; Basu, U.; Kuster, D.; Ramachandran, A.; Grudzien-Nogalska, E.; Towheed, A.; Wallace, D.C.; Kiledjian, M.; Temiakov, D.; Patel, S.S.; et al. Highly efficient 5′ capping of mitochondrial RNA with NAD+ and NADH by yeast and human mitochondrial RNA polymerase. Elife 2018, 7, e42179. [Google Scholar] [CrossRef] [PubMed]
- Grudzien-Nogalska, E.; Wu, Y.; Jiao, X.; Cui, H.; Mateyak, M.K.; Hart, R.P.; Tong, L.; Kiledjian, M. Structural and mechanistic basis of mammalian Nudt12 RNA deNADding. Nat. Chem. Biol. 2019, 15, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Hofer, K.; Li, S.; Abele, F.; Frindert, J.; Schlotthauer, J.; Grawenhoff, J.; Du, J.; Patel, D.J.; Jaschke, A. Structure and function of the bacterial decapping enzyme NudC. Nat. Chem. Biol. 2016, 12, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Imai, S. “Clocks” in the NAD World: NAD as a metabolic oscillator for the regulation of metabolism and aging. Biochim. Biophys. Acta 2010, 1804, 1584–1590. [Google Scholar] [CrossRef] [PubMed]
- Lin, H. Nicotinamide adenine dinucleotide: Beyond a redox coenzyme. Org. Biomol. Chem. 2007, 5, 2541–2554. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-J.; Guarente, L. Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr. Opin. Cell Biol. 2003, 15, 241–246. [Google Scholar] [CrossRef]
- Chini, C.C.S.; Tarrago, M.G.; Chini, E.N. NAD and the aging process: Role in life, death and everything in between. Mol. Cell Endocrinol. 2017, 455, 62–74. [Google Scholar] [CrossRef]
- Lakowicz, J.R.; Szmacinski, H.; Nowaczyk, K.; Johnson, M.L. Fluorescence lifetime imaging of free and protein-bound NADH. Proc. Natl. Acad. Sci. USA 1992, 89, 1271–1275. [Google Scholar] [CrossRef]
- Skala, M.C.; Riching, K.M.; Bird, D.K.; Gendron-Fitzpatrick, A.; Eickhoff, J.; Eliceiri, K.W.; Keely, P.J.; Ramanujam, N. In vivo multiphoton fluorescence lifetime imaging of protein-bound and free nicotinamide adenine dinucleotide in normal and precancerous epithelia. J. Biomed. Opt. 2007, 12, 024014. [Google Scholar] [CrossRef]
- Blacker, T.S.; Mann, Z.F.; Gale, J.E.; Ziegler, M.; Bain, A.J.; Szabadkai, G.; Duchen, M.R. Separating NADH and NADPH fluorescence in live cells and tissues using FLIM. Nat. Commun. 2014, 5, 3936. [Google Scholar] [CrossRef]
- Gordon Scott, T.; Spencer, R.D.; Leonard, N.J.; Weber, G. Emission Properties of NADH. Studies of Fluorescence Lifetimes and Quantum Efficiencies of NADH, AcPyADH, and Simplified Synthetic Models. J. Am. Chem. Soc. 1970, 92, 687–695. [Google Scholar] [CrossRef]
- Pergolizzi, G.; Butt, J.N.; Bowater, R.P.; Wagner, G.K. A novel fluorescent probe for NAD-consuming enzymes. Chem. Commun. 2011, 47, 12655–12657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovira, A.R.; Fin, A.; Tor, Y. Emissive Synthetic Cofactors: An Isomorphic, Isofunctional, and Responsive NAD+ Analogue. J. Am. Chem. Soc. 2017, 139, 15556–15559. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Sinkeldam, R.W.; Tor, Y. Emissive RNA alphabet. J. Am. Chem. Soc. 2011, 133, 14912–14915. [Google Scholar] [CrossRef] [PubMed]
- Noe, M.S.; Sinkeldam, R.W.; Tor, Y. Oligodeoxynucleotides containing multiple thiophene-modified isomorphic fluorescent nucleosides. J. Org. Chem. 2013, 78, 8123–8128. [Google Scholar] [CrossRef] [Green Version]
- Rovira, A.R.; Fin, A.; Tor, Y. Chemical Mutagenesis of an Emissive RNA Alphabet. J. Am. Chem. Soc. 2015, 137, 14602–14605. [Google Scholar] [CrossRef] [Green Version]
- Mizrahi, R.A.; Shin, D.; Sinkeldam, R.W.; Phelps, K.J.; Fin, A.; Tantillo, D.J.; Tor, Y.; Beal, P.A. A Fluorescent Adenosine Analogue as a Substrate for an A-to-I RNA Editing Enzyme. Angew. Chem. Int. Ed. 2015, 54, 8713–8716. [Google Scholar] [CrossRef] [Green Version]
- Halle, F.; Fin, A.; Rovira, A.R.; Tor, Y. Emissive Synthetic Cofactors: Enzymatic Interconversions of tzA Analogues of ATP, NAD+, NADH, NADP+, and NADPH. Angew. Chem. Int. Ed. 2018, 57, 1087–1090. [Google Scholar] [CrossRef]
- Feldmann, J.; Li, Y.; Tor, Y. Emissive Synthetic Cofactors: A Highly Responsive NAD+ Analogue Reveals Biomolecular Recognition Features. Chem. Eur. J. 2019, 25, 4379–4389. [Google Scholar] [CrossRef]
- Dieter, H.; Koberstein, R.; Sund, H. Nicotinamide 1, N6-ethenoadenine dinucleotide, a coenzyme for glutamate dehydrogenase. FEBS Lett. 1974, 47, 90–93. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.E.; Aizono, Y.; Sonenberg, M.; Swilocki, N.I. Further chemical characterization and biological activities of fluorescent 1, N6-ethenoadenosine derivatives. Bioorg. Chem. 1975, 4, 181–187. [Google Scholar] [CrossRef]
- Oei, S.L.; Griesenbeck, J.; Buchlow, G.; Jorcke, D.; Mayer-Kuckuk, P.; Wons, T.; Ziegler, M. NAD+ analogs substituted in the purine base as substrates for poly(ADP-ribosyl) transferase. FEBS Lett. 1996, 397, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Shrimp, J.H.; Hu, J.; Dong, M.; Wang, B.S.; MacDonald, R.; Jiang, H.; Hao, Q.; Yen, A.; Lin, H. Revealing CD38 cellular localization using a cell permeable, mechanism-based fluorescent small-molecule probe. J. Am. Chem. Soc. 2014, 136, 5656–5663. [Google Scholar] [CrossRef] [PubMed]
- Moreau, C.; Kirchberger, T.; Zhang, B.; Thomas, M.P.; Weber, K.; Guse, A.H.; Potter, B.V.L. Aberrant Cyclization Affords a C-6 Modified Cyclic Adenosine 5′-Diphosphoribose Analogue with Biological Activity in Jurkat T Cells. J. Med. Chem. 2012, 55, 1478–1489. [Google Scholar] [CrossRef] [PubMed]
- Buntz, A.; Wallrodt, S.; Gwosch, E.; Schmalz, M.; Beneke, S.; Ferrando-May, E.; Marx, A.; Zumbusch, A. Real-Time Cellular Imaging of Protein Poly(ADP-ribos) ylation. Angew. Chem. Int. Ed. 2016, 55, 11256–11260. [Google Scholar] [CrossRef] [Green Version]
- Leung, A.K. Poly(ADP-ribose): An organizer of cellular architecture. J. Cell. Biol. 2014, 205, 613–619. [Google Scholar] [CrossRef] [Green Version]
- Corda, D.; Di Girolamo, M. Functional aspects of protein mono-ADP-ribosylation. EMBO J. 2003, 22, 1953–1958. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J. Use of biotinylated NAD to label and purify ADP-ribosylated proteins. Methods Enzymol. 1997, 280, 255–265. [Google Scholar]
- Garcia Soriano, F.; Virag, L.; Jagtap, P.; Szabo, E.; Mabley, J.G.; Liaudet, L.; Marton, A.; Hoyt, D.G.; Murthy, K.G.; Salzman, A.L.; et al. Diabetic endothelial dysfunction: The role of poly(ADP-ribose) polymerase activation. Nat. Med. 2001, 7, 108–113. [Google Scholar] [CrossRef]
- Du, J.; Jiang, H.; Lin, H. Investigating the ADP-ribosyltransferase activity of sirtuins with NAD analogues and 32P-NAD. Biochemistry 2009, 48, 2878–2890. [Google Scholar] [CrossRef]
- Jiang, H.; Kim, J.H.; Frizzell, K.M.; Kraus, W.L.; Lin, H. Clickable NAD analogues for labeling substrate proteins of poly(ADP-ribose) polymerases. J. Am. Chem. Soc. 2010, 132, 9363–9372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Rosner, D.; Grzywa, M.; Marx, A. Chain-terminating and clickable NAD+ analogues for labeling the target proteins of ADP-ribosyltransferases. Angew. Chem. Int. Ed. 2014, 53, 8159–8162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallrodt, S.; Buntz, A.; Wang, Y.; Zumbusch, A.; Marx, A. Bioorthogonally Functionalized NAD+ Analogues for In-Cell Visualization of Poly(ADP-Ribose) Formation. Angew. Chem. Int. Ed. 2016, 55, 7660–7664. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.N.; Cheng, Q.; Chen, J.; Lam, A.T.; Lu, Y.; Dai, Z.; Pei, H.; Evdokimov, N.M.; Louie, S.G.; Zhang, Y. A ribose-functionalized NAD+ with unexpected high activity and selectivity for protein poly-ADP-ribosylation. Nat. Commun. 2019, 10, 4196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, C.; Ribeiro, M.; Scott, S.; Lonial, S. Daratumumab: Monoclonal antibody therapy to treat multiple myeloma. Drugs Today 2016, 52, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Si, Y.Q.; Sun, S.Y.; Pu, X.P.; Yang, Z.J.; Zhang, L.R.; Zhang, L.H.; Leung, F.P.; Lam, C.M.; Kwong, A.K.; et al. Design, synthesis and biological characterization of novel inhibitors of CD38. Org. Biomol. Chem. 2011, 9, 3246–3257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwong, A.K.; Chen, Z.; Zhang, H.; Leung, F.P.; Lam, C.M.; Ting, K.Y.; Zhang, L.; Hao, Q.; Zhang, L.H.; Lee, H.C. Catalysis-based inhibitors of the calcium signaling function of CD38. Biochemistry 2012, 51, 555–564. [Google Scholar] [CrossRef]
- Wang, S.; Zhu, W.; Wang, X.; Li, J.; Zhang, K.; Zhang, L.; Zhao, Y.J.; Lee, H.C.; Zhang, L. Design, synthesis and SAR studies of NAD analogues as potent inhibitors towards CD38 NADase. Molecules 2014, 19, 15754–15767. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.C.; Hallows, W.C.; Denu, J.M. Mechanisms and molecular probes of sirtuins. Chem. Biol. 2008, 15, 1002–1013. [Google Scholar] [CrossRef] [Green Version]
- Pesnot, T.; Kempter, J.; Schemies, J.; Pergolizzi, G.; Uciechowska, U.; Rumpf, T.; Sippl, W.; Jung, M.; Wagner, G.K. Two-step synthesis of novel, bioactive derivatives of the ubiquitous cofactor nicotinamide adenine dinucleotide (NAD). J. Med. Chem. 2011, 54, 3492–3499. [Google Scholar] [CrossRef]
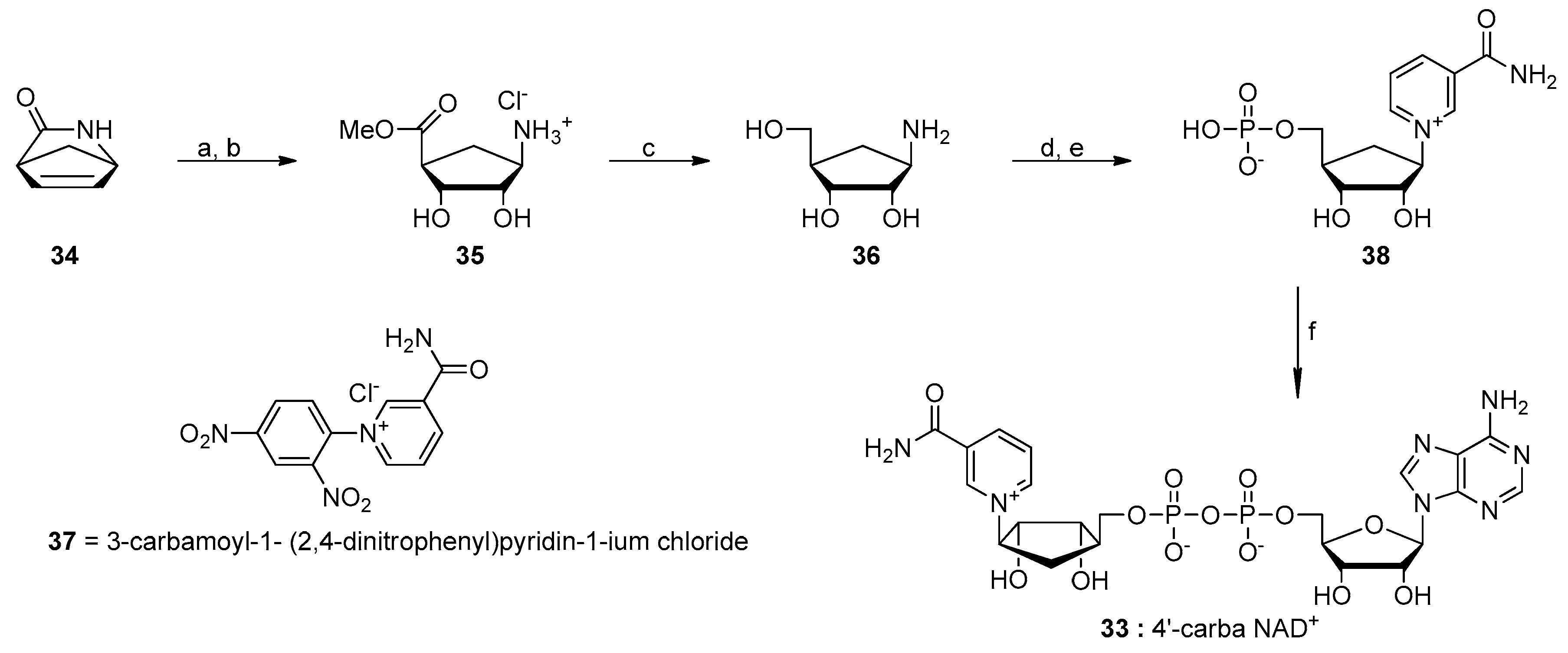
- Szczepankiewicz, B.G.; Dai, H.; Koppetsch, K.J.; Qian, D.; Jiang, F.; Mao, C.; Perni, R.B. Synthesis of carba-NAD and the structures of its ternary complexes with SIRT3 and SIRT5. J. Org. Chem. 2012, 77, 7319–7329. [Google Scholar] [CrossRef] [PubMed]
- Slama, J.T.; Simmons, A.M. Carbanicotinamide adenine dinucleotide: Synthesis and enzymological properties of a carbocyclic analogue of oxidized nicotinamide adenine dinucleotide. Biochemistry 1988, 27, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Zhang, X.N.; Nasertorabi, F.; Cheng, Q.; Pei, H.; Louie, S.G.; Stevens, R.C.; Zhang, Y. Facile chemoenzymatic synthesis of a novel stable mimic of NAD. Chem. Sci. 2018, 9, 8337–8342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, L.S.; Lee, H.W.; Jacobson, K.A.; Kim, H.O.; Shin, D.H.; Lee, J.A.; Gao, Z.G.; Lu, C.; Duong, H.T.; Gunaga, P.; et al. Structure-activity relationships of 2-chloro-N6-substituted-4′-thioadenosine-5′-uronamides as highly potent and selective agonists at the human A3 adenosine receptor. J. Med. Chem. 2006, 49, 273–281. [Google Scholar] [CrossRef]
- Langelier, M.F.; Zandarashvili, L.; Aguiar, P.M.; Black, B.E.; Pascal, J.M. NAD+ analog reveals PARP-1 substrate-blocking mechanism and allosteric communication from catalytic center to DNA-binding domains. Nat. Commun. 2018, 9, 844. [Google Scholar] [CrossRef]
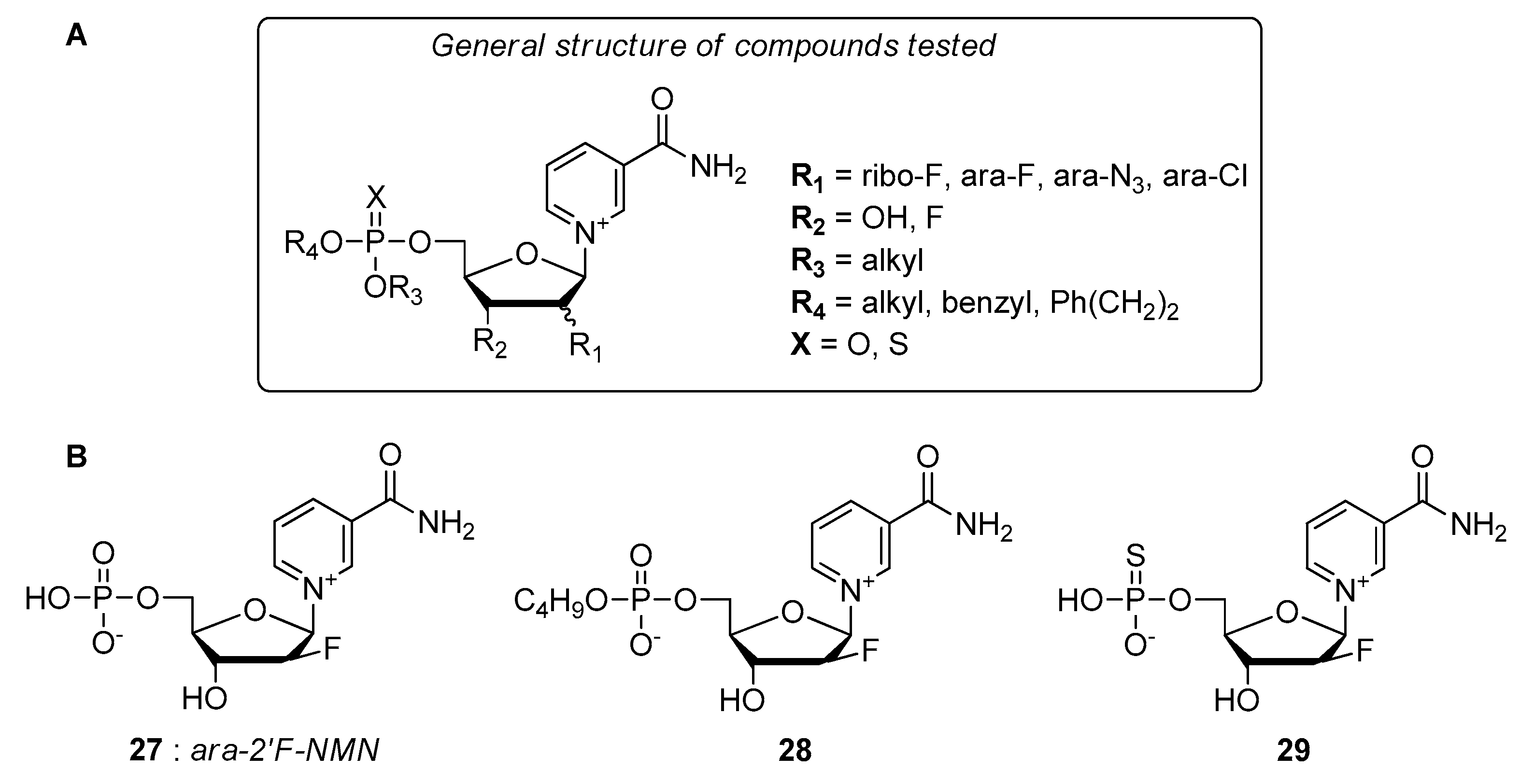
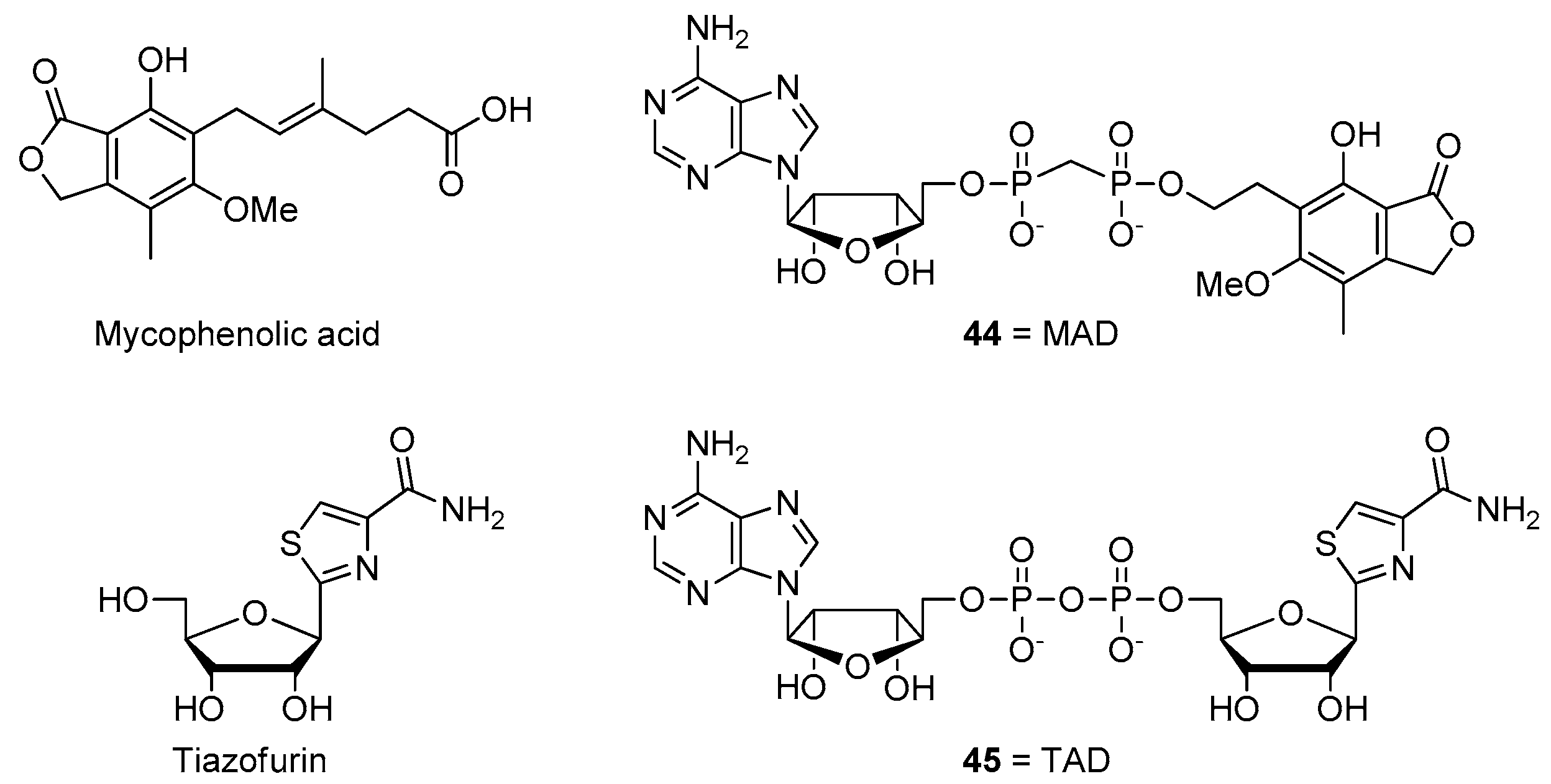
- Felczak, K.; Pankiewicz, K.W. Rehab of NAD(P)-dependent enzymes with NAD(P)-based inhibitors. Curr. Med. Chem. 2011, 18, 1891–1908. [Google Scholar] [CrossRef]
- Pankiewicz, K.W.; Felczak, K. From ribavirin to NAD analogues and back to ribavirin in search for anticancer agents. Heterocycl. Commun. 2015, 21, 249–257. [Google Scholar] [CrossRef]
- Chen, L.; Wilson, D.J.; Xu, Y.; Aldrich, C.C.; Felczak, K.; Sham, Y.Y.; Pankiewicz, K.W. Triazole-linked inhibitors of inosine monophosphate dehydrogenase from human and Mycobacterium tuberculosis. J. Med. Chem. 2010, 53, 4768–4778. [Google Scholar] [CrossRef] [Green Version]
- Felczak, K.; Chen, L.; Wilson, D.; Williams, J.; Vince, R.; Petrelli, R.; Jayaram, H.N.; Kusumanchi, P.; Kumar, M.; Pankiewicz, K.W. Cofactor-type inhibitors of inosine monophosphate dehydrogenase via modular approach: Targeting the pyrophosphate binding sub-domain. Bioorg. Med. Chem. 2011, 19, 1594–1605. [Google Scholar] [CrossRef]
- Felczak, K.; Pankiewicz, K.W. Synthesis of methylenebis(phosphonate) analogues of 2-, 4-, and 6-pyridones of nicotinamide adenine dinucleotide. Nucleosides Nucleotides Nucleic Acids 2011, 30, 512–523. [Google Scholar] [CrossRef]
- Dutta, S.P.; Crain, P.F.; McCloskey, J.A.; Chheda, G.B. Isolation and characterization of 1-beta-D-ribofuranosylpyridin-4-one-3-carboxamide from human urine. Life Sci. 1979, 24, 1381–1388. [Google Scholar] [CrossRef]
- Felczak, K.; Vince, R.; Pankiewicz, K.W. NAD-based inhibitors with anticancer potential. Bioorg. Med. Chem. Lett. 2014, 24, 332–336. [Google Scholar] [CrossRef] [PubMed]
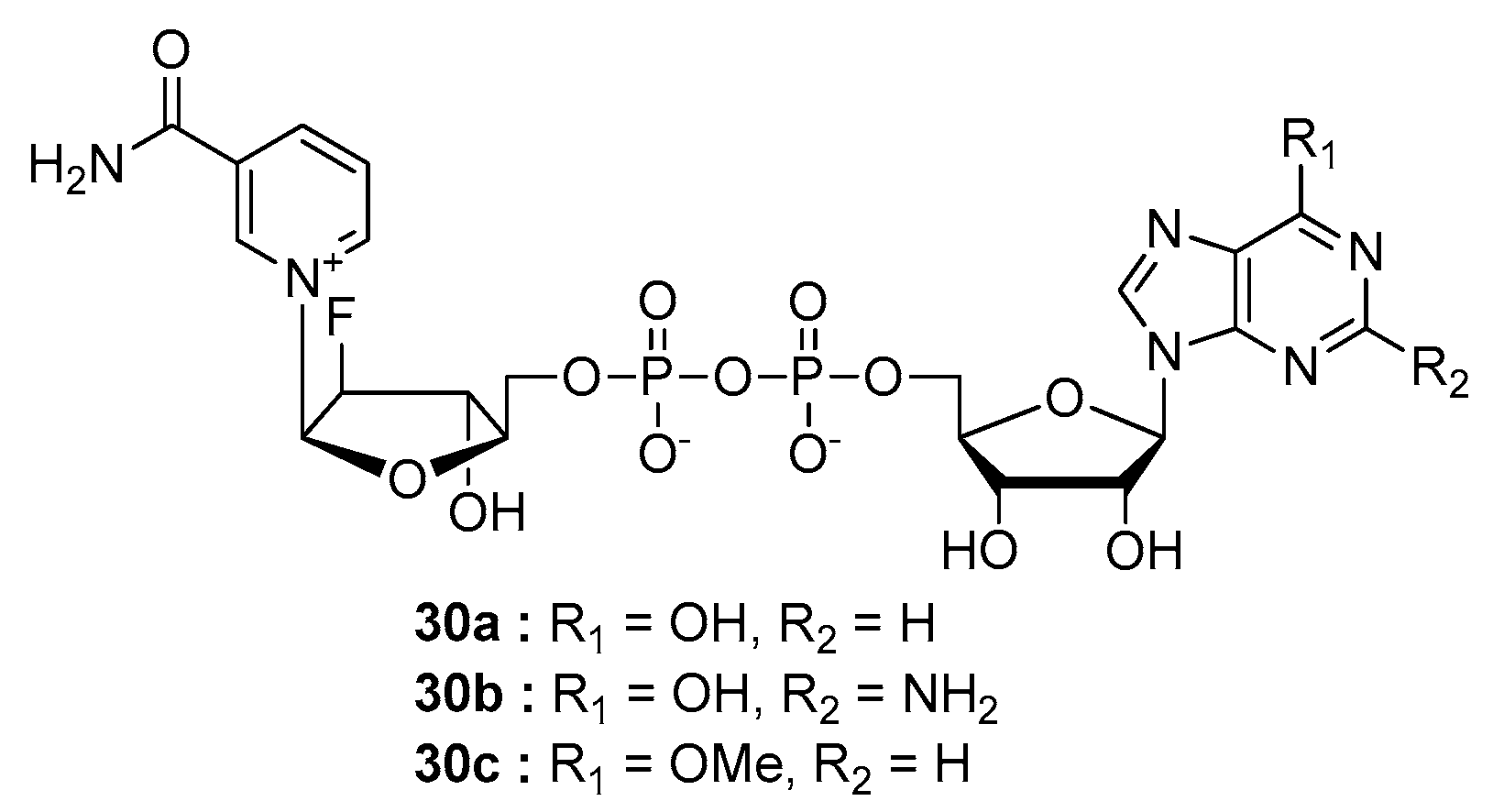
- Poncet-Montange, G.; Assairi, L.; Arold, S.; Pochet, S.; Labesse, G. NAD kinases use substrate-assisted catalysis for specific recognition of NAD. J. Biol. Chem. 2007, 282, 33925–33934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedeschi, P.M.; Bansal, N.; Kerrigan, J.E.; Abali, E.E.; Scotto, K.W.; Bertino, J.R. NAD+ Kinase as a Therapeutic Target in Cancer. Clin. Cancer Res. 2016, 22, 5189–5195. [Google Scholar] [CrossRef] [Green Version]
- Petrelli, R.; Felczak, K.; Cappellacci, L. NMN/NaMN adenylyltransferase (NMNAT) and NAD kinase (NADK) inhibitors: Chemistry and potential therapeutic applications. Curr. Med. Chem. 2011, 18, 1973–1992. [Google Scholar] [CrossRef]
- Petrelli, R.; Sham, Y.Y.; Chen, L.; Felczak, K.; Bennett, E.; Wilson, D.; Aldrich, C.; Yu, J.S.; Cappellacci, L.; Franchetti, P.; et al. Selective inhibition of nicotinamide adenine dinucleotide kinases by dinucleoside disulfide mimics of nicotinamide adenine dinucleotide analogues. Bioorg. Med. Chem. 2009, 17, 5656–5664. [Google Scholar] [CrossRef]
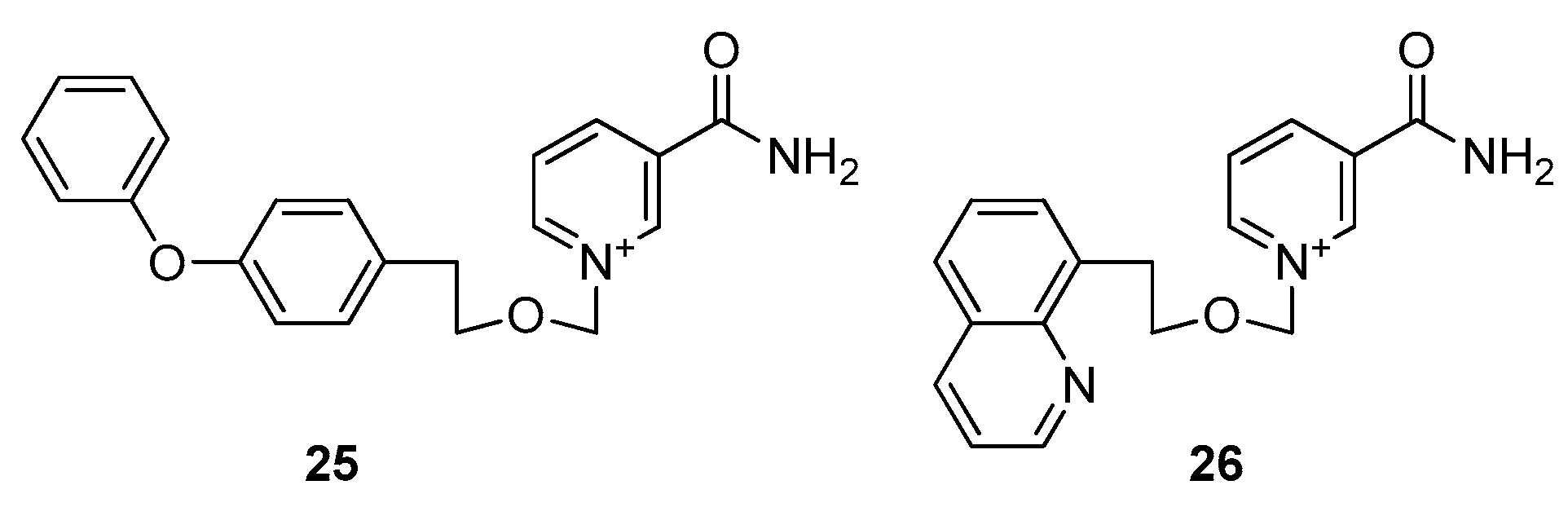
- Hsieh, Y.C.; Tedeschi, P.; Adebisi Lawal, R.; Banerjee, D.; Scotto, K.; Kerrigan, J.E.; Lee, K.C.; Johnson-Farley, N.; Bertino, J.R.; Abali, E.E. Enhanced degradation of dihydrofolate reductase through inhibition of NAD kinase by nicotinamide analogs. Mol. Pharm. 2013, 83, 339–353. [Google Scholar] [CrossRef] [Green Version]
- Tedeschi, P.M.; Lin, H.; Gounder, M.; Kerrigan, J.E.; Abali, E.E.; Scotto, K.; Bertino, J.R. Suppression of Cytosolic NADPH Pool by Thionicotinamide Increases Oxidative Stress and Synergizes with Chemotherapy. Mol. Pharm. 2015, 88, 720–727. [Google Scholar] [CrossRef] [Green Version]
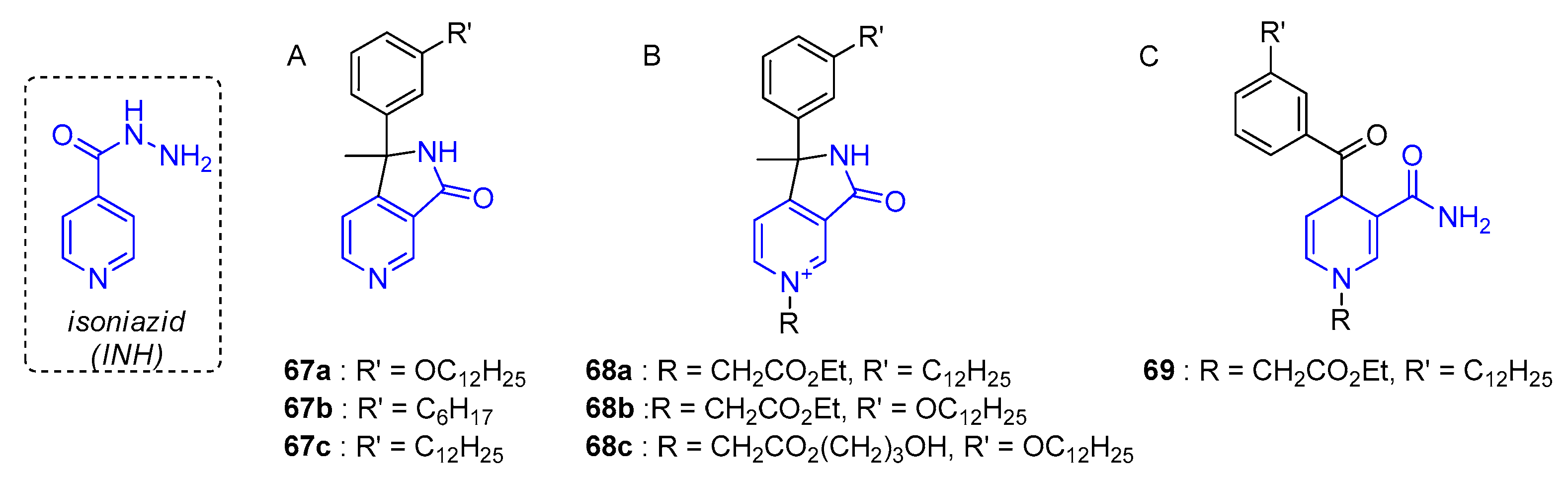
- Delaine, T.; Bernardes-Genisson, V.; Quemard, A.; Constant, P.; Meunier, B.; Bernadou, J. Development of isoniazid-NAD truncated adducts embedding a lipophilic fragment as potential bi-substrate InhA inhibitors and antimycobacterial agents. Eur. J. Med. Chem. 2010, 45, 4554–4561. [Google Scholar] [CrossRef]
- Delaine, T.; Bernardes-Genisson, V.; Quemard, A.; Constant, P.; Cosledan, F.; Meunier, B.; Bernadou, J. Preliminary investigations of the effect of lipophilic analogues of the active metabolite of isoniazid toward bacterial and plasmodial strains. Chem. Biol. Drug Des. 2012, 79, 1001–1006. [Google Scholar] [CrossRef]
- Liu, W.; Wu, S.; Hou, S.; Zhao, Z. Synthesis of phosphodiester-type nicotinamide adenine dinucleotide analogs. Tetrahedron 2009, 65, 8378–8383. [Google Scholar] [CrossRef]
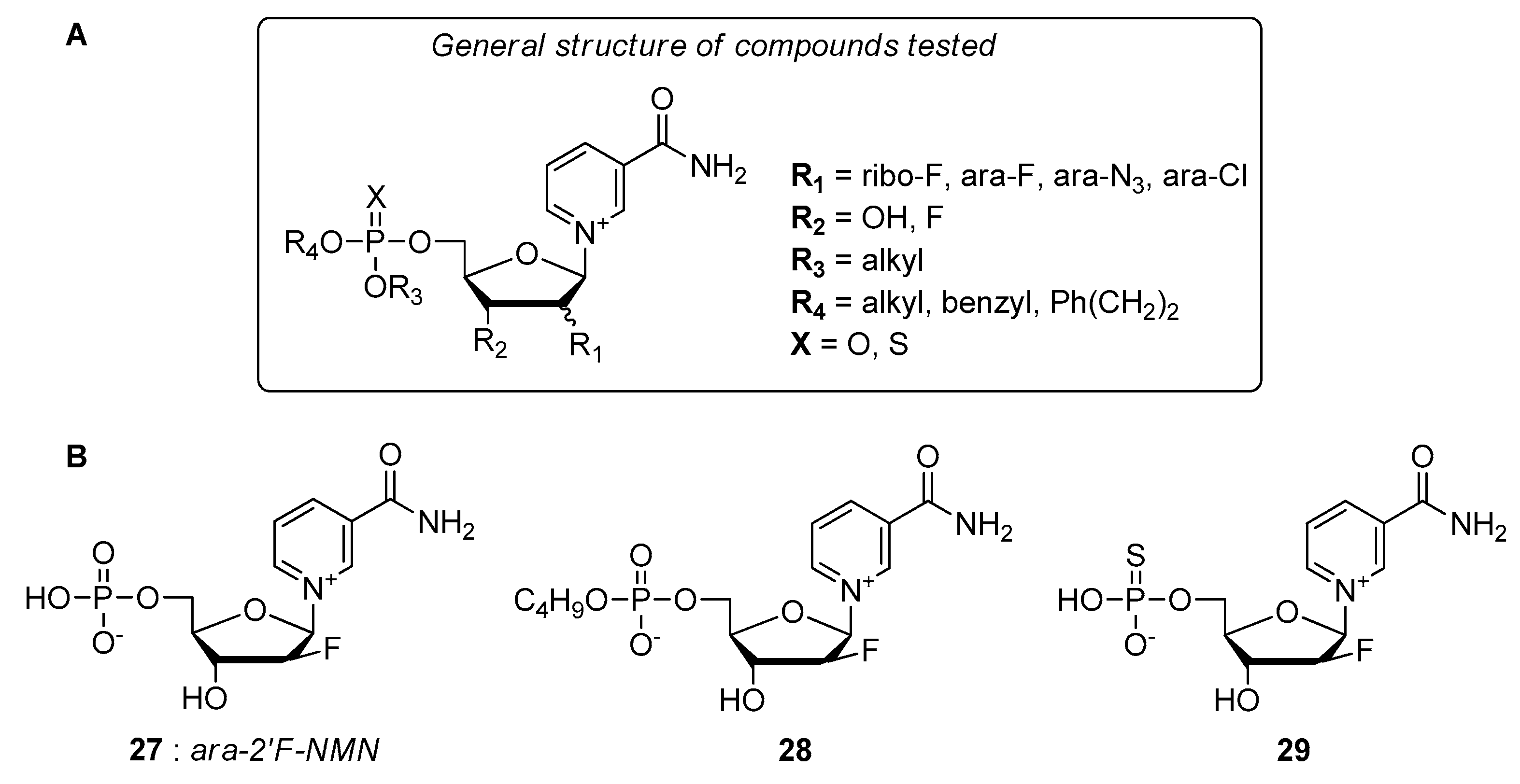
- Jain, P.; Slama, J.T.; Perez-Haddock, L.A.; Walseth, T.F. Nicotinic acid adenine dinucleotide phosphate analogues containing substituted nicotinic acid: Effect of modification on Ca2+ release. J. Med. Chem. 2010, 53, 7599–7612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trabbic, C.J.; Zhang, F.; Walseth, T.F.; Slama, J.T. Nicotinic Acid Adenine Dinucleotide Phosphate Analogues Substituted on the Nicotinic Acid and Adenine Ribosides. Effects on ReceptorMediated Ca2+ Release. J. Med. Chem. 2015, 58, 3593–3610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, C.E.; Arends, I.W.C.E.; Hollmann, F. Is Simpler Better? Synthetic Nicotinamide Cofactor Analogues for Redox Chemistry. ACS Catal. 2014, 4, 788–797. [Google Scholar] [CrossRef]
- Hou, S.; Liu, W.; Ji, D.; Wang, Q.; Zhao, Z.K. Synthesis of 1,2,3-triazole moiety-containing NAD analogs and their potential as redox cofactors. Tetrahedron Lett. 2011, 52, 5855–5857. [Google Scholar] [CrossRef]
- Hou, S.; Ji, D.; Liu, W.; Wang, L.; Zhao, Z.K. Identification of malic enzyme mutants depending on 1,2,3-triazole moiety-containing nicotinamide adenine dinucleotide analogs. Bioorg. Med. Chem. Lett. 2014, 24, 1307–1309. [Google Scholar] [CrossRef]
- Ji, D.; Wang, L.; Hou, S.; Liu, W.; Wang, J.; Wang, Q.; Zhao, Z.K. Creation of bioorthogonal redox systems depending on nicotinamide flucytosine dinucleotide. J. Am. Chem. Soc. 2011, 133, 20857–20862. [Google Scholar] [CrossRef]
- Ji, D.; Wang, L.; Liu, W.; Hou, S.; Zhao, K.Z. Synthesis of NAD analogs to develop bioorthogonal redox system. Sci. China Chem. 2013, 56, 296–300. [Google Scholar] [CrossRef]
- Liu, Y.; Feng, Y.; Wang, L.; Guo, X.; Liu, W.; Li, Q.; Wang, X.; Xue, S.; Zhao, Z.K. Structural Insights into Phosphite Dehydrogenase Variants Favoring a Non-natural Redox Cofactor. ACS Catal. 2019, 9, 1883–1887. [Google Scholar] [CrossRef]
- Lee, S.H.; Lee, H.J.; Won, K.; Park, C.B. Artificial electron carriers for photoenzymatic synthesis under visible light. Chem. Eur. J. 2012, 18, 5490–5495. [Google Scholar] [CrossRef]
- Zhang, H.; Zhong, H.; Zhang, S.; Shao, X.; Ni, M.; Cai, Z.; Chen, X.; Xia, Y. NAD tagSeq reveals that NAD+-capped RNAs are mostly produced from a large number of protein-coding genes in Arabidopsis. Proc. Natl. Acad. Sci. USA 2019, 116, 12072–12077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Alvin Chew, B.L.; Lai, Y.; Dong, H.; Xu, L.; Balamkundu, S.; Cai, W.M.; Cui, L.; Liu, C.F.; Fu, X.Y.; et al. Quantifying the RNA cap epitranscriptome reveals novel caps in cellular and viral RNA. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
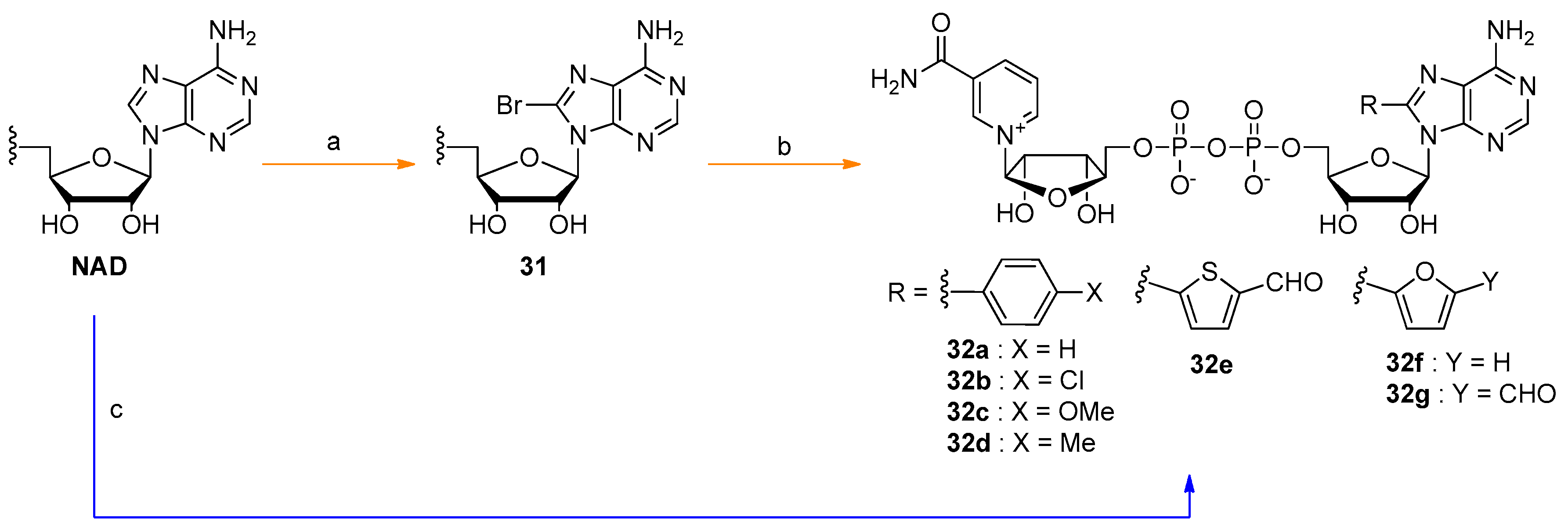
- Hofer, K.; Abele, F.; Schlotthauer, J.; Jaschke, A. Synthesis of 5′-NAD-Capped RNA. Bioconjug. Chem. 2016, 27, 874–877. [Google Scholar] [CrossRef] [PubMed]
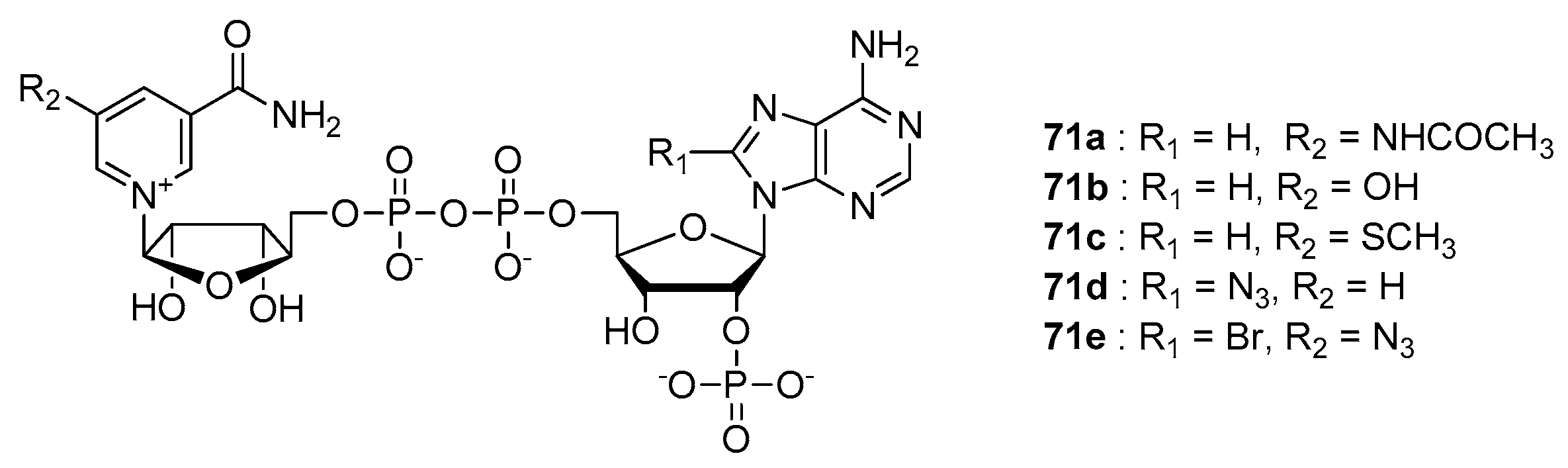
- Vvedenskaya, I.O.; Bird, J.G.; Zhang, Y.; Zhang, Y.; Jiao, X.; Barvik, I.; Krasny, L.; Kiledjian, M.; Taylor, D.M.; Ebright, R.H.; et al. CapZyme-Seq Comprehensively Defines Promoter-Sequence Determinants for RNA 5′ Capping with NAD+. Mol. Cell 2018, 70, 553–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grudzien-Nogalska, E.; Bird, J.G.; Nickels, B.E.; Kiledjian, M. “NAD-capQ” detection and quantitation of NAD caps. RNA 2018, 24, 1418–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mlynarska-Cieslak, A.; Depaix, A.; Grudzien-Nogalska, E.; Sikorski, P.J.; Warminski, M.; Kiledjian, M.; Jemielity, J.; Kowalska, J. Nicotinamide-Containing Di- and Trinucleotides as Chemical Tools for Studies of NAD-Capped RNAs. Org. Lett. 2018, 20, 7650–7655. [Google Scholar] [CrossRef] [PubMed]
- Huang, F. Efficient incorporation of CoA, NAD and FAD into RNA by in vitro transcription. Nucleic Acids Res. 2003, 31, e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Liu, B.; Liu, Y.; Sun, Y.; Liu, W.; Yu, D.; Zhao, Z.K. Escherichia coli Strain Designed for Characterizing In Vivo Functions of Nicotinamide Adenine Dinucleotide Analogues. Org. Lett. 2019, 21, 3218–3222. [Google Scholar] [CrossRef]





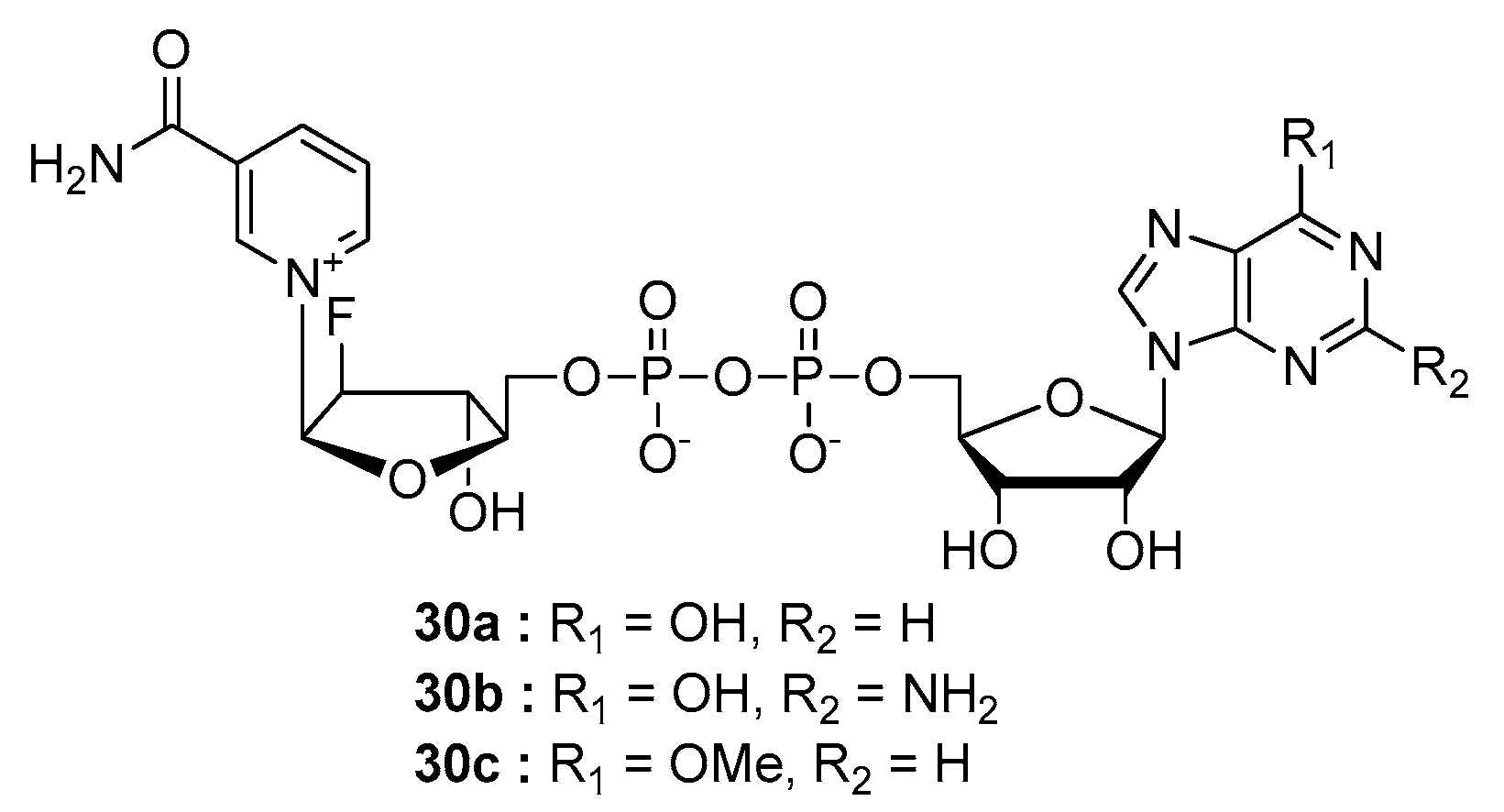


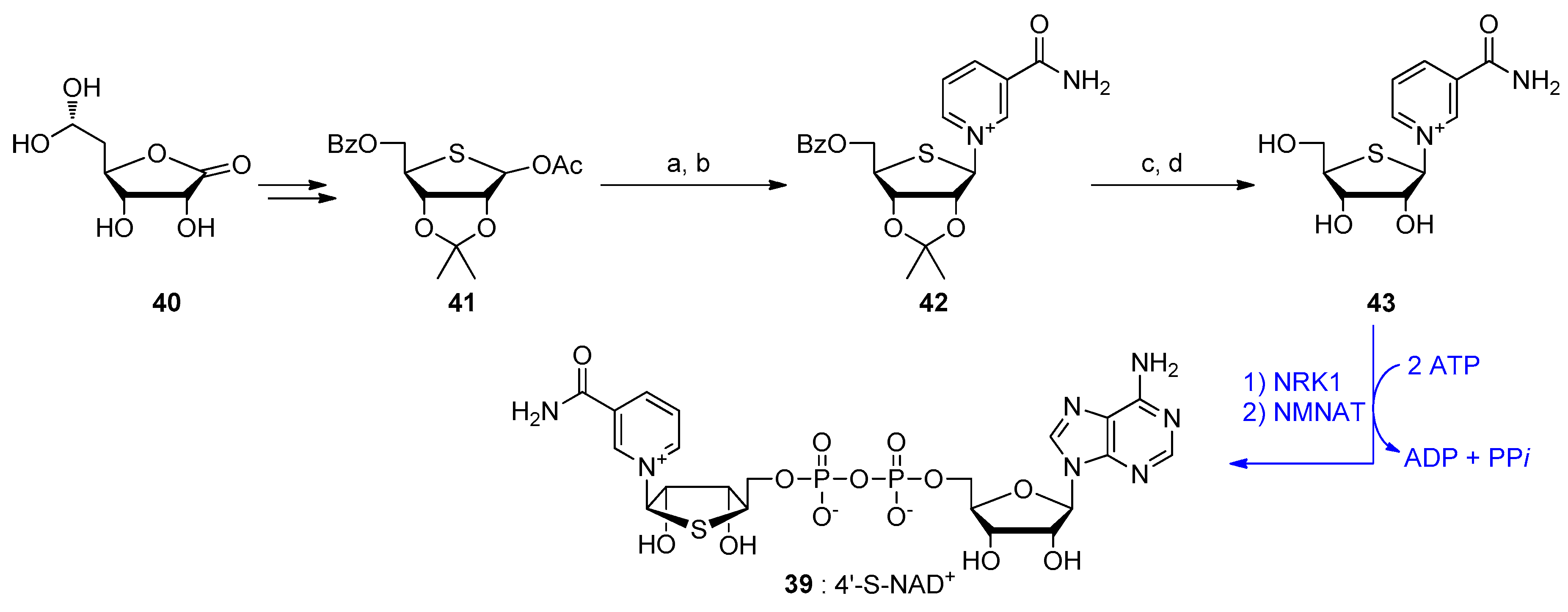

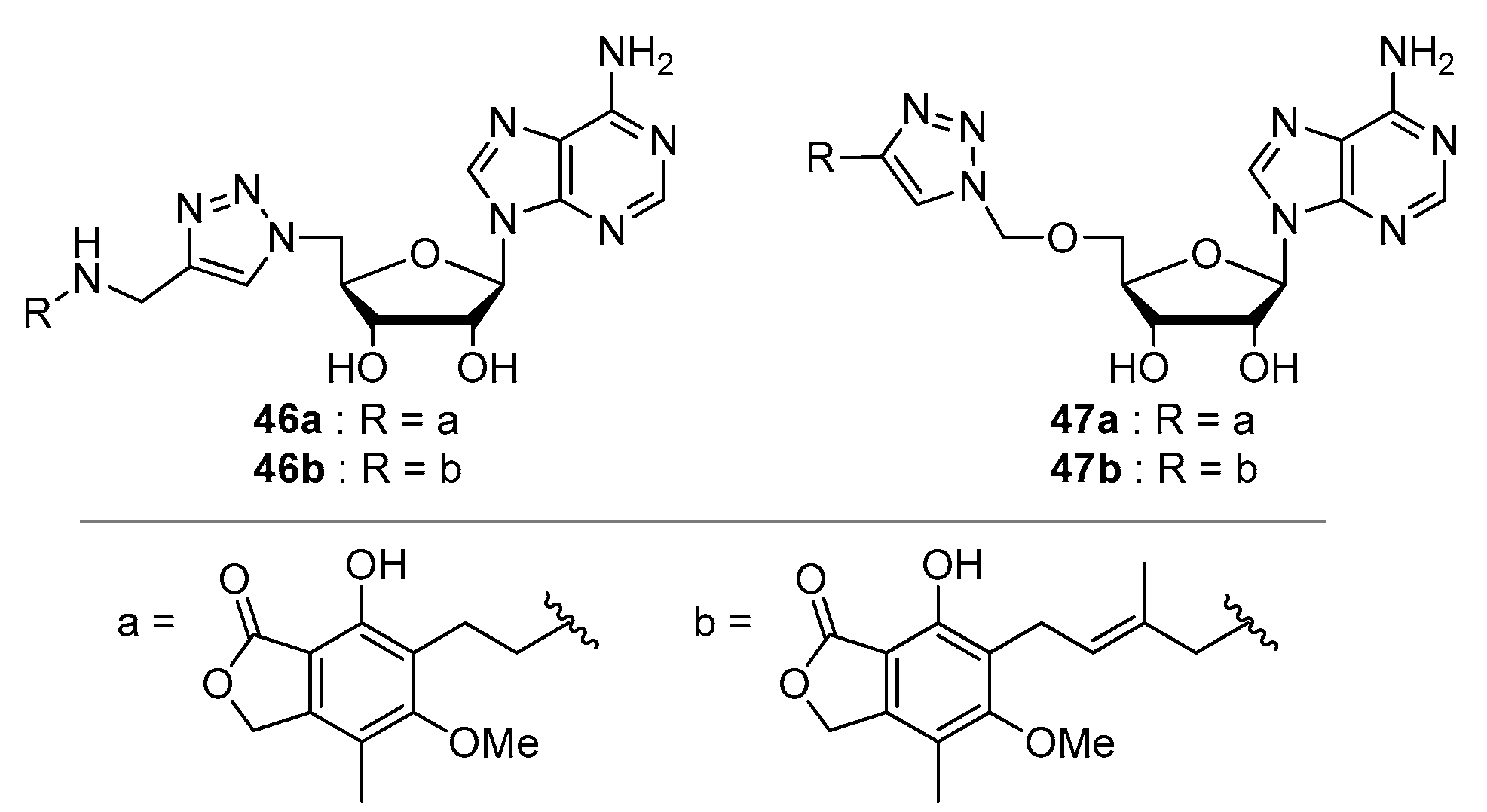






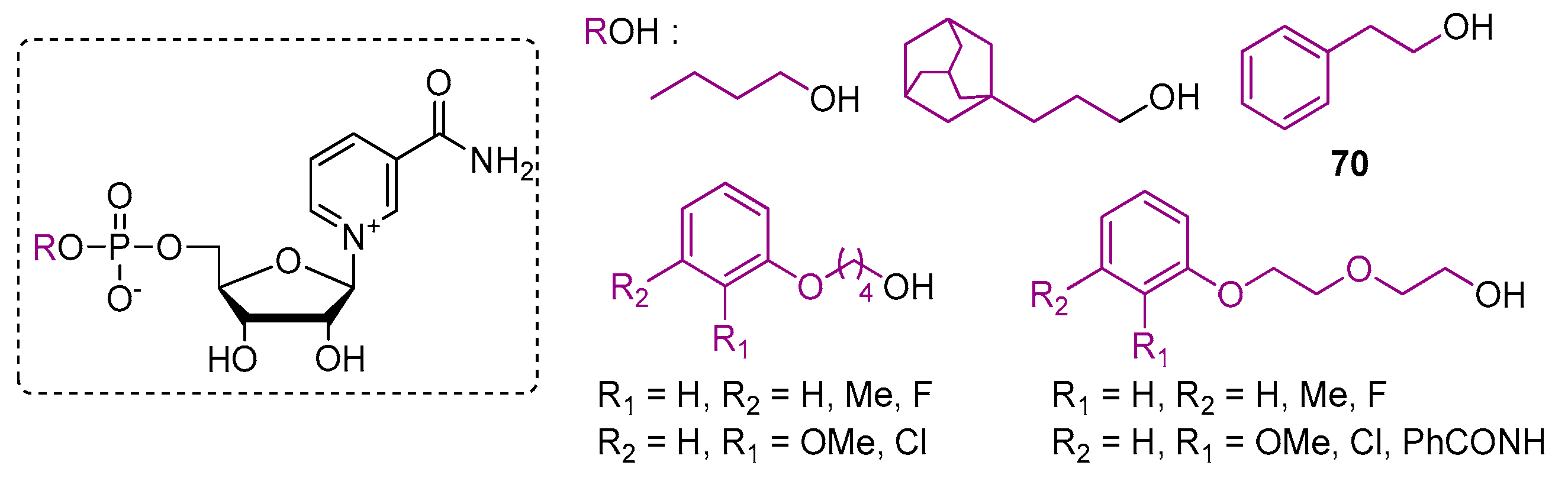


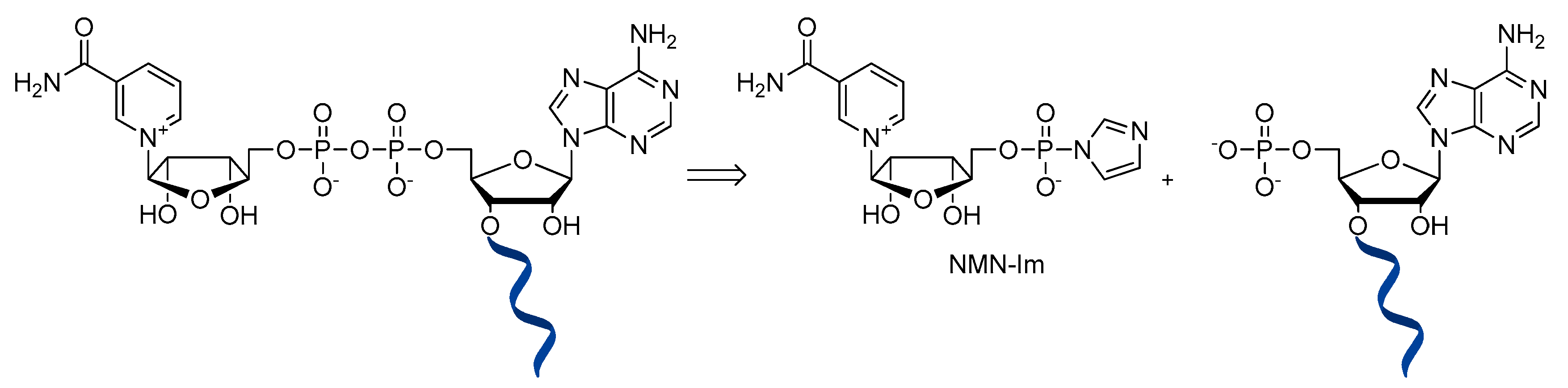

{kind=link}
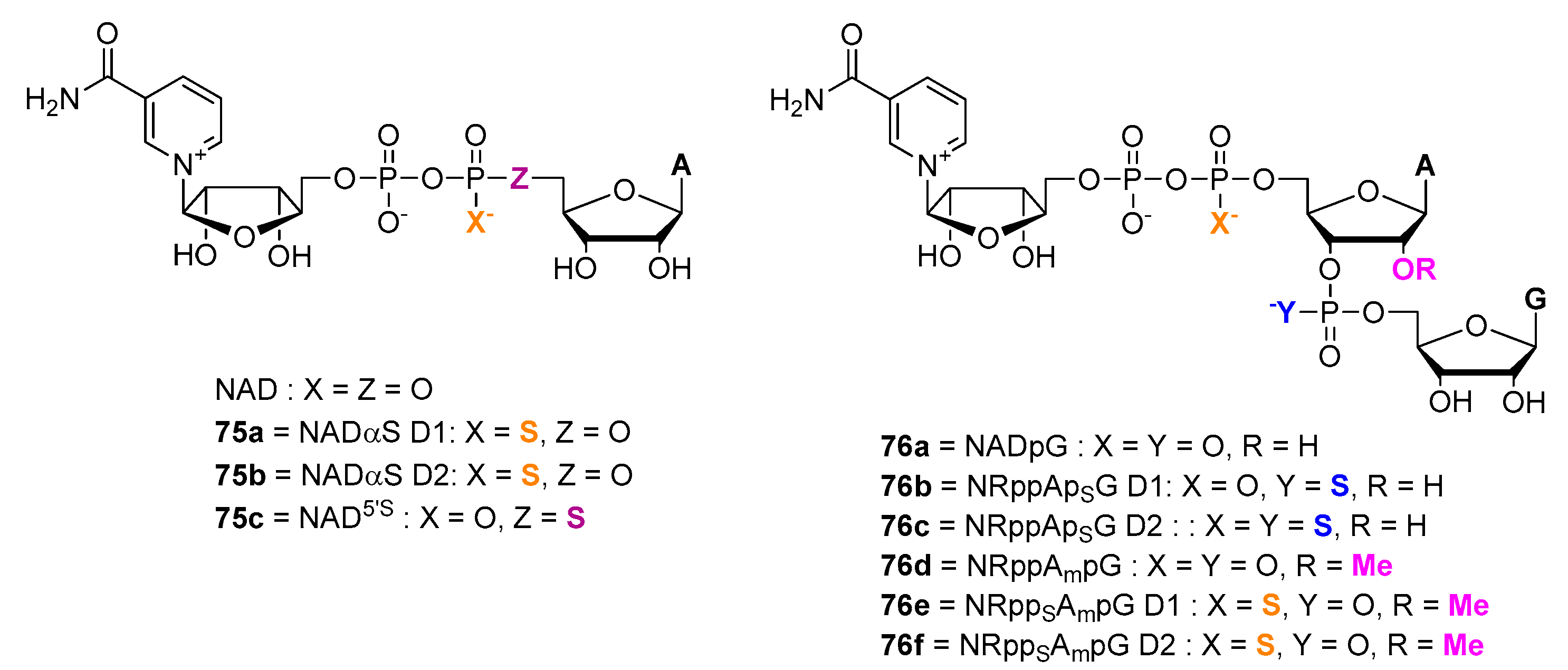
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
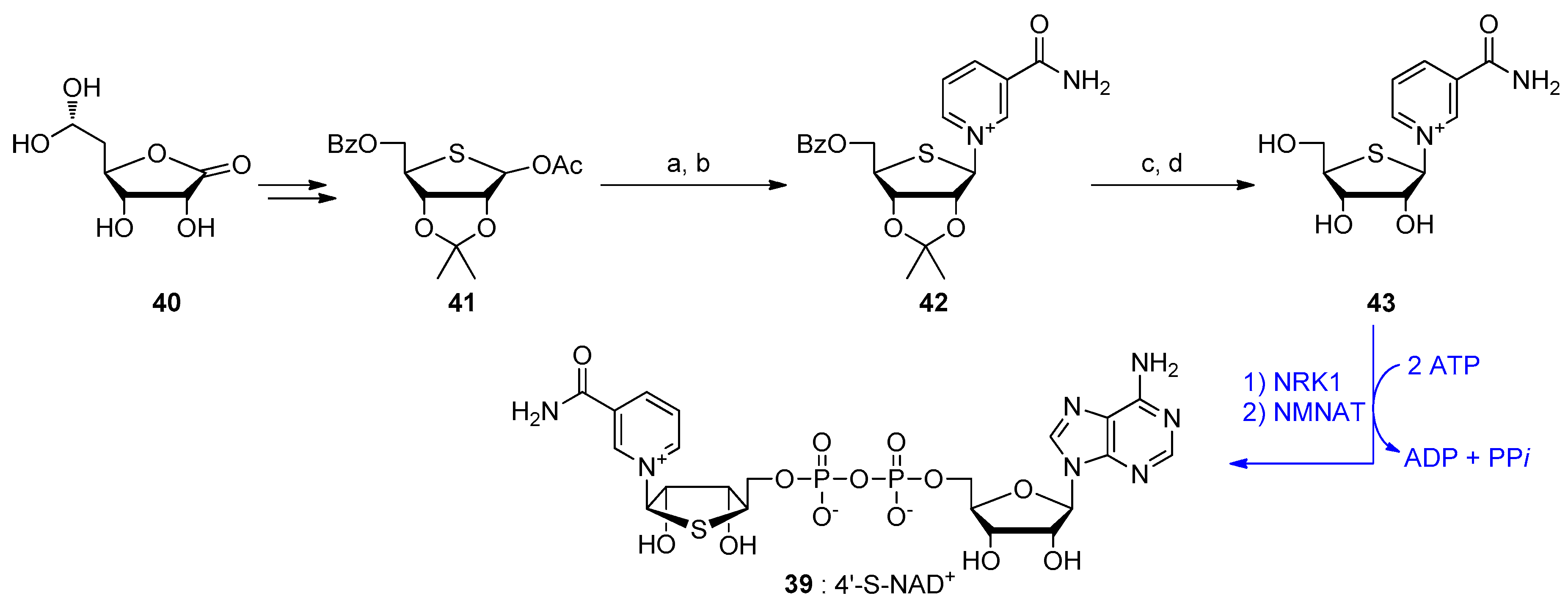{kind=link}
{kind=link}
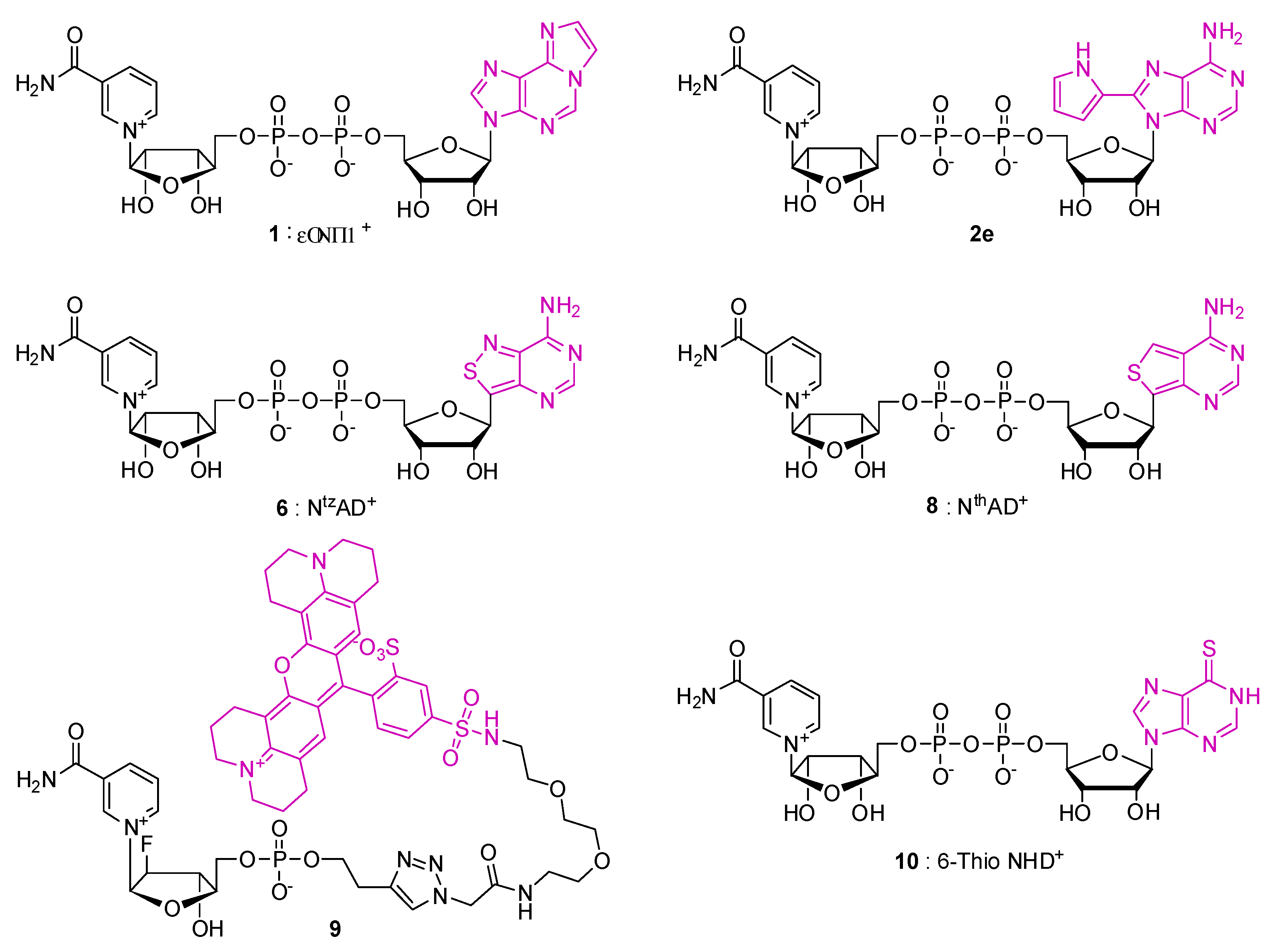
| Compound | Modified Nucleoside | λex a | λem a (Φ) | Enzymatic Activities Tested with NAD Analog b | Refs. |
|---|---|---|---|---|---|
| NAD+ | 259 | - | n/a | [31] | |
| NADH | 340 | 460 (0.02) | |||
| ε-NAD+ 1 | 1,N6-Ethenoadenosine (ε-A) | 300 | 415 | Glutamate dehydrogenase (+) NAD glycohydrolase (8%) NAD pyrophosphatase (54%) Alcohol dehydrogenase (3%) PARP (-) | [40,41,42] |
| 8-(Pyr)NAD+ 2e | 8-(2-pyrrolyl)adenosine | 300 | 410 (0.005) | NAD glycohydrolase (+) nucleotide pyrophosphatase (+) ADPR cyclase/cADPR hydrolase (+) | [32] |
| NtzAD+ 6 | isothiazolo[4,3-d]pyrimidine-riboside (tzA) | 336 | 411 (0.038) | Alcohol dehydrogenase (+) NAD glycohydrolase (+) Mono-ADP-ribose transferase (ART 5c/CTAd) (+) NAD+ kinaseEnzymatic synthesis from tzATP by NMNAT (+) PARP (+/-) | [33,38,39] |
| NtzADH | 336 | 412 (0.015) | Lactate dehydrogenase (+) NAD glycohydrolase (+) | ||
| NthAD+ 8 | thieno[3,4-d]pyrimidine-riboside (thA) | 341 | 431 (0.071) | Enzymatic synthesis from thATP by NMNAT (+) Alcohol dehydrogenase (+) NADase (+) Mono-ADP-ribose transferase (CTA) (+) PARP (-) | [39] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Depaix, A.; Kowalska, J. NAD Analogs in Aid of Chemical Biology and Medicinal Chemistry. Molecules 2019, 24, 4187. https://doi.org/10.3390/molecules24224187
Depaix A, Kowalska J. NAD Analogs in Aid of Chemical Biology and Medicinal Chemistry. Molecules. 2019; 24(22):4187. https://doi.org/10.3390/molecules24224187
Chicago/Turabian StyleDepaix, Anais, and Joanna Kowalska. 2019. "NAD Analogs in Aid of Chemical Biology and Medicinal Chemistry" Molecules 24, no. 22: 4187. https://doi.org/10.3390/molecules24224187
APA StyleDepaix, A., & Kowalska, J. (2019). NAD Analogs in Aid of Chemical Biology and Medicinal Chemistry. Molecules, 24(22), 4187. https://doi.org/10.3390/molecules24224187